

Abstract
The present studies sought to further understand how the anti-folate pemetrexed and the multi-kinase inhibitor sorafenib interact to kill tumor cells. Sorafenib activated SRC, and via SRC the drug combination activated ERK1/2. Expression of dominant negative SRC or dominant negative MEK1 abolished drug-induced ERK1/2 activation, together with drug-induced autophagy, acidic lysosome formation, and tumor cell killing. Protein phosphatase 2A is an important regulator of the ERK1/2 pathway. Fulvestrant resistant MCF7 cells expressed higher levels of the PP2A inhibitor SET/I2PP2A, had lower endogenous PP2A activity, and had elevated basal ERK1/2 activity compared with their estrogen dependent counterparts. Overexpression of I2PP2A blocked drug-induced activation of ERK1/2 and tumor cell killing. PP2A can be directly activated by ceramide and SET/I2PP2A can be inhibited by ceramide. Inhibition of the de novo ceramide synthase pathway blocked drug-induced ceramide generation, PP2A activation and tumor cell killing. Collectively these findings demonstrate that ERK1/2 plays an essential role downstream of SRC in pemetrexed and sorafenib lethality and that PP2A plays an important role in regulating this process.
Keywords: ERK, I2PP2A, PP2A, SRC, autophagy, ceramide, pemetrexed, sorafenib
Introduction
Chemotherapeutic targeting of individual cellular signaling pathways can be an effective method of cancer treatment, yet there are obstacles surrounding this approach. One example, resulting from prolonged administration of anti-cancer agents, is acquired drug resistance.1 One means of circumventing this obstacle is through use of multiple/combination chemotherapies with the intention to simultaneously inhibit many cell survival pathways at once.
There are two types of programmed cell death: Type I, apoptosis, is an energy dependent process that can occur via death receptor signaling, i.e., the extrinsic pathway, or through mitochondrial dysfunction, i.e., the intrinsic pathway.2-5 The second type of programmed cell death, type II, occurs via autophagy. Autophagy is a naturally occurring event that takes place in cells as a means of recycling proteins back into their amino acid building block forms. Autophagy can be induced by intracellular insults such as elevated levels of ROS, increases in intracellular Ca2+ levels, increases in ceramide levels, AMP accumulation, or energy and nutrient deprivation (see ref. 6 and references therein). Activation of autophagy in these circumstances can occur as a protective effort to restore homeostasis within the cell. However, prolonged activation of this signal without restoration of the cell to an unstressed state, has the potential to lead to autophagy becoming toxic.6-10
The anti-folate drug pemetrexed (abbreviated in this manuscript as “PTX”) (ALIMTA®) was FDA-approved for the treatment of advanced and metastatic non-small cell lung cancer (NSCLC) in 2004. It was developed as an inhibitor of thymidylate synthase (TS), however based on continued an anti-proliferative effect on cells in vitro in the presence of exogenous thymidine, preventing the cytotoxic effects of TS inhibition, it became apparent that pemetrexed has at least one secondary target.11-14 Subsequently, the folate-dependent enzyme, aminoimidazole-carboxamide ribonucleotide formyl-transferase (AICART), was shown to be a secondary target for pemetrexed.11,12 Inhibition of AICART results in elevated levels of ZMP. Intracellular accumulation of ZMP leads to the activation of AMP-activated protein kinase (AMPK) and downstream inhibition of mammalian target of rapamycin (mTOR).11,12,15 Inhibition of mTOR stimulates autophagy by enabling the association of ATG proteins required to initiate formation of the autophagosome.6-10 We have demonstrated previously that pemetrexed induces tumor cell death and autophagy in a dose-dependent fashion.6
Receptor tyrosine kinases (RTKs) are important regulators of a variety of cellular signaling cascades, e.g., ERK1/2, which stimulate cell growth and survival.16,17 For this reason, many diverse pharmacological inhibitors have been developed to target RTKs thereby blocking activation of signal transduction pathways, in turn, to promote growth inhibitory and pro-apoptotic effects in cells. Upon activation of an RTK, a conformational change occurs in the receptor’s intracellular domains enabling receptor trans-phosphorylation which in turn provides a docking site(s) for proteins with SRC homology 2 (SH2) domains.16-18 Among the many adaptor proteins that bind to the SH2 domains, and of particular interest to anti-cancer therapies, is the SRC family of non-receptor tyrosine kinases.19-21 Signaling by SRC has been linked to many oncogenic processes including tumor cell growth and invasion. Our group has shown that inhibition of PDGFRβ by sorafenib in human hepatoma cells resulted in enhanced phosphorylation of SRC Y416 and SRC-dependent activation of the CD95 death receptor, correlating with an induction of autophagy.22
Sorafenib (Bay 43–9006, Nexavar®) was FDA-approved for treatment of advanced renal carcinoma in 2005 and then for hepatic carcinoma in 2007. Sorafenib was originally developed as an inhibitor of RAF-1, an upstream activator of the ERK1/2 pathway, though recently has also been shown in some circumstances to paradoxically activate ERK1/2 (e.g., Rose et al., 2010).23 ERK1/2 signaling is generally thought to act as a protective signal against the toxic effects of therapeutic agents; agents that activate apoptotic pathways. Sorafenib was subsequently found to inhibit multiple RTKs such as platelet-derived growth factor (PDGFR), vascular endothelial growth factor receptors 1 and 2 (VEGFR1 and VEGFR2), c-Kit and FLT3.24-27 (and refs. therein). We have shown that in combination with pemetrexed, sorafenib interacted synergistically to kill cells of multiple cancer cell types including breast, brain, lung, and liver. In these tumor cell models, combination treatment resulted in a profound enhancement of autophagy and autophagy-dependent tumor cell killing.6
The present studies sought to define in further detail how pemetrexed (PTX) and sorafenib (SOR) interact to kill mammary carcinoma cells. More specifically, based on prior data linking sorafenib, PDGFRβ and SRC, we initially determined whether drug-induced SRC activation plays any role in the regulation of ERK1/2 and in pemetrexed/sorafenib toxicity. Subsequently we defined whether protein phosphatases, that inactivate ERK1/2, also play a role in drug combination toxicity.
Results
In mammary carcinoma cells the drug combination of sorafenib (SOR) and pemetrexed (PTX) increased levels of early autophagic vesicles, as judged using GFP-LC3 and the formation of punctae; an effect that was reduced upon knockdown of Beclin1 (Fig. 1A, data not shown). Upon treatment with PTX and SOR, carcinoma cells also demonstrated a time dependent enhancement, downstream of GFP-LC3 vesicularization, in lysosomal acidification (AVOs), arguing that autophagic flux was being stimulated (Fig. 1B). This correlated with an increase in LC3-II levels and a decrease in p62 levels (Fig. 1B, upper blot). Incubation of cells with 3-methyl adenine or chloroquine reduced the increase in lysotracker red staining (data not shown). The induction of lysosomal acidification was also blocked by knockdown of Beclin1 (Fig. 1C and D). Knockdown of Beclin 1 expression resulted in a reduction in drug-combination toxicity (Fig. 1E). In further agreement with lysosomal acidification playing a role in PTX and SOR lethality, treatment of cells with ammonium chloride, which neutralizes acidified endosomes, suppressed the induction of drug combination toxicity (data not shown).

Figure 1A-D. Pemetrexed and sorafenib induce autophagy, AVOs, and tumor cell killing that is suppressed by knockdown of Beclin1. (A) BT474 and MCF7F cells were transfected with a plasmid to express LC3-GFP in parallel with scrambled siRNA (siSCR) or to knockdown Beclin1 (siBeclin1). After 24h cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Twenty-four h later cells were examined under a fluorescent microscope. The mean number of LC3-GFP vesicles per cell was determined (n = 3, ± SEM) # p < 0.05 less than corresponding value in CMV transfected cells. (B) BT474 and MCF7F cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM) and 6h-24h later portions of cells were treated with lysotracker red to visualize acidic endosomes (AVOs). Images are a representative (n = 3). Upper blot: MCF7F cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Cells were isolated 12h later and immunoblotting performed for LC3-II and p62. (C) MCF7F cells were transfected with scrambled siRNA (siSCR) or to knockdown Beclin1. After 24h cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Six h and 24h later portions of cells were treated with lysotracker red. Images are representative (n = 3). (D) BT474 cells were transfected with scrambled siRNA (siSCR) or to knockdown Beclin1. Twenty-four h later cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Six h and 24h later portions of cells were treated with lysotracker red. Images are representative (n = 3).
Figure 1E.

BT474 and MCF7F cells were transfected to knockdown Beclin1 were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Cells were isolated 24h later and viability determined by trypan blue exclusion (n = 3, ± SEM) # p < 0.05 less than corresponding value in CMV transfected cells.
Sorafenib (SOR) is a multi-kinase inhibitor whose biologic actions have often been tied to inhibition of class III receptor tyrosine kinases and treatment of cells with SOR (3 μM) decreased PDGFRβ tyrosine phosphorylation by 88% ± 5%.6 During our studies examining PDGFRβ we also surveyed the expression and activity of other kinases whose functions can be modulated by this receptor, most notably SRC family non-receptor tyrosine kinases. Knockdown of PDGFRβ increased SRC Y416 phosphorylation almost 2-fold (Fig. 2A, upper left). Expression of a dominant negative form of SRC reduced drug-induced formation of GFP-LC3 punctate vesicles (Fig. 2A and B). And, expression of dominant negative SRC suppressed drug combination toxicity (Fig. 2C).
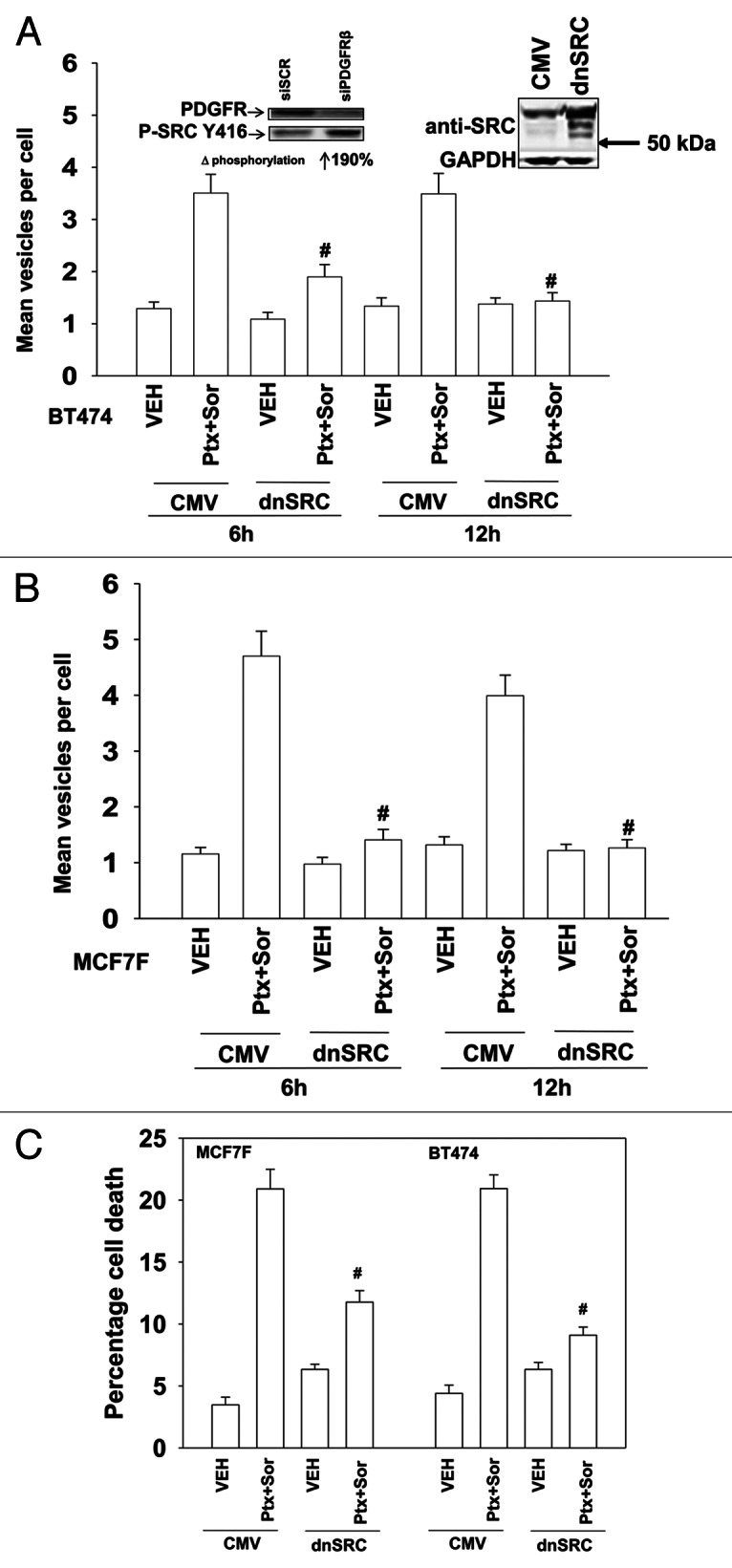
Figure 2. SRC signaling plays an essential role in pemetrexed and sorafenib toxicity. (A) and (B) BT474 and MCF7F cells were transfected to express LC3-GFP and with either empty vector (CMV) or with a plasmid to express dominant negative SRC (dnSRC). After 24h cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM), and 6h and 24h later cells were examined under a fluorescent microscope. The mean number of LC3-GFP vesicles per cell was determined (n = 3, ± SEM) # p < 0.05 less than corresponding value in CMV transfected cells. Upper panels: Knockdown of PDGFRβ increases SRC Y416 phosphorylation; increased total expression of c-SRC in cells expressing dominant negative c-SRC. (C) MCF7F and BT474 cells were transfected with either empty vector (CMV) or with a plasmid to express dominant negative SRC. After 24h cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Cells were isolated 24h later and viability determined by trypan blue exclusion (n = 3, ± SEM) # p < 0.05 less than corresponding value in CMV transfected cells.
There are multiple effector signaling pathways downstream of SRC family kinases, in particular the ERK1/2 pathway. Treatment of cells with PTX and SOR caused activation of ERK1/2 that was blocked by expression of dominant negative SRC (Fig. 3A). Expression of dominant negative MEK1 suppressed PTX and SOR lethality (Fig. 3B). Based on these cell viability findings, and data in Figures 1 and 2, we determined whether the ERK1/2 pathway was also regulating drug-induced autophagy. Expression of dominant negative MEK1 reduced the ability of PTX and SOR to induce formation of early autophagic vesicles and late acidic endosomes (Fig. 3C and D). Collectively our data argue that a pathway exists from PDGFRβ to SRC to ERK1/2 in the regulation of autophagy and tumor cell survival following PTX and SOR treatment.

Figure 3. SRC-MEK signaling promotes drug combination cell killing. (A) MCF7F cells were transfected with either empty vector (CMV) or with a plasmid to express dominant negative SRC. After 24h cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Cells were isolated 24h later and the levels of ERK1/2 phosphorylation determined (n = 3). (B) MCF7, MCF7F and BT474 cells were infected with either empty vector (CMV) virus or with a virus to express dominant negative MEK1. After 24h cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Cells were isolated 24h later and viability determined by trypan blue exclusion (n = 3, ± SEM) # p < 0.05 less than corresponding value in CMV transfected cells. (C) MCF7F cells were transfected to express LC3-GFP and infected with either empty vector (CMV) virus or with a virus to express dominant negative MEK1. After 24h cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Cells were examined 12h later under a fluorescent microscope. The mean number of LC3-GFP vesicles per cell was determined (n = 3, ± SEM) # p < 0.05 differential value than corresponding value in CMV transfected cells. (D) MCF7F and BT474 cells were infected with either empty vector (CMV) virus or with a virus to express dominant negative MEK1. After 24h cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Twenty-four h later portions of cells were treated with lysotracker red. Images are representative (n = 3).
Signaling through the ERK1/2 pathway is controlled at multiple levels not only by protein kinases but also by the actions of protein phosphatases, in particular protein phosphatase 2A (PP2A).29 A reduction in PP2A activity has been linked to increased tumorigenic potential and resistance to chemotherapy.30 Based on our prior data showing that fulvestrant resistant MCF7 cells (MCF7F) are more aggressive than parental MCF7 cells, we next determined whether PP2A activity levels and the expression of an endogenous inhibitor of PP2A, SET/I2PP2A influenced the ERK1/2-pathway -dependent regulation of pemetrexed and sorafenib toxicity.6,31 PP2A activity was lower in MCF7F compared with parental MCF7 cells that correlated with the MCF7F cells having higher basal ERK1/2 activity and higher SET/I2PP2A levels (Fig. 4A and B). Overexpression of I2PP2A in MCF7 cells increased basal levels of ERK1/2 activity and also suppressed the ability of the drug combination to cause activation of ERK1/2 above basal levels; indeed in the presence of I2PP2A drug treatment reduced ERK1/2 activity (Fig. 4B). We next determined the impact of I2PP2A expression on drug lethality. In agreement with the hypothesis that drug-induced activation of ERK1/2, rather not with a hypothesis that simply altered basal levels of ERK1/2 was responsible for drug lethality, expression of I2PP2A suppressed drug-induced killing (Fig. 4C). Thus drug-induced activation of ERK1/2 is required, not simply elevated basal levels of activity. These findings were also reflected in reduced GFP-LC3 vesicle formation (data not shown). Collectively these findings argue that PP2A activity status, as judged by I2PP2A expression, regulates ERK1/2 signaling that in turn regulates pemetrexed and sorafenib lethality in breast cancer cells.

Figure 4. SET/I2PP2A modulates the response of cells to pemetrexed and sorafenib exposure. (A) MCF7 and MCF7F cells were isolated and PP2A activity determined. Upper blot: MCF7 and MCF7F cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Cells were isolated 24 h later and the expression of I2PP2A determined. Changes in expression were normalized to GAPDH levels and presented as the –Fold change in I2PP2A levels (n = 3 ± SEM). * p < 0.05 greater than corresponding vehicle control; % p < 0.05 greater than corresponding value in MCF7 cells. (B) Upper blot: MCF7 and MCF7F cells were isolated and immunoblotting performed to determine the phosphorylation of ERK1/2. Changes in P-ERK1/2 levels were normalized to total ERK2 and presented as the –Fold change in P-ERK1/2 levels (n = 3 ± SEM). Lower blot: MCF7F cells were transfected with empty vector control (CMV) or a plasmid to express I2PP2A. Twenty-four h later cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Cells were isolated 24h later and the phosphorylation of ERK1/2 determined. Changes in levels were normalized to total ERK2 levels and presented as the -fold change in P-ERK1/2 levels (n = 3 ± SEM). * p < 0.05 greater than corresponding vehicle control; % p < 0.05 greater than corresponding value in vector control cells. (C) MCF7F cells were transfected with either an empty vector control (CMV) or plasmid to express I2PP2A. Twenty-four h later cells were treated with Vehicle (VEH), pemetrexed (PTX, 1 μM) and/or sorafenib (SOR, 3 μM). Cells were isolated 24h later and viability determined by trypan blue exclusion (n = 3, ± SEM).
Ceramide generation has been linked by some groups to the induction of autophagy, independently of any correlation to PP2A function.32 The protein SET/I2PP2A can bind to the lipid ceramide and as a result no-longer inhibits PP2A.31 PP2A can also be activated by ceramide independently of SET/I2PP2A.33 Thus, as our data in Figure 4 argued that PP2A activity regulated autophagy and cell viability, we next determined whether PTX and SOR treatment acts to promote PP2A activation and cell killing through a ceramide-dependent pathway. Knockdown of ceramide synthase 6 (LASS6) prevented PTX and SOR from activating PP2A (Fig. 5A). Treatment of cells with PTX and SOR increased C16:0; C24:1 and C24:0 dihydro-ceramide levels (Fig. 5B). Knockdown of LASS6 suppressed the production of C16 dihydro-ceramide but did not block the drug-stimulated levels of C24:1 or C24:0 dihydro-ceramides (Fig. 5B). Knockdown of LASS6 or treatment with an inhibitor of the de novo ceramide synthesis pathway, myriocin, suppressed PTX and SOR -induced autophagic GFP-LC3 vesicle formation (Fig. 5C, not shown). Inhibition of the de novo ceramide synthase pathway, via knockdown of LASS6 or use of myriocin, also suppressed drug combination lethality (Fig. 5D). These data argue that ceramide-dependent effects on PP2A activity, that in turn regulates ERK1/2, play a central role in regulation in the toxicity of this drug combination.

Figure 5. Ceramide-dependent regulation of PP2A function plays an important role in the toxicity of pemetrexed and sorafenib treatment. (A) MCF7F and BT474 cells were transfected with scrambled siRNA or an siRNA to knockdown LASS6 expression. Twenty-four h later cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Cells were isolated 24 h later and PP2A activity determined. Data are normalized, with vehicle control + vector control cell PP2A activity equal to 1.00 (n = 3 ± SEM). (B) BT474 cells were transfected with a scrambled siRNA or an siRNA to knockdown LASS6 expression. Twenty-four h later cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Cells were isolated after 12 h and prepared for mass spectrometric assessment of dihydro (DH)-ceramide levels as described in Methods (n = 2, 6 independent samples total, ± SEM). (C) BT474 cells were transfected to express LC3-GFP and with siRNA scrambled or to knockdown LASS6 expression. Twenty-four h later cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Twelve h and 24h later cells were examined under a fluorescent microscope. The mean number of LC3-GFP vesicles per cell was determined (n = 3, ± SEM). (D) BT474 cells were transfected with scrambled siRNA or an siRNA to knockdown LASS6 expression. Twenty-four h later cells were treated with vehicle or myriocin (MYR, 1 μM), as indicated. Thirty minutes later cells were treated with Vehicle (VEH) or pemetrexed (PTX, 1 μM) and sorafenib (SOR, 3 μM). Cells were isolated 24h later and viability determined by trypan blue exclusion (n = 3, ± SEM).
Discussion
The anti-folate pemetrexed elevates ZMP levels, thereby activating AMPK and inactivating mTOR, with an associated increase in autophagy. Sorafenib is a multi-kinase inhibitor that we, as well as others, have shown can stimulate autophagy through ER stress signaling and other less well defined mechanisms. The present studies sought to define the molecular mechanisms by which pemetrexed and sorafenib interact to kill tumor cells. Pemetrexed and sorafenib exposure caused an increase in the formation of early autophagic vesicles as well as late auto-lysosomes (AVOs, acidic endosomes). As previously noted by ourselves and other groups, autophagy can either act to protect cells from a toxic stress or can facilitate the toxicity of the stress all of which is based on the stimulus, its duration and the cell type being examined. In our system the formation of both autophagic vesicles and auto-lysosomes was dependent on expression of Beclin 1; and, knockdown of Beclin 1 or neutralization of acidic cellular compartments protected cells from drug combination lethality.
Sorafenib is an inhibitor of multiple kinases, including class III receptor tyrosine kinases such as PDGFR. Previously we had shown in GI tumor cells that sorafenib, via inhibition of PDGFRβ, caused activation of SRC family kinases that in turn phosphorylated and activated the death receptor CD95.24-27 In our present studies, sorafenib caused activation of SRC family kinases, and SRC signaling, in combination with pemetrexed treatment was essential for: the formation of autophagic vesicles; for activation of ERK1/2; and for drug-induced cell death. Drug combination-induced ERK1/2 signaling, downstream of SRC, was also found to be essential for increased formation of autophagic vesicles and for drug-induced cell death. The mechanisms by which SRC or ERK1/2 signaling promote autophagy are not fully understood, though there are multiple manuscripts showing ERK1/2 signaling stimulates autophagic vesicle formation with diverse cellular outcomes.34-37 It has been reported that AMPK activation, downstream of the pemetrexed target AICART in tumor cells can be enhanced by c-SRC signaling, providing a possible signaling pathway overlap between sorafenib and pemetrexed.38 It is also known that autophagy is regulated by the reversible phosphorylation of multiple ATG family genes and it is probable that ERK1/2 acts to alter ATG protein phosphorylation that in turn facilitates an increase in vesicle formation.
Studies examining the regulation of cell signaling processes by chemotherapeutic agents most often examine the actions of protein kinases. Fewer studies explore the actions of protein phosphatases, enzymes that have approximately an order of magnitude greater specific activity than kinases. PP2A is an important regulator of the ERK1/2 pathway and it is known that more advanced tumors can have lower PP2A activity. As such we determined whether the inhibitor of PP2A, SET/I2PP2A, and PP2A activity could play a role in the ERK1/2 dependent effects of pemetrexed and sorafenib treatment. MCF7F cells had ~50% lower PP2A activity as compared with parental MCF7 cells that correlated with MCF7F cells having a ~50% increase in SET/I2PP2A expression and having ~2-fold higher basal ERK1/2 activity. Pemetrexed and sorafenib did not activate ERK1/2 in MCF7F cells that overexpressed I2PP2A and in fact the drug treatment reduced ERK1/2 activity in these cells: thus a lack of ERK1/2 activation, rather than changes in basal activity levels, which was associated with a lack of drug combination lethality. More significantly, overexpression of SET/I2PP2A suppressed PP2A activity, blocked drug –induced autophagy and reduced tumor cell killing.
Forms of the bio-active lipid ceramide have been shown by a number of investigators to regulate PP2A activity.39-41 Some studies have argued for ceramide inhibiting SET/I2PP2A in a direct fashion and thereby activating PP2A while others have argued for ceramide directly activating PP2A through catalytic subunit binding.31,39-41 Treatment of cells with sorafenib and pemetrexed increased the levels of dihydro-ceramides. Knockdown of ceramide synthase 6 (LASS6) suppressed the drug-stimulated increase in C16:0 dihydro-ceramide but not the increases in other dihydro-ceramides. Knockdown of LASS6 or treatment with the small molecule inhibitor of the de novo ceramide synthesis pathway, myriocin, suppressed drug-induced autophagy and tumor cell killing. At present, there are no reports linking pemetrexed treatment to increases in dihydro-ceramide levels. Several reports from our laboratory have shown that sorafenib, in combination with histone deacetylase inhibitors, can increase dihydro-ceramide levels in a LASS6 dependent fashion, however the precise molecular mechanism(s) by which this ceramide dependent signaling pathway is stimulated still remain elusive.
At face value our findings with respect to changes in ceramide levels were a little surprising based on the biological responses (autophagy, PP2A activity, cell death) and the relatively low alterations in the amount of dihydro-ceramide being generated and the impact of LASS6 knockdown on only C16:0 dihydro-ceramide. It is possible that all forms of dihydro-ceramide whose levels are stimulated may play a role in autophagy/cell killing process in our system and that a lack of just one ceramide moiety, dh-C16:0, results in an inability of cells to stimulate autophagy and undergo cell death. The precise targets of dh-C16:0 ceramide, including PP2A and SET/I2PP2A will require studies beyond the scope of this manuscript.
In conclusion, pemetrexed and sorafenib cause a toxic form of autophagy associated with acidic endosome formation. Downstream from SRC family kinases activation of ERK1/2 plays a central role in the induction of autophagic and acidic endosome vesicles. Ceramide generation by the de novo ceramide synthesis pathway regulates PP2A activity and activation of PP2A plays a central role in increased levels of autophagy and tumor cell killing. Additional mechanistic studies will be required to understand how the drug combination increases dihydro-ceramide levels and the precise mode by which ceramide activates PP2A.
Materials and Methods
Materials
Sorafenib tosylate was purchased from Eton Bioscience Inc. Pemetrexed was purchased from LC Laboratories. Trypsin-EDTA, DMEM, RPMI, penicillin-streptomycin were purchased from GIBCOBRL (GIBCOBRL Life Technologies). All cells, except MCF7 and MCF7F, were purchased from the ATCC and were not further validated beyond that claimed by ATCC. Cells were re-purchased every ~6 mo. MCF7 cells were obtained from their primary source (U. Michigan, Ann Arbor). MCF7F cells were generated as noted in [Fan et al. (2006) Cancer Res. 66:11954–66]. Plasmids were purchased from Addgene. Commercially available validated short hairpin RNA molecules to knockdown RNA/protein levels were from Qiagen or were supplied by collaborators. Reagents and performance of experimental procedures were described in references 6, 11, 12, 24, 25 and 26.
Culture and in vitro exposure of cells to drugs
All cell lines were cultured at 37°C (5% (v/v CO2) in vitro using RPMI supplemented with dialyzed 5% (v/v) fetal calf serum and 10% (v/v) Non-essential amino acids. Cells growing in “complete” fetal calf serum that contains thymidine were gradually weaned into dialyzed serum lacking thymidine over 2 weeks and were then used for experimental analyses for the following 3 weeks before discarding. Cells were re-isolated in thymidine-less media as required. For short-term cell killing assays, immunoblotting studies, cells were plated at a density of 3 × 103 per cm2 (~2 × 105 cells per well of a 12-well plate) and 48h after plating treated with various drugs, as indicated. In vitro pemetrexed and sorafenib treatments were from 100 mM stock solutions of each drug and the maximal concentration of Vehicle (DMSO) in media was 0.02% (v/v). Cells were not cultured in reduced serum media during any study in this manuscript.
In vitro cell treatments, microscopy, SDS-PAGE and western blot analysis
For in vitro analyses of short-term cell death effects, cells were treated with Vehicle or pemetrexed/sorafenib for the indicated times in the figure legends. For apoptosis assays, cells were isolated at the indicated times, and subjected to trypan blue cell viability assay by counting in a light microscope.
For SDS PAGE and immunoblotting, cells were plated at 5 × 105 cells/cm2 and treated with drugs at the indicated concentrations and after the indicated time of treatment, lysed in whole-cell lysis buffer (0.5 M TRIS-HCl, pH 6.8, 2%SDS, 10% glycerol, 1% β-mercaptoethanol, 0.02% bromophenol blue), and the samples were boiled for 30 min. The boiled samples were loaded onto 10–14% SDS-PAGE and electrophoresis was run overnight (10–100 μg/lane based on the gel size. Proteins were electrophoretically transferred onto 0.22 μm nitrocellulose, and immunoblotted with various primary antibodies against different proteins. All immunoblots were visualized using an Odyssey Infrared Imager. For presentation, immunoblots were digitally assessed using the provided Odyssey Imager software (the data sets presented are the -fold increase ± SEM (n = 3) in expression of the indicated protein compared with GAPDH loading control; for phospho-proteins the -fold increase ± SEM (at least n = 3) is normalized to the total protein level of the indicated kinase or substrate). Errors are not numerically shown due to space restrictions in the figure panels; any indicated significant differences between the expression/phosphorylation levels of proteins are indicated by an asterisk or other annotation and have a p < 0.05.
Transfection of cells with siRNA or with plasmids
For plasmids
Cells were plated as described above and 24 h after plating, transfected. For mouse embryonic fibroblasts (2–5 μg) or other cell types (0.5 μg) plasmids expressing a specific mRNA (or siRNA) or appropriate vector control plasmid DNA was diluted in 50 μl serum-free and antibiotic-free medium (1 portion for each sample). Concurrently, 2 μl Lipofectamine 2000 (Invitrogen), was diluted into 50 μl of serum-free and antibiotic-free medium (1 portion for each sample). Diluted DNA was added to the diluted Lipofectamine 2000 for each sample and incubated at room temperature for 30 min. This mixture was added to each well/dish of cells containing 200 μl serum-free and antibiotic-free medium for a total volume of 300 μl, and the cells were incubated for 4 h at 37°C. An equal volume of 2 × medium was then added to each well. Cells were incubated for 48 h, then treated with pemetrexed/sorafenib.
Transfection for siRNA
Cells were plated in 60 mm dishes from a fresh culture growing in log phase as described above, and 24 h after plating transfected. Prior to transfection, the medium was aspirated and 1 ml serum-free medium was added to each plate. For transfection, 10 nM of the annealed siRNA, the positive sense control doubled stranded siRNA targeting GAPDH or the negative control (a “scrambled” sequence with no significant homology to any known gene sequences from mouse, rat or human cell lines) were used. Ten nM siRNA (scrambled or experimental) was diluted in serum-free media. Four μl Hiperfect (Qiagen) were added to this mixture and the solution was mixed by pipetting up and down several times. This solution was incubated at room temp for 10 min, then added drop-wise to each dish. The medium in each dish was swirled gently to mix, then incubated at 37°C for 2 h. One ml of 10% (v/v) serum-containing medium was added to each plate, and cells were incubated at 37°C for 48 h before re-plating (50 × 103 cells each) onto 12-well plates. Cells were allowed to attach overnight, then treated with pemetrexed/sorafenib (0–48 h). Trypan blue exclusion/TUNEL/flow cytometry assays and SDSPAGE/immunoblotting analyses were performed at the indicated time points.
Recombinant adenoviral vectors; infection in vitro
We generated and purchased previously described recombinant adenoviruses to modulate protein expression and to express constitutively activated and dominant negative proteins (Vector Biolabs, Philadelphia, PA). Cells were infected with these adenoviruses at an approximate m.o.i. of 50. Cells were further incubated for 24 h to ensure adequate expression of transduced gene products prior to drug exposures.
Analysis of PP2A activity
Cells were plated at 2 × 105 cells in 60 mm dishes in triplicate and cultured for 24 h. Cells were then either treated with vehicle or drugs for 12 h or transfected with control siRNA or siRNA against LASS6 (Qiagen) for 36 h, then treated with vehicle or drugs. After 12 h drug treatments, cells were washed twice with cold low phosphate lysis buffer to remove residual phosphatases present in the culture media, then harvested in 500 µl of fresh low phosphate lysis buffer. Samples were prepared according to the PP2A Phosphotase Activity Assay Kit protocol (R&D Systems) using filter-sterilized buffers prepared as described in the protocol. Three separate protein amounts for each condition were immunoprecipiated with a monoclonal antibody against PP2A coupled to sepharose beads provided in the kit. After immunoprecipitation, PP2A antibody-coupled sepharose beads were washed and bound protein cleaved using the manufacturer provided reagent at equal volumes for each sample. Samples were then transferred to a 96-well plate for the remainder of the protocol. After incubation with the detection substrate, samples were evaluated using a Malachite Green protocol for determination of Absorbance at 595nm using a Vector plate reader. Triplicate values obtained for each sample type, and 3 variable protein loading amounts, were averaged and plotted as fold-change in PP2A Activity relative to that of the si-control transfected vehicle-treated condition.
Microscopy for LysoTracker Red staining
Cells were plated in in 4 chambered microscopy slides at 2 × 104 cells per well, then cultured for 24 h. Cells were either exposed to drugs or transfected with DNA and/or siRNA at 1 µg or 20 pmol per well, respectively, using 1µl of Lipofectamine 2000 per reaction. Cells were cultured in antibiotic-free media for 36 h prior to treatment with vehicle or drugs for the indicated times. LysoTracker Red dye (Invitrogen) was diluted in fresh, pre-warmed 37°C medium at a final concentration of 15 nM. Culture media was aspirated from each sample, and the diluted LysoTracker Red stain was added to each cell sample and allowed to incubate at 37°C for 30 min. Post-staining, the dye-containing media was aspirated, cell samples were gently washed 2 times with PBS, then finally resuspended in 250 µl cold PBS per well. Cell samples were anyalyzed using a Zeiss Axiovert 200 fluorescent microscope at 40 × magnification. Twenty representative images were taken of each sample and samples were performed independently in triplicate. A representative image for each condition and time point was selected for figure illustration.
Microscopy for LC3-GFP expression
Cells were transfected with a plasmid to express an LC3-GFP fusion protein, and were then cultured for 24 h. Cells were then treated with drugs, as indicated/ LC3-GFP transfected cells were visualized at the indicated time points on the Zeiss Axiovert 200 microscope using the FITC filter. A minimum of 40 cells per condition per experiment were counted.
Determination of ceramide and dihydroceramide levels
Cells were plated at 2 × 105 cells in 60mm dishes in duplicate every day for 3 d for a total of 6 repeats per condition and cultured for 24 h prior to transfection. Cells were transfected with siRNA against LASS6 (Qiagen) using Lipofectamine 2000 as described above, cultured for 36 h, then treated with vehicle or drug combination for 24 h. Cells were then harvested in 550 µl of cold PBS and 50 µl taken for lysis and protein determination using the Bradford Assay (BioRad). Protein levels were normalized based on total protein levels for each sample. Cells were processed and subjected to quantitative mass spectrometry to determine the levels of ceramide and dihydro-ceramide species.27,28
Data analysis
Comparison of the effects of various treatments was performed using ANOVA and the Student’s t test. Differences with a p value of < 0.05 were considered statistically significant. Experiments shown are the means of multiple individual points (± SEM). Statistical examination of in vivo animal survival data utilized log rank statistical analyses between the different treatment groups.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest.
Acknowledgments
Support for the present study was provided; to P.D. from PHS grants (R01-DK52825; P01-CA104177; R01-CA108325; R01-CA141703; R01-CA150214), The Jim Valvano “V” foundation, and Department of Defense Award (DAMD17–03–1-0262 and W81XWH-10–1-0009); to S.G. from PHS grants (R01-CA63753; R01-CA77141) and a Leukemia Society of America grant 6405–97. P.D. is The Universal Inc. Professor in Signal Transduction Research.
Glossary
Abbreviations:
- ERK
extracellular regulated kinase
- MEK
mitogen activated extracellular regulated kinase
- PI3K
phosphatidyl inositol 3 kinase
- ca
constitutively active
- dn
dominant negative
- SOR
sorafenib
- PTX
pemetrexed
- PTX
pemetrexed
- ER
endoplasmic reticulum
- mTOR
mammalian target of rapamycin
- MAPK
mitogen activated protein kinase
- PDGFR
platelet derived growth factor receptor
- PTEN
phosphatase and tensin homologue on chromosome ten
- ROS
reactive oxygen species
- CMV
empty vector plasmid or virus
- si
small interfering
- SCR
scrambled
- PP
protein phosphatase
- IP
immunoprecipitation
- LASS
longevity assurance gene
- Ad
adenovirus
- VEH
vehicle
- AVO
acidic vesicular organelle
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/20562
References
- 1.Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–27. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 2.Hyer ML, Samuel T, Reed JC. The FLIP-side of Fas signaling. Clin Cancer Res. 2006;12:5929–31. doi: 10.1158/1078-0432.CCR-06-2098. [DOI] [PubMed] [Google Scholar]
- 3.Grinberg M, Sarig R, Zaltsman Y, Frumkin D. Grammatikakis N, Reuvany E, Gross A. tBid homo-oligomerizes in the mitochondrial membrane to induce apoptosis. J Biol Chem. 2002;277:12237–45. doi: 10.1074/jbc.M104893200. [DOI] [PubMed] [Google Scholar]
- 4.Esposti MD. The roles of Bid. Apoptosis. 2002;7:433–40. doi: 10.1023/A:1020035124855. [DOI] [PubMed] [Google Scholar]
- 5.Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7:1166–73. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- 6.Bareford MD, Park MA, Yacoub A, Hamed HA, Tang Y, Cruickshanks N, et al. Sorafenib enhances pemetrexed cytotoxicity through an autophagy-dependent mechanism in cancer cells. Cancer Res. 2011;71:4955–67. doi: 10.1158/0008-5472.CAN-11-0898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12(Suppl 2):1509–18. doi: 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]
- 8.Ishdorj G, Li L, Gibson SB. Regulation of autophagy in hematological malignancies: role of reactive oxygen species. Leuk Lymphoma. 2012;53:26–33. doi: 10.3109/10428194.2011.604752. [DOI] [PubMed] [Google Scholar]
- 9.Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012;441:523–40. doi: 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kung CP, Budina A, Balaburski G, Bergenstock MK, Murphy M. Autophagy in tumor suppression and cancer therapy. Crit Rev Eukaryot Gene Expr. 2011;21:71–100. doi: 10.1615/critreveukargeneexpr.v21.i1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Racanelli AC, Rothbart SB, Heyer CL, Moran RG. Therapeutics by cytotoxic metabolite accumulation: pemetrexed causes ZMP accumulation, AMPK activation, and mammalian target of rapamycin inhibition. Cancer Res. 2009;69:5467–74. doi: 10.1158/0008-5472.CAN-08-4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rothbart SB, Racanelli AC, Moran RG. Pemetrexed indirectly activates the metabolic kinase AMPK in human carcinomas. Cancer Res. 2010;70:10299–309. doi: 10.1158/0008-5472.CAN-10-1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jarmuła A. Antifolate inhibitors of thymidylate synthase as anticancer drugs. Mini Rev Med Chem. 2010;10:1211–22. doi: 10.2174/13895575110091211. [DOI] [PubMed] [Google Scholar]
- 14.Fleeman N, Bagust A, McLeod C, Greenhalgh J, Boland A, Dundar Y, et al. Pemetrexed for the first-line treatment of locally advanced or metastatic non-small cell lung cancer. Health Technol Assess. 2010;14(Suppl 1):47–53. doi: 10.3310/hta14Suppl1/07. [DOI] [PubMed] [Google Scholar]
- 15.Paglin S, Lee NY, Nakar C, Fitzgerald M, Plotkin J, Deuel B, et al. Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF-7 cells. Cancer Res. 2005;65:11061–70. doi: 10.1158/0008-5472.CAN-05-1083. [DOI] [PubMed] [Google Scholar]
- 16.Zwick E, Bange J, Ullrich A. Receptor tyrosine kinase signaling as a target for cancer intervention strategies. Endocr Relat Cancer. 2001;3:161–73. doi: 10.1677/erc.0.0080161. [DOI] [PubMed] [Google Scholar]
- 17.Caraglia M, Santini D, Bronte G, Rizzo S, Sortino G, Rini GB, et al. Predicting efficacy and toxicity in the era of targeted therapy: focus on anti-EGFR and anti-VEGF molecules. Curr Drug Metab. 2011;12:944–55. doi: 10.2174/138920011798062346. [DOI] [PubMed] [Google Scholar]
- 18.Stern DF. ERBB3/HER3 and ERBB2/HER2 duet in mammary development and breast cancer. J Mammary Gland Biol Neoplasia. 2008;13:215–23. doi: 10.1007/s10911-008-9083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906–9. doi: 10.1038/sj.onc.1208160. [DOI] [PubMed] [Google Scholar]
- 20.Baker CH, Trevino JG, Summy JM, Zhang F, Caron A, Nesbit M, et al. Inhibition of PDGFR phosphorylation and Src and Akt activity by GN963 leads to therapy of human pancreatic cancer growing orthotopically in nude mice. Int J Oncol. 2006;29:125–38. [PubMed] [Google Scholar]
- 21.Lu XL, Cao X, Liu XY, Jiao BH. Recent progress of Src SH2 and SH3 inhibitors as anticancer agents. Curr Med Chem. 2010;17:1117–24. doi: 10.2174/092986710790827861. [DOI] [PubMed] [Google Scholar]
- 22.Park MA, Reinehr R, Häussinger D, Voelkel-Johnson C, Ogretmen B, Yacoub A, et al. Sorafenib activates CD95 and promotes autophagy and cell death via Src family kinases in gastrointestinal tumor cells. Mol Cancer Ther. 2010;9:2220–31. doi: 10.1158/1535-7163.MCT-10-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rose A, Grandoch M, vom Dorp F, Rübben H, Rosenkranz A, Fischer JW, et al. Stimulatory effects of the multi-kinase inhibitor sorafenib on human bladder cancer cells. Br J Pharmacol. 2010;160:1690–8. doi: 10.1111/j.1476-5381.2010.00838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yacoub A, Hamed HA, Allegood J, Mitchell C, Spiegel S, Lesniak MS, et al. PERK-dependent regulation of ceramide synthase 6 and thioredoxin play a key role in mda-7/IL-24-induced killing of primary human glioblastoma multiforme cells. Cancer Res. 2010;70:1120–9. doi: 10.1158/0008-5472.CAN-09-4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker T, Mitchell C, Park MA, Yacoub A, Graf M, Rahmani M, et al. Sorafenib and vorinostat kill colon cancer cells by CD95-dependent and -independent mechanisms. Mol Pharmacol. 2009;76:342–55. doi: 10.1124/mol.109.056523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin AP, Park MA, Mitchell C, Walker T, Rahmani M, Thorburn A, et al. BCL-2 family inhibitors enhance histone deacetylase inhibitor and sorafenib lethality via autophagy and overcome blockade of the extrinsic pathway to facilitate killing. Mol Pharmacol. 2009;76:327–41. doi: 10.1124/mol.109.056309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sauane M, Su ZZ, Dash R, Liu X, Norris JS, Sarkar D, et al. Ceramide plays a prominent role in MDA-7/IL-24-induced cancer-specific apoptosis. J Cell Physiol. 2010;222:546–55. doi: 10.1002/jcp.21969. [DOI] [PubMed] [Google Scholar]
- 28.Le Stunff H, Giussani P, Maceyka M, Lépine S, Milstien S, Spiegel S. Recycling of sphingosine is regulated by the concerted actions of sphingosine-1-phosphate phosphohydrolase 1 and sphingosine kinase 2. J Biol Chem. 2007;282:34372–80. doi: 10.1074/jbc.M703329200. [DOI] [PubMed] [Google Scholar]
- 29.Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008;22:954–65. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- 30.Kalev P, Sablina AA. Protein phosphatase 2A as a potential target for anticancer therapy. Anticancer Agents Med Chem. 2011;11:38–46. doi: 10.2174/187152011794941172. [DOI] [PubMed] [Google Scholar]
- 31.Mukhopadhyay A, Saddoughi SA, Song P, Sultan I, Ponnusamy S, Senkal CE, et al. Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEB J. 2009;23:751–63. doi: 10.1096/fj.08-120550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang Q, Rao X, Kim CY, Freiser H, Zhang Q, Jiang Z, et al. Gamma-tocotrienol induces apoptosis and autophagy in prostate cancer cells by increasing intracellular dihydrosphingosine and dihydroceramide. Int J Cancer. 2012;130:685–93. doi: 10.1002/ijc.26054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cornell TT, Hinkovska-Galcheva V, Sun L, Cai Q, Hershenson MB, Vanway S, et al. Ceramide-dependent PP2A regulation of TNFalpha-induced IL-8 production in respiratory epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;296:L849–56. doi: 10.1152/ajplung.90516.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu J, Shao CJ, Chen FR, Ng HK, Chen ZP. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro-oncol. 2010;12:328–40. doi: 10.1093/neuonc/nop005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kandouz M, Haidara K, Zhao J, Brisson ML, Batist G. The EphB2 tumor suppressor induces autophagic cell death via concomitant activation of the ERK1/2 and PI3K pathways. Cell Cycle. 2010;9:398–407. doi: 10.4161/cc.9.2.10505. [DOI] [PubMed] [Google Scholar]
- 36.Sivaprasad U, Basu A. Inhibition of ERK attenuates autophagy and potentiates tumour necrosis factor-alpha-induced cell death in MCF-7 cells. J Cell Mol Med. 2008;12:1265–71. doi: 10.1111/j.1582-4934.2008.00282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shinojima N, Yokoyama T, Kondo Y, Kondo S. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin-induced autophagy. Autophagy. 2007;3:635–7. doi: 10.4161/auto.4916. [DOI] [PubMed] [Google Scholar]
- 38.Mizrachy-Schwartz S, Cohen N, Klein S, Kravchenko-Balasha N, Levitzki A. Up-regulation of AMP-activated protein kinase in cancer cell lines is mediated through c-Src activation. J Biol Chem. 2011;286:15268–77. doi: 10.1074/jbc.M110.211813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deng X, Gao F, May WS. Protein phosphatase 2A inactivates Bcl2’s antiapoptotic function by dephosphorylation and up-regulation of Bcl2-p53 binding. Blood. 2009;113:422–8. doi: 10.1182/blood-2008-06-165134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem. 2007;282:9777–88. doi: 10.1074/jbc.M608310200. [DOI] [PubMed] [Google Scholar]
- 41.Chalfant CE, Szulc Z, Roddy P, Bielawska A, Hannun YA. The structural requirements for ceramide activation of serine-threonine protein phosphatases. J Lipid Res. 2004;45:496–506. doi: 10.1194/jlr.M300347-JLR200. [DOI] [PubMed] [Google Scholar]