

Abstract
Pathogenic non-O1/non-O139 Vibrio cholerae strains can cause sporadic outbreaks of cholera worldwide. In this study, multilocus sequence typing (MLST) of seven housekeeping genes was applied to 55 non-O1/non-O139 isolates from clinical and environmental sources. Data from five published O1 isolates and 17 genomes were also included, giving a total of 77 isolates available for analysis. There were 66 sequence types (STs), with the majority being unique, and only three clonal complexes. The V. cholerae strains can be divided into four subpopulations with evidence of recombination among the subpopulations. Subpopulations I and III contained predominantly clinical strains. PCR screening for virulence factors including Vibrio pathogenicity island (VPI), cholera toxin prophage (CTXΦ), type III secretion system (T3SS), and enterotoxin genes (rtxA and sto/stn) showed that combinations of these factors were present in the clinical isolates with 85.7% having rtxA, 51.4% T3SS, 31.4% VPI, 31.4% sto/stn (NAG-ST) and 11.4% CTXΦ. These factors were also present in environmental isolates but at a lower frequency. Five strains previously mis-identified as V. cholerae serogroups O114 to O117 were also analysed and formed a separate population with V. mimicus. The MLST scheme developed in this study provides a framework to identify sporadic cholera isolates by genetic identity.
Introduction
Vibrio cholerae is best known for its capacity to cause pandemic cholera and continues to be a major health threat, highlighted by a recent cholera outbreak in Haiti [1], [2]. With the exception of the O139 Bengal strain, which is known to be a derivative of the 7th pandemic causing epidemic cholera, all major outbreaks and pandemic cholera have been of the O1 serogoup [3], [4]. However, many other O serogroups have been reported for cases of V. cholerae associated with diarrhoea in various parts of the world and have been linked with sporadic outbreaks of cholera-like diseases [5]–[10]. Some strains are distributed globally, for example toxigenic O141 strains have been isolated from diverse geographical regions including the USA, Spain, Taiwan and India [8]. Non-O1/non-O139 sporadic outbreaks have been quite common in Bangladesh and India [7], [11]–[13] and have also occurred in Peru [5], Thailand [14] and more recently the USA [9], [10].
Symptoms of infection due to pathogenic non-O1/non-O139 V. cholerae strains range from mild gastroenteritis to violent diarrhoea which resembles cholera elicited by the pandemic O1 V. cholerae strains [15]. However, in most cases, patients suffer a less severe form of the disease than those infected by O1 V. cholerae strains [16]–[18].
The presence of cholera toxin (CTX) and toxin co-regulated pilus (TCP) is characteristic of pandemic isolates and both were initially found only in O1 toxigenic clones. However, many non-O1/non-O139 strains have been found to carry one or both virulence factors [12], [19]–[21]. Some strains such as the O141 strains carry both Vibrio Pathogenicity Island (VPI) and cholera toxin phage (CTXΦ) [8]. Other strains carry only VPI or CTXΦ, or VPI plus a partial CTX prophage [22]–[26]. However, none of the isolates studied by Sharma et al. [7] has the VPI or CTXΦ. Only two isolates, one O8 clinical and one O11 environmental, of the 34 isolates from 17 serogroups examined by Chakraborty et al. [13] contained tcpA and ctx genes. Other virulence-associated genes such as the rtxA and the sto/stn genes, which encode the repeat-like toxin (RtxA) - a cytotoxin [27] and the heat-stable enterotoxin (NAG-ST), respectively [28], [29], have also been observed in some of these strains [8], [30]–[32]. More recent studies showed that a type III secretion system (T3SS) is present in some non-O1/non-O139 isolates and appears to be an important virulence factor for these isolates. The T3SS translocates a number of T3SS effectors to the host cell such as VopF and VopE which interfere with host cell signalling pathways [33], [34]. A functional T3SS has been shown to be essential for the pathogenicity of the non-O1/non-O139 strain AM-19226 [35]. Environmental isolates have also been reported to carry one or more of the virulence factors [30], [31], [36], [37].
Strain description up to now primarily uses serogrouping based on O antigen, and serogroup has been used as a major indicator in assessing virulence potential. There are over 200 serogroups [38] and at least 19 have been reported to be associated with sporadic cholera including O10, O12, O26, O31, O37, O53 and O75 [12], [13], [15], [36], [39]–[41]. Some isolates from these non-O1/non-O139 sporadic cholera cases have been characterised by DNA fingerprinting [7], [13] and gene sequencing [39]. However, the genetic background of the clinical and environmental isolates is diverse and a sequence-based study of relationships of the isolates is much needed. In this study, multilocus sequence typing (MLST) was applied to 35 clinical and 20 environmental non-O1/non-O139 V. cholerae isolates, including 15 isolates from sporadic cases in Bangladesh from 1998 to 2001, to examine their relationships to each other and with the O1 toxigenic and pandemic strains. There were no seven-gene MLST schemes available when this study was commenced, although one has been published recently [42].
Materials and Methods
Bacterial Isolates Used
The 55 V. cholerae isolates used were either clinical isolates or environmental isolates (Table 1). Fifteen of the 55 isolates were sporadic isolates from Bangledesh from 1998 to 2001 while others were from various countries with the majority being O antigen reference strains and were from the collection by T. Shimada, National Institute of Health, Japan.
Table 1. Vibrio cholerae and other Vibrio spp. isolates analysed in this study.
| Strain Name | Original Strain Name | Category | Serogroup | Location (year) | VPI† | T3SS§ | CTX | rtxA | NAG-ST | adk | gyrB | mdh | metE | pntA | purM | pyrC | ST | Clonal Complex (CC)# | Subpopulation¶ |
| M2565 | AQ 24151 | Clinical | O94 | Bangladesh (2001) | − | + | − | + | − | 26 | 5 | 2 | 1 | 31 | 19 | 45 | 50 | CC2 | I |
| M2549 | AO 823 | Clinical | O12 | Bangladesh (1999) | − | − | − | + | − | 12 | 23 | 2 | 49 | 30 | 1 | 40 | 9 | Singleton | I |
| M553 | 141-94 | Environmental | O70 | Germany (1994) | + | − | − | + | − | 17 | 5 | 14 | 50 | 31 | 14 | 23 | 22 | Singleton | I |
| M1121 | 208 | Clinical | O27 | Thailand (1992) | + | − | − | + | − | 19 | 9 | 12 | 23 | 9 | 1 | 5 | 26 | Singleton | I |
| M1576 | Clinical | O124 | India (1981) | − | − | − | + | + | 2 | 7 | 8 | 22 | 39 | 1 | 45 | 31 | Singleton | I | |
| M1052 | 334-72 | Clinical | O26 | Phillipines (1972) | − | − | − | + | − | 24 | 5 | 14 | 50 | 31 | 14 | 45 | 39 | Singleton | I |
| M2559 | AQ 9921 | Clinical | O2 | Bangladesh (2001) | − | + | − | + | − | 26 | 23 | 2 | 50 | 31 | 1 | 45 | 43 | Singleton | I |
| M2547 | AN 26575 | Clinical | O44 | Bangladesh (1998) | − | − | − | + | − | 26 | 3 | 1 | 50 | 14 | 18 | 45 | 44 | Singleton | I |
| M2561 | AQ 13192 | Clinical | O145 | Bangladesh (2001) | − | + | − | + | − | 26 | 5 | 1 | 8 | 14 | 14 | 45 | 45 | Singleton | I |
| M2560 | AQ 11489 | Clinical | O15 | Bangladesh (2001) | − | + | − | + | − | 26 | 5 | 14 | 50 | 31 | 14 | 2 | 46 | Singleton | I |
| M535 | #82 | Environmental | O1 | Thailand (1992) | − | + | − | + | + | 26 | 5 | 14 | 50 | 31 | 15 | 45 | 47 | Singleton | I |
| M2564 | AQ 18953 | Clinical | O24 | Bangladesh (2001) | − | − | − | + | − | 27 | 5 | 9 | 21 | 30 | 6 | 45 | 51 | Singleton | I |
| M2556 | AP 20403 | Clinical | ND | Bangladesh (2000) | − | + | − | + | − | 26 | 5 | 2 | 1 | 31 | 17 | 45 | 49 | CC2 | II |
| M536 | #35 | Environmental | O1 | Thailand (1992) | − | + | − | + | + | 1 | 15 | 19 | 15 | 24 | 5 | 14 | 3 | Singleton | II |
| M1562 | AU256 | Environmental | O110 | Japan (1992) | − | + | − | + | + | 1 | 41 | 21 | 4 | 2 | 16 | 43 | 4 | Singleton | II |
| M1095 | 27973 | Clinical | O77 | India (1976) | + | − | − | + | + | 1 | 42 | 4 | 60 | 20 | 1 | 12 | 5 | Singleton | II |
| M2563 | AQ 16457 | Clinical | O24 | Bangladesh (2001) | − | − | − | + | − | 12 | 23 | 2 | 6 | 30 | 17 | 40 | 10 | Singleton | II |
| M1105 | 984-81 | Clinical | O89 | India (1981) | − | + | − | + | + | 12 | 5 | 3 | 33 | 31 | 18 | 26 | 11 | Singleton | II |
| M555 | 905-93 | Environmental | O97 | Argentina (1993) | + | − | − | + | + | 13 | 40 | 17 | 46 | 3 | 9 | 39 | 12 | Singleton | II |
| M559 | 179-94 | Environmental | O52 | Denmark (1994) | − | − | − | − | − | 14 | 1 | 14 | 44 | 6 | 9 | 39 | 13 | Singleton | II |
| M1071 | 1154-74 | Clinical | O49 | India (1974) | − | − | − | + | − | 14 | 14 | 4 | 53 | 22 | 17 | 35 | 14 | Singleton | II |
| M1098 | 1421-77 | Clinical | O80 | India (1977) | + | + | − | + | + | 14 | 23 | 6 | 47 | 11 | 1 | 36 | 15 | Singleton | II |
| 12129 | Genome* | Environmental | O1 | Australia (1985) | + | + | − | + | − | 14 | 31 | 20 | 39 | 16 | 1 | 13 | 16 | Singleton | II |
| M1118 | 577-88 | Clinical | O105 | China (1988) | + | + | + | − | − | 14 | 38 | 14 | 43 | 8 | 21 | 11 | 17 | Singleton | II |
| M560 | 190-94 | Environmental | O39 | Denmark (1994) | − | + | − | + | − | 14 | 6 | 14 | 7 | 3 | 20 | 47 | 18 | Singleton | II |
| M548 | ATCC33653 | Environmental | O25 | Germany (1993) | + | + | − | + | + | 14 | 6 | 29 | 11 | 25 | 1 | 24 | 19 | Singleton | II |
| VL426 | Genome | Environmental | non-O1/non-O139 | UK | − | − | − | + | − | 15 | 29 | 16 | 18 | 10 | 1 | 22 | 20 | Singleton | II |
| M1049 | 169-68 | Clinical | O22 | Phillipines (1968) | − | − | − | + | − | 18 | 5 | 14 | 40 | 17 | 1 | 1 | 24 | Singleton | II |
| M1051 | 14438-62 | Clinical | O24 | Phillipines (1962) | + | − | − | + | − | 19 | 23 | 14 | 12 | 1 | 1 | 6 | 25 | Singleton | II |
| RC385 | Genome | Environmental | O135 | Chesapeake bay (1998) | − | − | − | + | + | 2 | 10 | 18 | 45 | 4 | 8 | 48 | 27 | Singleton | II |
| MZO-2 | Genome | Clinical | O14 | Bangladesh (2001) | − | − | − | + | − | 2 | 23 | 15 | 42 | 18 | 13 | 10 | 28 | Singleton | II |
| AM-19226 | Genome | Clinical | O39 | Bangladesh (2001) | + | + | − | + | − | 2 | 5 | 13 | 19 | 6 | 6 | 5 | 29 | Singleton | II |
| 1587 | Genome | Clinical | O12 | Peru (1994) | + | − | − | + | − | 2 | 6 | 29 | 2 | 25 | 1 | 3 | 30 | Singleton | II |
| M556 | 921-93 | Environmental | O74 | Argentina (1993) | − | − | − | + | − | 22 | 39 | 10 | 38 | 3 | 17 | 15 | 36 | Singleton | II |
| M1078 | 1463-76 | Clinical | O57 | India (1976) | − | − | − | + | + | 24 | 5 | 1 | 6 | 39 | 18 | 2 | 38 | Singleton | II |
| M1036 | 8394-62 | Clinical | O7 | India (1962) | − | + | − | − | + | 24 | 5 | 14 | 55 | 30 | 1 | 44 | 40 | Singleton | II |
| M1575 | 345-81 | Clinical | O123 | India (1981 | − | − | − | − | + | 24 | 5 | 14 | 55 | 7 | 1 | 45 | 41 | Singleton | II |
| M1593 | 243-93 | Clinical | O141 | India (1993) | + | + | + | + | + | 25 | 5 | 22 | 16 | 21 | 8 | 11 | 42 | Singleton | II |
| V51 | Genome | Clinical | O141 | US | + | + | + | + | − | 25 | 5 | 22 | 16 | 21 | 8 | 11 | 42 | Singleton | II |
| TM11079-80 | Genome | Environmental | O1 | Brazil (1980) | + | − | − | + | − | 26 | 5 | 14 | 54 | 28 | 14 | 45 | 48 | Singleton | II |
| M549 | 1085-93 | Environmental | O37 | Germany (1993) | − | − | − | + | − | 4 | 23 | 28 | 13 | 25 | 1 | 9 | 65 | Singleton | II |
| MZO-3 | Genome | Clinical | O37 | Bangladesh (2001) | − | − | − | + | − | 4 | 23 | 28 | 13 | 25 | 1 | 9 | 65 | Singleton | II |
| M563 | 370-94 | Environmental | O81 | Korea (1994) | − | + | − | − | + | 5 | 26 | 14 | 34 | 2 | 4 | 47 | 66 | Singleton | II |
| M2552 | AO 21097 | Clinical | O27 | Bangladesh (1999) | − | + | − | + | − | 6 | 4 | 14 | 35 | 15 | 3 | 46 | 67 | Singleton | II |
| M1618 | N1 (V523) | Environmental | non-O1/non-O139 | Australia (1977) | + | − | + | + | + | 7 | 11 | 4 | 17 | 23 | 1 | 35 | 68 | Singleton | II |
| M2129 | ATCC 25872 | Clinical | O37 | Czechoslovakia (1965) | + | − | + | + | + | 7 | 11 | 4 | 17 | 23 | 1 | 35 | 68 | Singleton | II |
| M2130 | S-21 | Clinical | O37 | Sudan (1968) | + | − | + | + | + | 7 | 11 | 4 | 17 | 23 | 1 | 35 | 68 | Singleton | II |
| V52 | Genome | Clinical | O37 | Sudan | + | − | + | + | − | 7 | 11 | 4 | 17 | 23 | 1 | 35 | 68 | Singleton | II |
| M1086 | 981-75 | Clinical | O65 | India (1975) | + | − | − | + | − | 7 | 12 | 14 | 37 | 12 | 1 | 36 | 74 | Singleton | II |
| CT5369-93 | Genome | Environmental | non-O1/non-O139 | − | − | − | + | − | 7 | 24 | 30 | 59 | 3 | 1 | 45 | 76 | Singleton | II | |
| M562 | 369-94 | Environmental | O10 | Korea (1994) | − | − | − | + | − | 8 | 30 | 27 | 36 | 3 | 9 | 39 | 78 | Singleton | II |
| TMA21 | Genome | Environmental | non-O1/non-O139 | Brazil (1993) | + | + | − | + | − | 9 | 25 | 3 | 41 | 6 | 1 | 5 | 79 | Singleton | II |
| N16961 | Genome | 7th pandemic | O1 | Bangladesh (197X) | + | − | + | + | − | 7 | 11 | 4 | 37 | 12 | 1 | 20 | 69 | CC1 | III |
| M2140 | SIMP/77 | Australian toxigenic | O1 | Australia (1977) | + | − | + | + | + | 7 | 11 | 4 | 37 | 12 | 1 | 37 | 70 | CC1 | III |
| M2141 | M4287/77 | Australian toxigenic | O1 | Australia (1977) | + | − | + | + | + | 7 | 11 | 4 | 37 | 12 | 1 | 37 | 70 | CC1 | III |
| BX330286 | Genome | Australian toxigenic | O1 | Australia (1986) | + | − | + | + | + | 7 | 11 | 4 | 37 | 12 | 1 | 37 | 70 | CC1 | III |
| M802 | M66-2/Makassar 759 (Genome) | pre-7th pandemic | O1 | Indonesia (1937) | + | − | + | + | − | 7 | 11 | 4 | 37 | 12 | 1 | 38 | 71 | CC1 | III |
| M1616 | O395 (Genome) | 6th pandemic | O1 | India (1965) | + | − | + | − | + | 7 | 11 | 4 | 9 | 12 | 1 | 38 | 73 | CC1 | III |
| M796 | 4808 | US Gulf Coast | O1 | USA (1978) | + | − | + | + | + | 7 | 2 | 4 | 37 | 12 | 1 | 38 | 75 | CC1 | III |
| M2562 | AQ 14875 | Clinical | O49 | Bangladesh (2001) | − | + | − | + | − | 1 | 1 | 4 | 48 | 29 | 1 | 41 | 1 | CC3 | III |
| M2553 | AP 2007 | Clinical | O49 | Bangladesh (2000) | − | + | − | + | − | 1 | 1 | 4 | 48 | 29 | 17 | 41 | 2 | CC3 | III |
| M2554 | AP 9172 | Clinical | O49 | Bangladesh (2000) | − | + | − | + | − | 1 | 1 | 4 | 48 | 29 | 17 | 41 | 2 | CC3 | III |
| M2555 | AP 14558 | Clinical | O49 | Bangladesh (2000) | − | + | − | + | − | 1 | 1 | 4 | 48 | 29 | 17 | 41 | 2 | CC3 | III |
| M2558 | AQ 5961 | Clinical | O49 | Bangladesh (2001) | + | + | − | + | − | 1 | 1 | 4 | 48 | 29 | 17 | 41 | 2 | CC3 | III |
| M1619 | N19 (V58) | Environmental | non-O1/non-O139 | Australia (1977) | + | − | + | + | + | 1 | 8 | 15 | 5 | 13 | 1 | 7 | 6 | Singleton | III |
| M1035 | B4202-64 | Clinical | O5 | Phillipines (1964) | − | + | − | − | + | 11 | 27 | 11 | 56 | 19 | 1 | 4 | 8 | Singleton | III |
| M1092 | 113-79 | Clinical | O73 | India (1979) | − | + | − | + | − | 11 | 27 | 11 | 56 | 19 | 1 | 4 | 8 | Singleton | III |
| 623-39 | Genome | Environmental | non-O1/non-O139 | Bangladesh (2002) | + | − | − | + | − | 20 | 41 | 5 | 3 | 18 | 2 | 4 | 32 | Singleton | III |
| M550 | 1089-93 | Environmental | O99 | Germany (1993) | − | + | − | + | − | 3 | 28 | 11 | 56 | 19 | 12 | 4 | 54 | Singleton | III |
| M561 | 366-94 | Environmental | O74 | Korea (1994) | − | − | − | + | − | 7 | 36 | 7 | 10 | 38 | 1 | 42 | 77 | Singleton | III |
| M554 | Environmental | O83 | Germany (1994) | + | − | − | + | + | 10 | 13 | 25 | 57 | 34 | 10 | 25 | 7 | Singleton | IV | |
| M1565 | AU105 | Environmental | O113 | Japan (1995) | − | + | − | + | − | 16 | 33 | 26 | 31 | 34 | 11 | 26 | 21 | Singleton | IV |
| M1563 | AU291 | Environmental | O111 | Japan (1993) | − | + | − | − | + | 17 | 5 | 25 | 32 | 36 | 11 | 29 | 23 | Singleton | IV |
| M1093 | 429-79 | Clinical | O75 | India (1979) | + | − | − | − | − | 21 | 37 | 23 | 58 | 35 | 7 | 28 | 33 | Singleton | IV |
| M557 | 928-93 | Environmental | O6 | Argentina (1993) | − | − | − | − | − | 22 | 32 | 24 | 20 | 33 | 9 | 27 | 34 | Singleton | IV |
| M558 | 929-93 | Environmental | O66 | Argentina (1993) | − | − | − | − | − | 22 | 32 | 24 | 20 | 5 | 1 | 27 | 35 | Singleton | IV |
| M1560 | AU124 | Environmental | O108 | Japan (1990) | + | − | − | − | − | 23 | 34 | 26 | 57 | 37 | 10 | 25 | 37 | Singleton | IV |
| M2548 | AN 28767 | Clinical | O37 | Bangladesh (1998) | − | − | − | − | − | 26 | 3 | 1 | N/A‡ | 14 | 18 | N/A | |||
| M2550 | AO 13987 | Clinical | O8 | Bangladesh (1999) | N/A | N/A | N/A | N/A | N/A | N/A | 1 | 11 | N/A | N/A | N/A | N/A | |||
| M2551 | AO 21097 | Clinical | O27 | Bangladesh (1999) | − | − | − | − | − | 34 | 41 | 33 | 58 | 26 | 31 | N/A | |||
| M547 | ATCC33653 | V. mimicus | 31 | 18 | 41 | 52 | 40 | 23 | 33 | 56 | |||||||||
| M1566 | 246-79 | V. mimicus | O114 | USA (1979) | 32 | 20 | 39 | 29 | 44 | 30 | 19 | 58 | |||||||
| M1567 | 523-80 | V. mimicus | O115 | USA (1980) | 32 | 21 | 38 | 26 | 46 | 25 | 34 | 59 | |||||||
| M1115 | 559-88 | V. mimicus | O101 | China (1988) | 32 | 22 | 42 | 27 | 41 | 27 | 18 | 60 | |||||||
| M1569 | 381-82 | V. mimicus | O117 | Japan (1982) | 33 | 19 | 35 | 30 | 42 | 29 | 31 | 61 | |||||||
| M1568 | 980-78 | V. mimicus | O116 | USA (1978) | 34 | 17 | 34 | 28 | 43 | 31 | 16 | 62 | |||||||
| RC341 | Genome | V. metecus | 28 | 35 | 31 | 14 | 27 | 22 | 8 | 52 | |||||||||
| M552 | O103 | Germany (1993) | 37 | 42 | 43 | 47 | 27 | 34 | 50 | 95 | |||||||||
| RC586 | Genome | V. parilis | 29 | 35 | 33 | 51 | 26 | 32 | 21 | 53 | |||||||||
| VM223 | Genome | V. mimicus | Brazil | 30 | 16 | 36 | 62 | 40 | 24 | 30 | 55 | ||||||||
| MB451 | Genome | V. mimicus | Bangladesh | 31 | 22 | 40 | 24 | 41 | 28 | 17 | 57 | ||||||||
| SX-4 | Genome | V. mimicus | China (2009) | 32 | 21 | 38 | 26 | 46 | 25 | 34 | 59 | ||||||||
| VM573 | Genome | V. mimicus | USA (1990s) | 32 | 21 | 38 | 26 | 46 | 25 | 34 | 59 | ||||||||
| VM603 | Genome | V. mimicus | Brazil (1990s) | 35 | 19 | 37 | 25 | 45 | 26 | 32 | 63 | ||||||||
| CMCP6 | Genome | V. vulnificus | 36 | 43 | 32 | 61 | 32 | 33 | 49 | 64 |
Genome strains 1587 (GenBank Accession No. AAUR00000000); 623-39 (Accession No. AAWG00000000); AM-19226 (Accession No. AATY00000000); BX330286 (Accession No. ACIA00000000); CMCP6 (Accession No. AE016795.3); CT5369-93 (Accession No. ADAL00000000); M66-2 (Accession No. CP001233); MB451 (Accession No. ADAF00000000); MZO-2 (Accession No. AAWF00000000); MZO-3 (Accession No. AAUU00000000); N16961 (Accession No. AE003852); M1616/O395 (Accession No. CP000626); RC341 (Accession No. ACZT00000000); RC385 (Accession No. AAKH00000000); RC586 (Accession No. ADBD00000000); SX-4 (Accession No. ADOO00000000); TM11079-80 (Accession No. ACHW00000000); TMA21 (Accession No. ACHY00000000); V51 (Accession No. AAKI00000000); V52 (Accession No. AAKJ00000000); VL426 (Accession No. ACHV00000000); VM223 (Accession No. ADAJ00000000); VM573 (Accession No. ACYV00000000); VM603 (Accession No. ACYU00000000); 12129 (Accession No. ACFQ00000000).
Two primer pairs were used as previously described in Tay et al. [44] to amplify divergent tcpA genes.
Two primer pairs were used for typing the type III secretion system (T3SS), one for vcsC2 and one for vcsV2.
N/A: Data not available.
Clonal complex (CC) as defined by eBURST analysis.
Subpopulations are derived from STRUCTURE analysis.
MLST Genes and Primers
Seven genes were selected from the 26 housekeeping genes that we used previously to determine the evolutionary relationships of the pandemic clones [43]. Three criteria were used to select seven genes: 1) both chromosomes are represented with five (gyrB, mdh, adk, metE, purM) and two (pntA and pyrC) genes from chromosomes one and two, respectively; 2) the genes are evenly distributed around the chromosomes; 3) These genes are present in related Vibrio species. In order to design primers that will be able to identify all of V. cholerae strains, primers were based on conserved regions among the sequences of more closely related Vibrio spp. The primers for the seven housekeeping genes (gyrB, mdh, adk, metE, purM, pntA and pyrC) are shown in Table 2.
Table 2. Primers used in this study.
| Gene | Gene Product | Direction* | Oligonucleotide Sequence (5' à 3') | Reference |
| adk | adenylate kinase | F | CATCATTCTTCTCGGTGCTC | This study |
| R | AGTGCCGTCAAACTTCAGGTA | |||
| gyrB | DNA gyrase subunit B | F | GTACGTTTCTGGCCTAGTGC | This study |
| R | GGGTCTTTTTCCTGACAATC | |||
| metE | methionine synthase | F | CGGGTGACTTTGCTTGGT | This study |
| R | CAGATCGACTGGGCTGTG | |||
| mdh | malate dehydrogenase | F | ATGAAAGTCGCTGTTATTGG | This study |
| R | GCCGCTTGGCCCATAGAAAG | |||
| R | TAGCTTGATAGGTTGGG | This study | ||
| pntA | pyridine nucleotide transhydrogenase | F | CTTTGATGGAAAAACTCTCA | |
| R | GATATTGCCGTCTTTTTCTT | This study | ||
| F | GGCCAGCCCAAAATCCT | |||
| purM | phosphoribosyl-formylglycinamide cyclo-ligase | F | GGTGTCGATATTGATGCAGG | This study |
| R | GGAATGTTTTCCCAGAAGCC | |||
| pyrC | Dihydroorotase | F | ATCATGCCTAACACGGTTCC | This study |
| R | TTCAAACACTTCGGCATA | |||
| ctxAB | cholera toxin | F | CTCAGACGGGATTTGTTAGGCACG | [69] |
| R | TCTATCTCTGTAGCCCCTATTACG | |||
| rtxA | repeat like toxin | F | GCGATTCTCAAAGAGATGC | [27] |
| R | CACTCATTCCGATAACCAC | |||
| sto/stn (NAG-ST) | heat-stable toxin | F | CCTATTCATTAGCATAATG | [12] |
| R | CCAAAGCAAGCTGGATTGC | |||
| tcp | toxin co-regulated pili | F1 | GTGACTGAAAGTCATCTCTTC | [44] |
| R1 | AATCCGACACCTTGTTGGTA | |||
| F2 | ATATGCAATTATTAAAACAGC | |||
| R2 | TTATTATTACCCGTTGTCGG | |||
| vcsC2 | inner membrane protein | F | GGAAAGATCTATGCGTCGACGTTACCGATGCTATGGGT | [12] |
| R | CATATGGAATTCCCGGGATCCATGCTCTAGAAGTCGGTTGTTTCGGTAA | |||
| vcsV2 | ATPase | F | ATGCAGATCTTTTGGCTCACTTGATGGG | [12] |
| R | ATGCGTCGACGCCACATCATTGCTTGCT |
PCR Assay and DNA Sequencing
Each PCR reaction included 2.5 µl of DNA template (approx. 20 ng), 0.5 µl (30 pmol/µl) of each forward and reverse primer (Table 2), 0.5 µl 10 mM dNTPs, 5 µl 10x PCR buffer (500 mM KCl, 100 mM Tris-HCl pH 9.0, 1% Triton® X-100 and 15 mM MgCl2), 0.25 µl (1.25 U) Taq polymerase (Promega) and MilliQ water to a total volume of 50 µl. PCR cycles were performed in a Hybaid PCR Sprint Thermocycler (Thermo Analysis Biocompany, Hybaid Limited, UK) with the following conditions: initial DNA denaturation for 2 min at 94°C; followed by DNA denaturation for 15 sec at 94°C, primer annealing for 30 sec at 50°C and polymerization for 1 min 30 sec at 72°C for 35 cycles, with a final extension of 5 min at 72°C. PCR products were verified on EtBr stained agarose gels before purification using sodium acetate/ethanol precipitation. The PCR sequencing reactions contained BigDye™ and were done as recommended by the manufacturer (Applied Biosystems). We sequenced both the 5′ and 3′ ends of the amplicons. Unincorporated dye terminators were removed by ethanol precipitation. The reaction products were separated and detected by gel electrophoresis using Automated DNA Sequence Analyzer ABI3730 (Applied Biosystems) at the Ramaciotti Centre (University of New South Wales, Sydney, Australia).
PCR Detection of Virulence Genes
We tested for the presence of the tcpA gene using two sets of primers as previously used to amplify divergent tcpA genes [44] and two primer pairs for the T3SS, one for vcsC2 and one for vcsV2 [12]. For both VPI and T3SS, if one primer pair was positive, we interpreted it as a positive result, respectively. We used only one primer pair for the other genes (ctxAB, rtxA and sto/stn) (Table 2).
Bioinformatic Analysis
The PHRED-PHRAP-CONSED [45] program package was utilised for sequence editing. ClustalW [46] and MULTICOMP [47] were used for multiple sequence alignment and comparison. PHYLIP [48] was used to generate phylogenetic trees and bootstrap values. SplitsTree version 3.2 [49] was used to create a network structure using the Neighbour-net algorithm and uncorrected “p“ distance [50]. The ratio of recombination (ρ) to mutation (θ) in different loci was estimated using a composite likelihood program LDhat version 2.2 [51].
Sequence variants of the seven genes were designated as alleles and the combination of seven alleles constitutes an allelic profile. Isolates with identical allelic profiles were assigned to the same sequence type (ST). The STs were analysed using eBURST [52] to determine the presence of clonal complexes (CCs). Clonal complexes are groups of closely related STs, which shares six loci to at least one other ST within the group, and are descendants of a recent common ancestor. The founder of a CC is defined as the ST that differs from the largest number of other STs at only a single locus.
STRUCTURE version 2.2 [53], which implements a Bayesian approach for analysis of MLST data, was used to assess the possibility that the isolates were derived from a finite number of prior “cryptic” populations with varying degrees of admixture. The number of populations, K, was determined under the “admixture” model and in each simulation run, the Markov Chain Monte Carlo (MCMC) simulation of 30,000 iterations gave the posterior probability of K following a burn-in of 10,000 iterations. Different values of K were run multiple times and the K value that generated the highest posterior probability was used as the probable number of ancestral populations. The assignment of an isolate to a particular population was done under the linkage model.
The GenBank accession numbers are KC894962-KC895395. Sequences are deposited into the PubMLST database using the Bacterial Isolate Genome Sequence platform [54] and are accessible at http://pubmlst.org/vcholerae/.
Results and Discussion
MLST Scheme for V. cholerae
We developed a new MLST scheme for V. cholerae that was based on fragments of seven housekeeping genes. The gene fragment sequences ranged in length from 416 bp –591 bp, for a total of 3,217 bp for concatenated sequences. We applied this MLST scheme to 55 non-O1/non-O139 V. cholerae strains representing 43 O antigen types and one strain of unknown O type (not tested). Most O antigens were represented by a single isolate but six types were represented by two to six isolates. As detailed below, an additional five V. mimicus strains, which were initially identified as V. cholerae, were included. Two other presumptive V. cholerae isolates, M552 and M2551, were found to be of other Vibrio species based on MLST.
Twenty-seven different O antigens were found in the 35 clinical isolates. Fifteen were isolated in Bangladesh between 1998 and 2001, including nine O antigens (one isolate was not typed), of which two were O24 isolates from the same year and five were O49 isolates over a 2 year period. The 20 environmental isolates represented 18 different O antigens.
Sequence Variation
PCR and sequencing of the seven housekeeping genes were initially done for 63 presumptive V. cholerae isolates. Sequences were obtained for all seven genes for 60 isolates. For strain M2548, two genes (metE and pyrC) failed and for strain M2551, one gene (pyrC) failed. For M2550, four genes (pyrC, adk, pntA and metE) failed to amplify a product and the strain was excluded from further analysis. Sequence data from 17 genome sequenced strains (excluding pandemic and related genome strains that were already represented) were also included for comparison. The most conserved gene was purM with maximum and average pairwise percentage differences of 2.94 and 0.78%, respectively while the most variable gene was pyrC with the highest maximum and average pairwise percentage differences of 16.48% and 6.50%, respectively.
Allelic Profiles, Sequence Types and Clonal Complexes
There were 66 STs, six of which were represented by multiple isolates, while the remaining STs were found in a single isolate. The STs with most isolates were ST2 and ST68, both of which contained four isolates. We used the definition of six out of seven shared alleles for a CC and identified only three CCs. CC1 was the largest containing seven STs and also included the two pandemic clones. The remaining two CCs were CC2 (ST49 and ST50) and CC3 (ST1 and ST2), all containing clinical isolates. The founder of CC1 was ST71 which was represented by M66-2, a pre-seventh pandemic strain, while CC2 and CC3 contained only two member STs and the founders could not be determined.
Four of the 17 genome strains had the same ST as a strain sequenced in this study. V51 was identical to M1593. Both were of O141 serogroup but were isolated from different locations in different years and were members of a known widespread toxigenic clone [8]. Genome strain V52 was identical to three other strains, M1618, M2129 and M2130. These strains were all O37 clinical isolates. Both V52 and M2130 were isolated in Sudan while M2129 was from Czechoslovakia. Interestingly, M1618 was an environmental isolate from Australia. The genome strain MZO-3 was identical to M549, both of which were of O37 serogroup. We previously sequenced 26 genes including the seven MLST genes from M549 [43]. The additional 19 genes were also identical to those of MZO-3. Lastly, genome strain BX330286, an O1 isolate, was found to be of the same ST as two other isolates, M2140 and M2141. All three isolates were from Australia and are toxigenic.
M2551 was the most divergent and clearly belonged to a different species. M552 was most closely related to RC341, the sole member of a new Vibrio species, V. metecus, proposed by Haley et al. [55]. The percentage DNA difference for the seven genes is 3.72% between M552 and RC341 which was far lower than that (8.1%) between M552 and the closest V. cholerae strain MZO-2.
Phylogenetic Relationships and Population Structure of V. cholerae
We performed a number of analyses to determine relationships and groupings of the isolates. A neighbour joining (NJ) tree was constructed using V. vulnificus strain CMCP6 as an outgroup to show the overall relationships (Figure 1). The NJ tree gave good bootstrap values only for branches with species-level differences. There was low bootstrap support for most branches, which may be due to recombination as V. cholerae is known to have a high level of recombination [43], [56]. A bifurcating tree may not be the best representation for their relationships, so a SplitsTree was constructed and showed an extensive network structure (Figure 2), consistent with high levels of recombination.
Figure 1. Phylogenetic relationships of Vibrio cholerae isolates based on neighbour-joining tree. ST is indicated in bracket after strain name for V. cholerae.
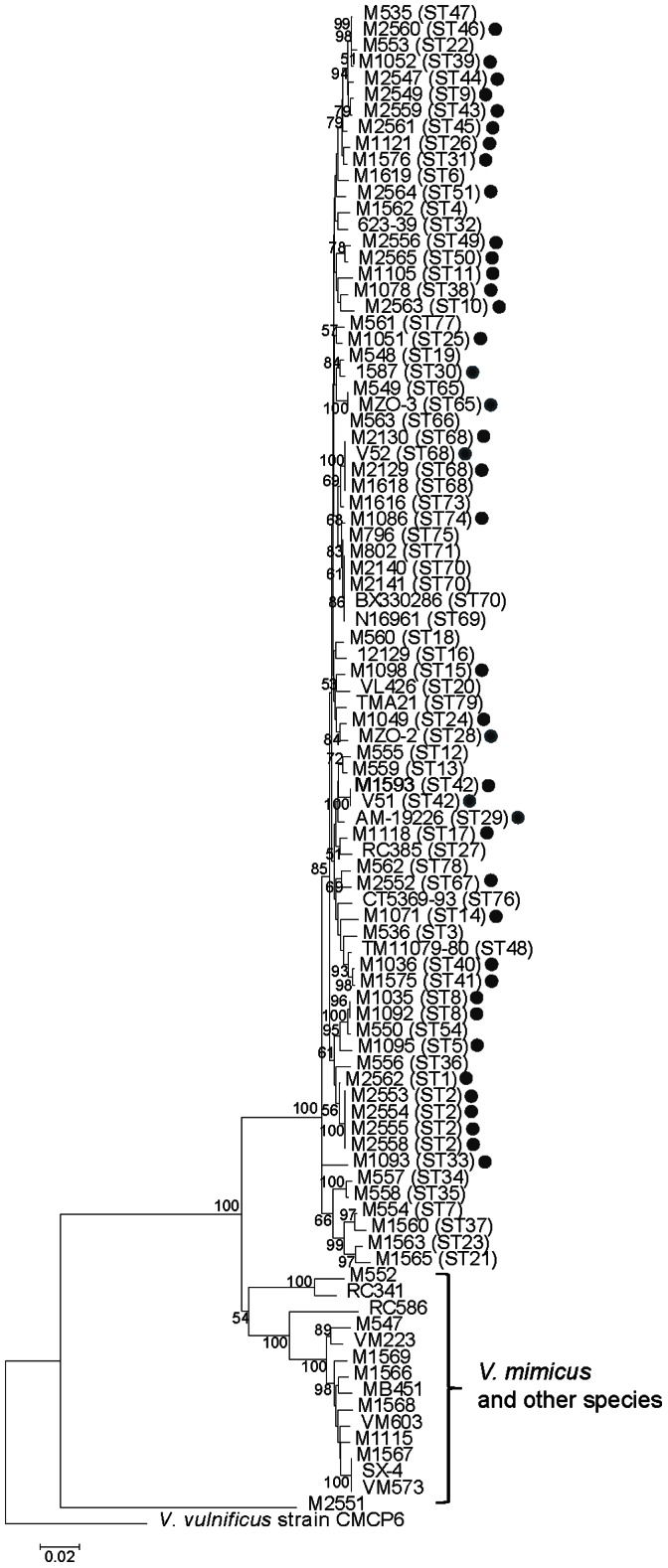
Non-O1/non-O139 V. cholerae strains from clinical sources are marked with a dot. Vibrio vulnificus strain CMCP6 was used as an outgroup. Bootstrap values, if greater than 50%, are presented at nodes of the neighbour joining trees.
Figure 2. Neighbour-net network of Vibrio cholerae isolates analysed in this study.

Four subpopulations (I to IV) as determined by STRUCTURE analysis are indicated with curly brackets. Isolates associated with 6th, pre-7th and 7th pandemics are indicated with an arrow.
We further assessed the frequency of recombination using the composite likelihood of recombination rate and mutation rate using LDhat [51]. The estimated rates of recombination (ρ) and mutation (θ) and their ratios for each gene are shown in Table 3. The ρ/θ ratio ranged from 0.465 (in metE) to 5.871 (in adk) and averaged 2.427 over the seven genes, showing significant levels of recombination. The average rate was much higher than these for E. coli [57] and Salmonella [58] with an average of 1.531 and 1.258 respectively. We also used the counting method of Feil et al. [59] to determine the nature of the changes observed in the three clonal complexes (CC1-CC3), in which the single allele difference between STs within a clonal complex was attributed to either mutation if the difference was a single base or recombination otherwise. There were five recombinational events and one mutational event in total, giving a ratio of 5 to 1. However, since there were only three CCs and only a few member STs within a CC, this estimate was likely to be unreliable. In the study of Salim et al. [43], 26 housekeeping genes were sequenced from the pandemic and closely related toxigenic strains, which provided another dataset to estimate the ratio of recombination to mutation. Together with strains added in the current study (see the section on the relationship of M1086 with pandemic lineages below), the 26 housekeeping gene data revealed 14 recombinational events and six mutational events within the two pandemic lineages (excluding the seven recombinational events between the two lineages, see the section on the relationship of M1086 with pandemic lineages below). This gave a recombination to mutation ratio of 2.333 to 1, which was similar to the estimate by the composite likelihood method above. This ratio can be compared with the estimates using the same method of other organisms including Y. pseudotuberculosis of a ratio of 1∶1 [60], Neisseria meningitidis of 10∶1 and Staphylococcus aureus of 1∶15 [61]. The results suggest that V. cholerae has a weakly clonal population structure.
Table 3. Estimates of mutation (θ) and recombination (ρ) rates.
| ρ | θ | ρ/θ | |
| adk | 0.089 | 0.015 | 5.871 |
| gyrB | 0.060 | 0.023 | 2.578 |
| mdh | 0.017 | 0.019 | 0.882 |
| metE | 0.017 | 0.036 | 0.465 |
| pntA | 0.053 | 0.020 | 2.622 |
| purM | 0.061 | 0.015 | 3.956 |
| pyrC | 0.031 | 0.050 | 0.619 |
| Mean | 0.047 | 0.026 | 2.427 |
We then used the Bayesian statistics tool, STRUCTURE to determine the population structure of the 66 V. cholerae STs (77 isolates). The most striking finding is that the V. cholerae isolates fell into four subpopulations that were designated as subpopulations I, II, III and IV (Figure 3). These were most clearly seen in the STRUCTURE plot (Figure 3), which we used to allocate isolates to the four subpopulations. The four subpopulations were also well demarcated in the SplitsTree (Figure 2).
Figure 3. STRUCTURE analysis of Vibrio cholerae isolates including genome strains.
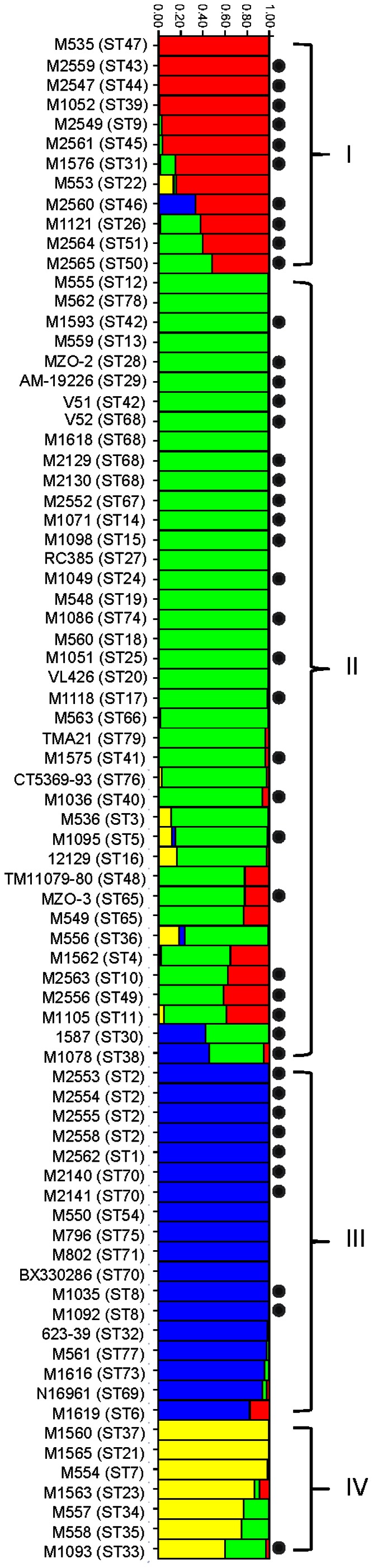
The four subpopulations are colour-coded with red, green, blue and yellow for subpopulation I, II, III and IV respectively. Each isolate has been allocated to a subpopulation. Isolates were identified by strain name with ST in brackets on the left. Mosaic colours for an isolate indicate mixed population origin from respective populations of matching colour. Y-axis represents percentage of population assignment. Non-O1/non-O139 V. cholerae strains from clinical sources are marked with a dot.
Subpopulation I contained 12 STs (12 isolates). Subpopulations II and III contained 34 STs (39 isolates) and 13 STs (19 isolates), respectively. Subpopulation IV contained seven STs (7 isolates). Subpopulations I and III were predominantly clinical isolates. Subpopulation III contained the pandemic clones and closely related isolates. Five isolates from sporadic cases from Bangladesh also belonged to this subpopulation which will be discussed below. Subpopulation II contained slightly more clinical isolates than environmental isolates. All except one isolate in subpopulation IV were of environmental origin although the subpopulation was small with only seven isolates. The majority of the genome sequenced strains fell into subpopulation II. Thus, more genomes should be sequenced from the other subpopulations to assess overall genomic diversity of the species.
The ancestry of each isolate/ST was estimated as the sum of probable sources from each of the presumed ancestral subpopulations over all polymorphic nucleotides. Thirty-one STs had no imports from other populations while 36 isolates (35 STs) contained alleles from other ancestral subpopulations (Figure 3); with 23 and 13 isolates having imports from one and two other ancestral subpopulations, respectively. The proportion of nucleotides from one or more minority subpopulations varied from 1% to 51%. Therefore, there were considerable gene flows between subpopulations.
It is interesting to note that the subpopulation assignment had two anomalies. ST68 (M1618, M2129, M2130 and V52) and ST74 (M1086) were closely related to pandemic strains (see below) but were assigned to subpopulation II rather than subpopulation III contrary to our expectations. This may be due to the different number of informative bases in different genes which influenced the subpopulation assignment.
Comparison with Environmental Isolates Typed by an Alternative MLST Scheme
Since the completion of this study, an MLST scheme has been applied to a large set of environmental isolates [42]. The two schemes share only two genes, which means that a direct seven-gene sequence based comparison was not feasible. However, we can compare the populations based on strains common to the two studies. There were 12 non-O1/non-O139 genome sequenced strains from Chun et al. [62] which we added to the environmental dataset. A STRUCTURE analysis divided this set of isolates into five subpopulations (data not shown). The genome sequenced strains all fell into one subpopulation which was the largest subpopulation. We also compared the isolates using the two genes (mdh and gyrB) common to the two studies (Figure S1). There were no clear demarcations of the subpopulations that were designated for the isolates analysed in this study. All except subpopulation IV were distributed across the NJ tree. The distribution of isolates in the tree showed that the two studies complemented each other in extending the genetic diversity.
Detection of Virulence Associated Genes in Sporadic Cholera Isolates
Previous studies have shown that sporadic cholera isolates may carry VPI or both VPI and CTX factors [12], [19]–[21]. Rtx toxin, T3SS and NAG-ST have also been reported to contribute to their virulence [27]–[29]. We used PCR to screen isolates for the presence of genes encoding these virulence factors. Excluding the genome strains, there were 35 clinical isolates. The most common virulence factor carried was RtxA at 85.7%, followed by T3SS at 51.4%, VPI and NAG-ST, each at 31.4% and CTX at 11.4%. All isolates carried at least one of these five virulence factors and one strain (M1593, O141) was positive for all five factors. Three isolates were positive for both VPI and CTX and an additional seven isolates were only positive for VPI. Clearly, the pathogenic mechanisms for non-O1/non-O139 strains were variable and diverse. For the environmental isolates, the most common virulence factor carried was also RtxA at 70%, followed by NAG-ST at 40%, T3SS and VPI each at 35% and CTX at 10%. Therefore, the number of virulence factors carried by environmental isolates was generally lower. This was also reflected in 15% of the environmental isolates not carrying any of the five virulence factors. This observation suggests that some strains isolated from the environment may not have a diarrhoeagenic potential in humans, while others do have the potential to cause diarrhoea.
Previous studies have reported that many strains carry VPI and/or CTX, including O27 [26], O37 [63], O44 [26], O64 [64], O65 [24] and O141 [8], [65] strains. Some other strains carry only VPI or CTXΦ or VPI plus a partial CTX prophage [22]–[24]. This study showed a similar picture for distribution of VPI and CTX, with 31.4% of the clinical isolates carrying VPI, although all our CTX positive isolates were also positive for VPI.
The virulence factors appeared to be quite mobile. Within the same ST, there were differences in the presence of the virulence factors. One of the four ST2 isolates was VPI positive. Our ST42 strain M2556 was negative for both VPI and CTX while the genome sequenced strain, V51, of the same ST carried both VPI and CTX. Of the two ST8 isolates, one carried only RtxA while the other carried only NAG-ST.
Further Resolution of Relationships of M1086 and Pandemic and Closely Related Isolates based on 26 Housekeeping Genes
M1086 (ST74) was closest to V52 (ST68) together with three other ST68 strains (M1618, M2129 and M2130). Note that of the four ST68 isolates, V52 and M2130 (original name S-21) were isolated in the same year (1968) and from the same location (Sudan) and were probably from the same outbreak. M2129 was an O37 strain isolated in 1965 from Czechoslovakia while M1618 (unknown serogroup) was isolated from Australia in 1977 from an environmental source.
The NJ tree from the MLST data presented an anomaly regarding the relationship of the pandemic and closely related strains. V52 and M1086 were closely related but were placed on different branches, well separated on the seven gene MLST tree from the pandemic strains. However, a previous analysis based on 26 housekeeping genes showed that the two pandemic clones grouped together [43] and this grouping has been confirmed by genome sequence data [56], [62]. The genome study by Chun et al. [62] defined two clades with the 7th pandemic lineage as phylocore genotype (PG) clade 1 and the 6th pandemic lineage as PG clade 2 [62]. The genome tree placed the O37 strain V52 in PG clade 2.
We sequenced 19 M1086 genes to complete the 26 gene set that Salim et al. [43] sequenced previously for the pandemic and closely related strains for comparison (see Salim et al. for the details of the 19 genes), and used the V52 genome sequence to represent ST66. A minimal spanning tree was constructed based on allelic difference rather than nucleotide sequence difference, which placed M1086 in the 7th pandemic lineage as the earliest diverged strain in that lineage (Figure 4). We denoted difference by one base as mutation and by two or more bases as recombination. M1086 differed from the ancestral alleles of that lineage by recombination in five genes (gppA, hmpA, metG, pepN and pyrC) and by mutation in one gene (gyrB), with the remaining 21 genes identical. Interestingly, three of the seven MLST genes showed a difference with one gene (gyrB) by mutation and two genes (mdh and pyrC) by recombination, which distorted the true relationship of the strain to the 7th pandemic lineage when seven genes were used. M1086 carried the O65 O antigen, and can thus be considered as a pre-7th pandemic strain that has the O1 antigen replaced, if we assume the ancestral strain was O1. M1086 was positive for VPI but negative for CTX which may have been lost. M1086 had also lost mannose-sensitive haemagglutinin pilin gene based on PCR detection (data not shown). Both pandemic lineages can now be seen to have at least one occasion of O antigen replacement in their early divergence, in the 6th pandemic lineage by O37 and the 7th pandemic lineage by O65.
Figure 4. Relationships of M1086 and pandemic related strains based on minimum spanning tree (MST).

MST was constructed using allelic difference of 26 housekeeping (hk) genes which are either resulted from recombination (r) or mutation (m). Events were marked on the branches with gene symbol (r or m) or for multiple genes as number of hk genes affected. M1086 and V52 were analysed in this study. See Salim et al. [43] for details of others. Changes of Vibrio pathogenicity island (VPI) and cholera toxin (CTX) are also indicated. Sequence data were M1086 from this study, V52 from Chun et al. [62] and others from Salim et al. [43]. Strain names, year of isolation and place of isolation were shown except for the pandemic strains and Australian and US Gulf toxigenic isolates.
Diversity of Sporadic Cholera Isolates from Bangladesh Region
We included in this study 18 non-O1/non-O139 isolates of 11 different serogroups isolated over four years from Bangladesh, with complete MLST data for 15 isolates from nine different serogroups. MLST data showed that none of these isolates was closely related to pandemic strains or derivatives of the pandemic clones by O antigen switching. MLST showed that the five O49 isolates from two different years were closely related, with four typed as ST2 and one as ST1. The two STs differed in one locus due to a single base change and were grouped together in CC3. Two isolates, typed as ST50 and ST49, belonged to CC2. One isolate belonged to serogroup O94 while the other was not typed for serogroup and may also be an O94 serogroup isolate. Two O24 isolates (M2563 and M2564) had two different STs differing by five loci, suggesting that they were not closely related. The remaining six isolates of different O serogroups had unique STs and differed from each other by three to six loci. These results suggest that there were multiple clones of V. cholerae causing sporadic cholera in Bangladesh with some being more prevalent.
Relationship with V. mimicus Strains
Five strains M1568, M1569, M1566, M1115 and M1567 were grouped together with our known V. mimicus strain M547 and five genome sequenced V. mimicus strains (VM603, VM573, VM223 MB451 and SX-4) [55], [66]–[68] (Figure 1), showing that these five “V. cholerae” strains were in fact V. mimicus that were initially misidentified. Maximum and average pairwise differences amongst the 11 isolates were 3.42% and 1.94%, respectively, while the maximum and average pairwise differences between V. cholerae and V. mimicus were 11.49% and 8.21%. Thus, the two species were well separated by sequence variation. Sucrose fermentation is commonly used to distinguish these species and we found them to be sucrose-negative as expected for V. mimicus.
Two of the five V. mimicus genome strains, VM573 (Clinical, USA, 1990s), VM223 (Environmental, Brazil) and SX-4 (Clinical, China, 2009) were identical to our M1567 (Clinical, USA, 1980) for all seven MLST genes and belong to ST59. All three strains were VPI+ and CTX+, suggesting that this ST is widespread as a cholera agent.
We also performed a STRUCTURE analysis with the 11 V. mimicus strains included and as expected, they were separated as a population of their own (data not shown). Interestingly, the V. mimicus population did not show any evidence of obtaining genes from the V. cholerae subpopulations and vice versa, which implies that there is clear species boundary for recombination. Given the high recombination rate in V. cholerae, this observation is surprising. However, it may be due to the small number of V. mimicus isolates sampled and/or the method of detection. There were small segments in the pyrC gene that were evidently of recombinant origin. A 60 bp segment in V. mimicus strains M547 and M1568 was likely to have been imported from V. cholerae which was not detected by STRUCTURE.
Conclusions
The identification of non-epidemic V. cholerae has primarily been based on O antigen as there were no real alternatives. MLST provided a framework to identify sporadic cholera isolates by their genetic characteristics. Sporadic cholera isolates are diverse. However, some were isolated in multiple locations or years, suggesting that these strains with repeated isolations are important pathogens and may have epidemic or pandemic potential. Much larger scale sampling will be required to monitor non-O1/non-O139 sporadic cholera clones.
We have shown in this study that the V. cholerae population can be divided into four subpopulations. Subpopulations I and III consisted of mainly clinical isolates, with pandemic clones belonging to subpopulation III. The ratio of recombination rate to mutation rate is 2.427, suggesting that V. cholerae is weakly clonal.
There was considerable variability in the proportion of the non-O1/non-O139 clinical isolates to carry virulence factors, with 85.7%, 51.4%, 31.4%, 31.4% and 11.4% of these isolates carrying RtxA, T3SS, VPI, NAG-ST and CTX, respectively. Only one of these isolates had all five virulence genes. These virulence genes were also present in the environmental isolates but at a lower frequency.
V. mimicus was well separated from V. cholerae by population structure analysis. One ST including two genome sequenced strains carrying VPI and CTX, is an important lineage of V. mimicus as a human pathogen.
Supporting Information
Phylogenetic relationships of Vibrio cholerae isolates based on neighbour-joining tree using concatenated sequences of two genes ( mdh and gyrB ) common between this study and the study by Keymer et al . [42]. Vibrio vulnificus strain CMCP6 was used as an outgroup.
(PDF)
Acknowledgments
We thank Dr Toshio Shimada from the National Institute of Infectious Diseases, Japan for providing us Vibrio cholerae strains. We thank Gordon Stevenson for technical assistance.
Funding Statement
This study was supported by an Australian Research Council discovery project grant. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Chin CS, Sorenson J, Harris JB, Robins WP, Charles RC, et al. (2011) The origin of the Haitian cholera outbreak strain. N Engl J Med 364: 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Talkington D, Bopp C, Tarr C, Parsons MB, Dahourou G, et al. (2011) Characterization of toxigenic Vibrio cholerae from Haiti, 2010–2011. Emerging Infect Dis 17: 2122–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reeves PR, Lan R (1998) Cholera in the 1990's. Br Med Bull 54: 611–623. [DOI] [PubMed] [Google Scholar]
- 4. Safa A, Nair GB, Kong RY (2010) Evolution of new variants of Vibrio cholerae O1. Trends Microbiol 18: 46–54. [DOI] [PubMed] [Google Scholar]
- 5. Dalsgaard A, Albert MJ, Taylor DN, Shimada T, Meza R, et al. (1995) Characterization of Vibrio cholerae non-O1 serogroups obtained from an outbreak of diarrhea in Lima, Peru. J Clin Microbiol 33: 2715–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dalsgaard A, Forslund A, Bodhidatta L, Serichantalergs O, Pitarangsi C, et al. (1999) A high proportion of Vibrio cholerae strains isolated from children with diarrhoea in Bangkok, Thailand are multiple antibiotic resistant and belong to heterogenous non-O1, non-O139 O-serotypes. Epidemiol Infect 122: 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sharma C, Thungapathra M, Ghosh A, Mukhopadhyay AK, Basu A, et al. (1998) Molecular analysis of non-O1, non-O139 Vibrio cholerae associated with an unusual upsurge in the incidence of cholera-like disease in Calcutta, India. J Clin Microbiol 36: 756–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dalsgaard A, Serichantalergs O, Forslund A, Lin W, Mekalanos J, et al. (2001) Clinical and environmental isolates of Vibrio cholerae serogroup O141 carry the CTX phage and the genes encoding the toxin-coregulated pili. J Clin Microbiol 39: 4086–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Onifade TJ, Hutchinson R, Van Zile K, Bodager D, Baker R, et al. (2011) Toxin producing Vibrio cholerae O75 outbreak, United States, March to April 2011. Eurosurveillance 16: 19870. [PubMed] [Google Scholar]
- 10. Tobin-D'Angelo M, Smith AR, Bulens SN, Thomas S, Hodel M, et al. (2008) Severe diarrhea caused by cholera toxin-producing Vibrio cholerae serogroup O75 infections acquired in the Southeastern United States. Clin Infect Dis 47: 1035–1040. [DOI] [PubMed] [Google Scholar]
- 11. Rahman MH, Biswas K, Hossain MA, Sack RB, Mekalanos JJ, et al. (2008) Distribution of genes for virulence and ecological fitness among diverse Vibrio cholerae population in a cholera endemic area: tracking the evolution of pathogenic strains. DNA Cell Biol 27: 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chatterjee S, Ghosh K, Raychoudhuri A, Chowdhury G, Bhattacharya MK, et al. (2009) Incidence, virulence factors, and clonality among clinical strains of non-O1, non-O139 Vibrio cholerae isolates from hospitalized diarrheal patients in Kolkata, India. J Clin Microbiol 47: 1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chakraborty S, Garg P, Ramamurthy T, Thungapathra M, Gautam JK, et al. (2001) Comparison of antibiogram, virulence genes, ribotypes and DNA fingerprints of Vibrio cholerae of matching serogroups isolated from hospitalised diarrhoea cases and from the environment during 1997–1998 in Calcutta, India. J Med Microbiol 50: 879–888. [DOI] [PubMed] [Google Scholar]
- 14. Bagchi K, Echeverria P, Arthur JD, Sethabutr O, Serichantalergs O, et al. (1993) Epidemic of diarrhea caused by Vibrio cholerae non-O1 that produced heat-stable toxin among Khmers in a camp in Thailand. J Clin Microbiol 31: 1315–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cariri FA, Costa AP, Melo CC, Theophilo GN, Hofer E, et al. (2010) Characterization of potentially virulent non-O1/non-O139 Vibrio cholerae strains isolated from human patients. Clin Microbiol Infect 16: 62–67. [DOI] [PubMed] [Google Scholar]
- 16. Ko WC, Chuang YC, Huang GC, Hsu SY (1998) Infections due to non-O1 Vibrio cholerae in southern Taiwan: predominance in cirrhotic patients. Clin Infect Dis 27: 774–780. [DOI] [PubMed] [Google Scholar]
- 17. Blake PA, Weaver RE, Hollis DG (1980) Diseases of humans (other than cholera) caused by Vibrios. Annu Rev Microbiol 34: 341–367. [DOI] [PubMed] [Google Scholar]
- 18.Morris J (1994) Non-O1 group Vibrio cholerae strains not associated with epidemic disease. In: Wachsmuth I, Blake P, Olsvik Ø, editors. Vibrio cholerae and cholera Molecular to globular perspectives. Washington, DC: ASM Press. 103–115.
- 19. Jagadeeshan S, Kumar P, Abraham WP, Thomas S (2009) Multiresistant Vibrio cholerae non-O1/non-O139 from waters in South India: resistance patterns and virulence-associated gene profiles. J Basic Microbiol 49: 538–544. [DOI] [PubMed] [Google Scholar]
- 20. Bhowmick TS, Das M, Ruppitsch W, Stoeger A, Pietzka AT, et al. (2009) Detection of virulence-associated and regulatory protein genes in association with phage typing of human Vibrio cholerae from several geographical regions of the world. J Med Microbiol 58: 1160–1167. [DOI] [PubMed] [Google Scholar]
- 21. Lee YL, Hung PP, Tsai CA, Lin YH, Liu CE, et al. (2007) Clinical characteristics of non-O1/non-O139 Vibrio cholerae isolates and polymerase chain reaction analysis of their virulence factors. J Microbiol Immunol Infect 40: 474–480. [PubMed] [Google Scholar]
- 22. Boyd EF, Heilpern AJ, Waldor MK (2000) Molecular analyses of a putative CTXphi precursor and evidence for independent acquisition of distinct CTX(phi)s by toxigenic Vibrio cholerae . J Bacteriol 182: 5530–5538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chakraborty S, Mukhopadhyay AK, Bhadra RK, Ghosh AN, Mitra R, et al. (2000) Virulence genes in environmental strains of Vibrio cholerae . Appl Environ Microbiol 66: 4022–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li M, Shimada T, Morris JG, Sulakvelidze A, Sozhamannan S (2002) Evidence for the emergence of non-O1 and non-O139 Vibrio cholerae strains with pathogenic potential by exchange of O-antigen biosynthesis regions. Infect Immun 70: 2441–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li M, Kotetishvili M, Chen Y, Sozhamannan S (2003) Comparative genomic analysis of the Vibrio pathogenicity island and cholera toxin prophage regions in nonepidemic serogroup strains of Vibrio cholerae . Appl Environ Microbiol 69: 1728–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mukhopadhyay AK, Chakraborty S, Takeda Y, Nair GB, Berg DE (2001) Characterization of VPI pathogenicity island and CTXphi prophage in environmental strains of Vibrio cholerae . J Bacteriol 183: 4737–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin W, Fullner KJ, Clayton R, Sexton JA, Rogers MB, et al. (1999) Identification of a Vibrio cholerae RTX toxin gene cluster that is tightly linked to the cholera toxin prophage. Proc Natl Acad Sci USA 96: 1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arita M, Takeda T, Honda T, Miwatani T (1986) Purification and characterization of Vibrio cholerae non-O1 heat-stable enterotoxin. Infect Immun 52: 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ogawa A, Kato J, Watanabe H, Nair BG, Takeda T (1990) Cloning and nucleotide sequence of a heat-stable enterotoxin gene from Vibrio cholerae non-O1 isolated from a patient with traveler's diarrhea. Infect Immun 58: 3325–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rivera IN, Chun J, Huq A, Sack RB, Colwell RR (2001) Genotypes associated with virulence in environmental isolates of Vibrio cholerae . Appl Environ Microbiol 67: 2421–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Faruque SM, Chowdhury N, Kamruzzaman M, Dziejman M, Rahman MH, et al. (2004) Genetic diversity and virulence potential of environmental Vibrio cholerae population in a cholera-endemic area. Proc Natl Acad Sci USA 101: 2123–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Theophilo GN, Rodrigues Ddos P, Leal NC, Hofer E (2006) Distribution of virulence markers in clinical and environmental Vibrio cholerae non-O1/non-O139 strains isolated in Brazil from 1991 to 2000. Rev Inst Med Trop Sao Paulo 48: 65–70. [DOI] [PubMed] [Google Scholar]
- 33. Dziejman M, Serruto D, Tam VC, Sturtevant D, Diraphat P, et al. (2005) Genomic characterization of non-O1, non-O139 Vibrio cholerae reveals genes for a type III secretion system. Proc Natl Acad Sci USA 102: 3465–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Alam A, Miller KA, Chaand M, Butler JS, Dziejman M (2011) Identification of Vibrio cholerae type III secretion system effector proteins. Infect Immun 79: 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shin OS, Tam VC, Suzuki M, Ritchie JM, Bronson RT, et al. (2011) Type III secretion is essential for the rapidly fatal diarrheal disease caused by non-O1, non-O139 Vibrio cholerae . MBio 2: e00106–00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bag PK, Bhowmik P, Hajra TK, Ramamurthy T, Sarkar P, et al. (2008) Putative virulence traits and pathogenicity of Vibrio cholerae Non-O1, Non-O139 isolates from surface waters in Kolkata, India. Appl Environ Microbiol 74: 5635–5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Faruque SM, Kamruzzaman M, Meraj IM, Chowdhury N, Nair GB, et al. (2003) Pathogenic potential of environmental Vibrio cholerae strains carrying genetic variants of the toxin-coregulated pilus pathogenicity island. Infect Immun 71: 1020–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shimada T, Arakawa E, Itoh K, Okitsu T, Matsushima A, et al. (1994) Extended serotyping scheme for Vibrio cholerae . Curr Microbiol 28: 175–178. [Google Scholar]
- 39. O'Shea YA, Reen FJ, Quirke AM, Boyd EF (2004) Evolutionary genetic analysis of the emergence of epidemic Vibrio cholerae isolates on the basis of comparative nucleotide sequence analysis and multilocus virulence gene profiles. J Clin Microbiol 42: 4657–4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fraga SG, Pichel M, Costagliola M, Cecilia M, Jurquiza V, et al. (2007) Environment and virulence factors of Vibrio cholerae strains isolated in Argentina. J Appl Microbiol 103: 2448–2456. [DOI] [PubMed] [Google Scholar]
- 41. Ottaviani D, Leoni F, Rocchegiani E, Santarelli S, Masini L, et al. (2009) Prevalence and virulence properties of non-O1 non-O139 Vibrio cholerae strains from seafood and clinical samples collected in Italy. Int J Food Microbiol 132: 47–53. [DOI] [PubMed] [Google Scholar]
- 42. Keymer DP, Boehm AB (2011) Recombination shapes the structure of an environmental Vibrio cholerae population. Appl Environ Microbiol 77: 537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Salim A, Lan R, Reeves PR (2005) Vibrio cholerae Pathogenic Clones. Emerging Infect Dis 11: 1758–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tay CY, Reeves PR, Lan R (2008) Importation of the major pilin TcpA gene and frequent recombination drive the divergence of the Vibrio Pathogenicity Island in Vibrio cholerae . FEMS Microbiol Lett 289: 210–218. [DOI] [PubMed] [Google Scholar]
- 45. Gordon D, Abajian C, Green P (1998) CONSED - A graphical tool for sequence finishing. Genome Res 8: 195–202. [DOI] [PubMed] [Google Scholar]
- 46. Thompson JD, G HD, Gibson TJ (1994) CLUSTAL W- Impoving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specifc gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reeves PR, Farnell L, Lan R (1994) MULTICOMP: a program for preparing sequence data for phylogenetic analysis. Bioinformatics 10: 281–284. [DOI] [PubMed] [Google Scholar]
- 48. Felsenstein J (1989) PHYLIP-phylogeny inference package. Cladistics 5: 164–166. [Google Scholar]
- 49. Huson DH (1998) SplitsTree: A Program for Analyzing and Visualizing Evolutionary Data. Bioinformatics 14: 68–73. [DOI] [PubMed] [Google Scholar]
- 50. Bryant D, Moulton V (2004) Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol 21: 255–265. [DOI] [PubMed] [Google Scholar]
- 51. McVean G, Awadalla P, Fearnhead P (2002) A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160: 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG (2004) eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol 186: 1518–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jolley KA, Maiden MC (2010) BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11: 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Haley BJ, Grim CJ, Hasan NA, Choi SY, Chun J, et al. (2010) Comparative genomic analysis reveals evidence of two novel Vibrio species closely related to V. cholerae . BMC Microbiol 10: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Feng L, Reeves PR, Lan R, Ren Y, Gao C, et al. (2008) A recalibrated molecular clock and independent origins for the cholera pandemic clones. PloS One 12: e4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wirth T, Falush D, Lan R, Colles F, Mensa P, et al. (2006) Sex and virulence in Escherichia coli: an evolutionary perspective. Mol Microbiol 60: 1136–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Didelot X, Bowden R, Street T, Golubchik T, Spencer C, et al. (2011) Recombination and population structure in Salmonella enterica . PLoS Genet 7: e1002191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Feil EJ, Smith JM, Enright MC, Spratt BG (2000) Estimating recombinational parameters in Streptococcus pneumoniae from multilocus sequence typing data. Genetics 154: 1439–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ch'ng SL, Octavia S, Xia Q, Duong A, Tanaka MM, et al. (2011) Population Structure and Evolution of Pathogenicity of Yersinia pseudotuberculosis . Appl Environ Microbiol 77: 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Spratt BG (2004) Exploring the concept of clonality in bacteria. Methods in molecular biology 266: 323–352. [DOI] [PubMed] [Google Scholar]
- 62. Chun J, Grim CJ, Hasan NA, Lee JH, Choi SY, et al. (2009) Comparative genomics reveals mechanism for short-term and long-term clonal transitions in pandemic Vibrio cholerae . Proc Natl Acad Sci USA 106: 15442–15447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nandi B, Nandy RK, Vicente AC, Ghose AC (2000) Molecular characterization of a new variant of toxin-coregulated pilus protein (TcpA) in a toxigenic non-O1/Non-O139 strain of Vibrio cholerae . Infect Immun 68: 948–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ghosh G, Nandy RK, Dasgupta SK, Nair GB, Hall RH, et al. (1997) A search for cholera toxin (CT), toxin coregulated pilus (TCP), the regulatory element ToxR and other virulence factors in non-O1/non-O139 Vibrio cholerae. . Microb Pathog 22: 199–208. [DOI] [PubMed] [Google Scholar]
- 65. Boyd EF, Waldor MK (2002) Evolutionary and functional analyses of variants of the toxin-coregulated pilus protein TcpA from toxigenic Vibrio cholerae non-O1/non-O139 serogroup isolates. Microbiology 148: 1655–1666. [DOI] [PubMed] [Google Scholar]
- 66. Hasan NA, Grim CJ, Haley BJ, Chun J, Alam M, et al. (2010) Comparative genomics of clinical and environmental Vibrio mimicus . Proc Natl Acad Sci USA 107: 21134–21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Thompson CC, Vicente AC, Souza RC, Vasconcelos AT, Vesth T, et al. (2009) Genomic taxonomy of Vibrios. BMC Evol Biol 9: 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang D, Wang H, Zhou Y, Zhang Q, Zhang F, et al. (2011) Genome sequencing reveals unique mutations in characteristic metabolic pathways and the transfer of virulence genes between V. mimicus and V. cholerae . PLoS One 6: e21299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shirai H, Nishibuchi M, Ramamurthy T, Bhattacharya SK, Pal SC, et al. (1991) Polymerase chain reaction for detection of the cholera enterotoxin operon of Vibrio cholerae . J Clin Microbiol 29: 2517–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phylogenetic relationships of Vibrio cholerae isolates based on neighbour-joining tree using concatenated sequences of two genes ( mdh and gyrB ) common between this study and the study by Keymer et al . [42]. Vibrio vulnificus strain CMCP6 was used as an outgroup.
(PDF)