

Abstract
The SMN2 transgenic mouse, Tg(SMN2)89Ahmb, has emerged as the most widely used in spinal muscular atrophy (SMA) research. Here we clone the genomic integration site of the transgene and demonstrate it to be in intron 4 of the metabotropic glutamate receptor 7 (mGluR7) gene. We found that the integration of this transgene significantly reduced both mGluR7 mRNA and protein levels (24% and 9%, respectively). To determine if phenotypes associated with mGluR7 knockout mice were present in Tg(SMN2)89Ahmb containing mice, we subjected mice homozygous for the transgene to open field and seizure susceptibility tests. When compared to wild type FVB/N mice, Tg(SMN2)89Ahmbtg/tg mice exhibited significantly longer times in finding a safe wall-adjacent square (+54s if Smn+/+, +90s if Smn+/−) as well as a significantly higher frequency of generalized seizure in response to a subthreshold dose of pentylenetrazol (0.11 vs 0.45). These findings aid in explaining the sudden unexpected death that occurs within SMA mouse colonies that contain a homozygous Tg(SMN2)89Ahmb transgene. This should be taken into account in pre-clinical studies that utilize this transgene, especially in therapy-treated SMA mice that have extended survival.
Keywords: Mouse, Spinal muscular atrophy, SMA, Metabotropic glutamate receptor 7, Seizure, Neurodegeneration
Introduction
Proximal spinal muscular atrophy (SMA) is a neurodegenerative disorder that affects 1 in every 10,000 live births. It is the leading hereditary cause of infant mortality and has a carrier frequency of 1 in 40 (Crawford and Pardo, 1996; Melki et al., 1990). SMA is characterized by progressive loss of lower motor neurons which generates a functional interruption of the neuromuscular junction, directly resulting in muscle atrophy. As disease advances, terminal endpoints are often associated with pulmonary complications, although patients can survive for many years in an intubated state (Zerres and Rudnik-Schoneborn, 1995).
Genetically SMA is caused by loss of function mutations in the survival motor neuron (SMN1) gene (Lefebvre et al., 1995). SMN plays a housekeeping function in RNA processing, and for this reason the complete absence of SMN protein is lethal to all cell types (Pellizzoni et al., 1998; Schrank et al., 1997). However, during a recent point in evolution the SMN gene was duplicated, generating two distinct copies, SMN1 and SMN2. The two genes are nearly identical except for a critical C to T nucleotide transition that causes SMN2’s exon 7 to be excluded from ~90% of the transcripts, referred to hereafter as Δ7SMN. Importantly, the ~10% of full length transcripts that contain exon 7 (FL-SMN) are enough to compensate for the loss of SMN1 in all cell types except motor neurons, causing the SMA phenotype.
Since mice lack a duplicated Smn genomic region, they cannot naturally replicate the disease. To confirm SMN2’s modifying ability and model SMA in mice, two separate SMN2 transgenic models were developed and distributed to the research community (Hsieh-Li et al., 2000; Monani et al., 2000). Despite having comparable SMN2 copies, the mouse developed by the Burghes Laboratory (Tg(SMN2)89Ahmbtg/tg;Smn−/−) survived up to 5 days postnatally, while the mouse developed in the Li Laboratory (Tg(SMN2)2Hungtg/o; SmnΔ7/Δ7) survived until 14 days of age (Gogliotti et al., 2010; Hsieh-Li et al., 2000). The Burghes Laboratory mouse reaches the survival equivalency of the Li mouse when a high-copy Δ7SMN cDNA transgene is combined with it (commonly referred to as the “delta 7” mouse, JAX #005025) (Le et al., 2005). Tg(SMN2)89Ahmb containing mice are the most commonly used in SMA research and to date are the standard models for pre-clinical drug studies.
We explain the decreased survival in the Burghes mouse by characterizing the expression and genomic structure of its SMN2 transgene, Tg(SMN2)89Ahmb. Additionally, we cloned and mapped the integration site of Tg(SMN2)89Ahmb to intron 4 of the metabotropic glutamate receptor 7 (mGluR7) gene. This integration reduces mGluR7 levels creating a hypomorphic allele, which on an FVB/N background, causes a heightened fear response and increased seizure susceptibility.
Materials and Methods
Animal maintenance and survival analysis
All mice used in this study were obtained from The Jackson Laboratory. All animals were maintained in a controlled animal facility at 20°C and 50% humidity, with a photoperiod of 12h light/12h dark where they were monitored daily for health as described previously (Heier and DiDonato, 2009). Mice were provided food and water ad labium (Harlan 6% crude fat diet at CMRC). For survival analysis, the day of birth was considered to be postnatal day (PND) zero. If pups were born after 3pm in the afternoon, their birth was considered to be the following morning; however none were noted in this study. Functional death as an endpoint was defined as mice which were not able to right themselves within 1 minute when placed on their back. At both CMRC and JAX lab facilities, functional death as an endpoint was applied to mice only on PND5, all other days pups were found dead in their cage. All maintenance and procedures were approved and performed in accordance with the Children’s Memorial Research Center’s or The Jackson Laboratories Institutional Animal Care and Use Committee regulations. The strains of mice used were FVB/N (JAX strain 1800), hybrid Tg(SMN2)tg/tg;Smn mice (JAX strain 5024), fully congenic Tg(SMN2)tg/tg;Smn mice (JAX strain 7944) and Tg(SMN2)2Hungtg/tg;Smn−/− mice (JAX strain 5058).
Genome scans
The genetic backgrounds of mice used in Figure 1B were determined at The Jackson laboratory using 172 single nucleotide polymorphisms (SNPs) distributed throughout the genome that were informative between FVB/N and C57Bl6 genetic backgrounds. Samples used specifically in the experiments reported here were assessed at the DartMouse™ Speed Congenic Core Facility at Dartmouth Medical School. DartMouse uses the Illumina, Inc. (San Diego, CA) GoldenGate Genotyping Assay to interrogate 1449 SNPs spread throughout the genome. Of these SNPs, 387 (26.7%) were informative (polymorphic across all three backgrounds, FVB/N, C57 Bl/6 and 129SvJ) and such a drop out is typical in a three strain analysis. The raw SNP data were analyzed using DartMouse’s proprietary SNaP-Map™ and Map-Synth™ software, allowing the determination for each mouse of the genetic background at each SNP location.”
Figure 1.

Survival of control and severe SMA mice. Kaplan-Meier analysis of control and SMA mice, (Tg(SMN2)89Ahmbtg/tg; Smn−/−) bred at two different facilities, CMRC and The Jackson Laboratory (JAX). The median survival at both sites is ≤ 1 day of age. B) Schematic results of genome scan of Strain 5024 reveals portions of the genome that are fixed for FVB/N (66%) still segregating (16%) or fixed for C57BL/6 (13%) alleles and 5% of genome was uninformative (retrospective data from 2006). Markers were selected to distinguish FVB/N from C57Bl/6. C) Schematic results of a three strain analysis genome scan (FVB/N vs C57 vs 129) representative of samples of strain 5024 used in this study. The fixed genetic contributions are 70% FVB/N, 17.2 % C57Bl/6 and 1% 129SvJ with the remaining genome being uninformative (5%) or still segregating (6.8%).
Expression analysis
RNA was prepared from tissues using Trizol reagent (Invitrogen) in accordance with manufacturer’s instructions. Samples were DNase treated (Roche TurboDNase kit) and cDNA was prepared using an Invitrogen Superscript II kit. The resulting cDNA was diluted to an initial RNA concentration of 100ng/μl. Real-Time PCR of FL-SMN was performed using 1.5μl cDNA as described previously (Heier et al., 2007). Samples were run in triplicate and experiments repeated three times. The average of the three runs was combined with the averages obtained from mice of the same genotype and used for final analysis.
mGluR7 transcript levels were determined using cDNA obtained from six week old Tg(SMN2)89Ahmbtg/tg (N=5) and wild type FVB/N (N=5) male mice. To ensure identical sampling regions, brains were placed in a “brain mold” (Braintree) with the optic lobe in slot 1 and the cerebellum in slot 14; the tissue from slots 4–6 was used for RNA. cDNA was prepared and used to co-amplify Gapdh, which served as an internal control and mGluR7 exons3–6 using primers in exon 3 (5′-ATGGTCCTGTCTTTGCGTTC) and exon 6 (5′-TCATGTGATGAAGGGCATGT). Densitometry was performed on the amplification products using a Microtek 1000XL scanner and Openlab 5.0 software. To quantify mGluR7 protein levels, brains from E12.5, P1, P5, P17 and 6–8 week old mice were dissected and halved down the central fissure into left and right hemispheres (N=5/genotype/time point for hybrid strain JAX 5024 and N=3/genotype/time point for congenic strain JAX 7949). RIPA buffer was used to prepare protein lysate which was quantified by Lowry Assay (Bio-Rad). Each sample (5μg) was resolved on a NuPage Novex 4–12% Bis-Tris gel and transferred to nitrocellulose membrane using the iBlot Gel Transfer System (Invitrogen). Membranes were blocked in Oddesey blocking buffer and probed with mGluR7 antibody (1:500,Abcam) and anti-tubulin (Developmental Hybridoma Bank). Protein was visualized using 1:5,000 LiCor fluorescent secondary (GAM-600, GAR-800) and quantified on the Oddesey western blotting system and software (v.3.0).
Immunoflourescence
To establish mGluR7 localization, 6-week old mice with and without the SMN2 transgene were perfused with 4% paraformaldehyde (EMS). Dissected brains were post fixed for 1 hour in 4% PFA at room temperature and then cryoprotected in 30% sucrose at 4°C overnight. Brains were separated down the central fissure, embedded in OCT and cryosectioned (12μ). Sections were permeabilized at room temperature (RT) for 30 min in a 0.5% trition, 4% goat solution and then blocked for 30 min in a 10% goat, 1% BSA solution free of detergent. Primary mGluR7 antibody (Upstate) was used at 1:100 in blocking buffer for 1hr at RT and a 1:2,500 secondary 594-GAR dilution was used for 2 hr at RT. A final 1:10,000 Dapi/PBS wash was for 5 min at RT labelled nuclei. Images were acquired using a Leica Spinning Disk Confocal microscope (Olympus) and processed in Photoshop.
Femoral motor axon counts
To quantify motor axon number, 16-week old mice with and without the Tg(SMN2)89Ahmb transgene were anesthetized with pentobarbital, perfused with a 0.1M CaCo2, 2% PFA, 2% gluteraldehyde solution and then post fixed in the same solution overnight at 4°C. The motor branch of the femoral nerve was removed, embedded in resin and processed for touluine blue. Myelinated motor axons were then quantified in a blinded fashion. Final axon numbers were analyzed via one-way ANOVA.
STS content mapping and integration
PCR primer sets were designed using the original PAC 215P15 clone as a template, the “x-kb” designation refers to their approximate reference position on PAC215P15. Additional primers were added to encompass historical marking sites Ag1CA, C212 and region 2F (DiDonato et al., 1994; Melki et al., 1994). PCR was performed using DNA (500ng) from each genotype. Once a region was identified where the Tg(SMN2)89Ahmb was absent, a series of three progressively forward primers with a known region were designed. Three rounds of nested PCR were then conducted, first with one of 4 random 4bp oligos, second with an adapter primer specific to the random primers and finally with a universal primer specific to the adapter (random adapter and universal primers provided in the SeeGene Genome Walking kit). Products were resolved on an agarose gel, excised, purified, subsequently cloned into pCR4.0 (Invitrogen) and sequenced using M13 forward primer.
Clone #1B1B contained elements of the transgene and the murine genome. A blast of the NCBI non-redundant database identified the genomic DNA sequence to be from intron 4 of the mGluR7 gene on chromosome 6. The integration was verified by PCR using a forward primer from the known region of the transgene (5′-GGTCTGTTCTACAGCCACAGC) and a mGluR7 reverse primer (5′-CTGACCTACCAGGGATGAGG). The addition of a forward mGluR7 primer (5′-CCCAGGTGGTTTATAGACTCAGA) allows one to determine if mice are wild type (400bp), hemizygous (300bp and 400bp) or homozygous (300bp) for the Tg(SMN2)89Ahmb transgene.
Open field analysis
The open field grid was divided into 25 equal squares (5×5) surrounded by a wall. Squares adjacent to the wall were deemed safe squares and those in the middle were deemed danger squares (Fig. 6A). Mice were chosen at random, as all observers were blinded to their genotypes. Mice were then placed in the center square and monitored for a 5 minute test period. Their path was traced by one observer who made note of the number of beams crossed while another monitored for myoclonic events. It was initially believed the stress of the open field might trigger seizure events, however none were observed. The mice were also monitored for the time taken to exit the 9 internal squares exposed to danger, and find one of the 16 safe wall-adjacent squares. Statistical significance was determined using a one-way ANOVA and Student’s t-test (Graphpad Prism 4.0). Males and females of each genotype were combined after analysis revealed no sex specific differences.
Figure 6.

Open field analysis of Tg(SMN2)89Ahmbtg/tg mice. A) Left: Depiction of the open field apparatus. Exterior squares were wall adjacent and deemed safe squares while center squares were exposed and deemed danger squares. Center: Depiction of average FVB/N behavior in the open field. Right: Depiction of abnormal Tg(SMN2)89Ahmbtg/tg mice in the open field. B) Quantification of the time taken for each genotype to reach a safe square. Tg(SMN2)89Ahmb mice took significantly longer to find a safe square within a 95% confidence interval (CI) if Smn was homozygous and a 99% CI if Smn was heterozygous as compared to wild type mice. C) Quantification of exploratory behavior represented by the number of lines on the grid (beams) crossed. Mice carrying the transgene that were also heterozygous for Smn (Tg(SMN2)89Ahmbtg/tg, Smn+/−) explored significantly less than wild type mice. D) Comparison of the time to reach a safe square for FVB/N mice compared to Smn+/− mice. E) Quantification of exploratory behavior of FVB/N mice compared to Smn+/− mice. Values are mean ±SEM.
Seizure susceptibility
An initial dose response study was conducted to determine a sub-threshold dose of PTZ for 12 week-old mice. This was critical as FVB/N mice are hypersensitive to seizures and their PTZ threshold changes with age. A final dose of 30mg/kg was identified as a dose that caused less than 10% of control mice to seize at 11–12 weeks of age.
Again mice were chosen at random from one of the 6 blinded groups. They were dosed and placed within a container lined with padding. Tonic, myoclonic, and tonic-clonic events were noted over a 10 minute period of observation. Generalized seizures were almost always preceded by several myoclonic events (which occurred even in the absence of any generalized seizures), thus the time to the first myoclonic event and the time to generalized seizure were recorded. Statistical significance was determined by one-way ANOVA.
Results
Survival analysis of severe SMA mice containing Tg(SMN2)89Ahmb
The SMN2 transgenic line of mice originally reported by Monani et al. (2000) were submitted to The Jackson Laboratory (Tg(SMN2)89Ahmbtg/tg; Smn+/−; JAX 005024). Intercrosses of this line consistently produced SMA mice that lived up to 2 days of age with a median survival of 1 day (N=13) when SMN2 was hemizygous on the null background (Tg(SMN2)89Ahmbtg/0;Smn−/−) and up to eight days with a median life span of 5 days (N=57) when SMN2 was homozygous (Tg(SMN2)89Ahmbtg/tg;Smn−/−) (Le et al., 2005; Monani et al., 2000). To validate the previously reported results after importation to The Jackson Laboratory, we set up a series of intercrosses and analyzed the F1 pups for genotype and survival at two separate facilities, CMRC and JAX labs. As expected, between the two facilities ~1/4 of the animals born had the SMA genotype, SMN2tg/tg;Smn−/−, (CMRC 12/52 and JAX 23/132). However, at the CMRC facility, 75% (9/12) of the SMA pups that were observed on the day of birth (PND0), were either still born or found dead and partially cannibalized at the first morning observation. Of the three remaining SMA pups, one died after 24 hrs and the other 2 survived to 5 days of age. These PND5 SMA pups were euthanized as they met criteria for functional death as an endpoint by their inability to right themselves within one minute when placed on their back. Similar to CMRC, of the 23 SMA pups born at the JAX lab, 10 were found still born, 7 died at PND1, 4 died at PND4 and 2 died at PND5 (Fig. 1A). In contrast to SMA pups, no controls at CMRC were still born or died within the first 5 days of life and at the JAX lab facility, 2/109 controls were still born or died within the first day of life. Overall, the median survival of severe SMA pups at both sites was ≤1 day of age and indicates a shift of this SMA line to a phenotype that is more severe than originally reported (Le et al., 2005; Monani et al., 2000).
To gain a better sense of the genetic background since it might influence survival, we retrospectively analyzed a low resolution genome scan that was performed in 2006 using 40 mice and 172 markers distributed throughout the genome. This scan was representative of the distribution line that was available to the research community at that time. As was known, strain 5024 is a genetic hybrid. Analysis of that 2006 scan indicated that the line was predominantly homozygous for FVB/N (66%) with portions of the genome still segregating (16%) or fixed for C57Bl/6 (13%) with ~5% of the genome being uninformative with the markers that were used (Fig. 1B). We performed a new, higher resolution genome scan (1,172 SNPs) using samples from mice derived from the lines used to generate the above data and those of the subsequent studies reported herein (Fig. 1C). In general, the results were consistent between the two studies. On average, the mice used in these studies were 70% homozygous for FVB/N. The increase was primarily due to the entirety of Chrm12 and a large portion of Chrm18 now being fixed in FVB/N (Fig. 1C).
Expression analysis of Tg(SMN2)89Ahmb
There are a number of external factors that contribute to the expression of a transgene, including its proximity to heterochromatin and its location relative to other genes or regulatory elements. As a first step to analyze the expression pattern of Tg(SMN2)Ahmb89 we performed standard RT-PCR analysis of tissues derived from JAX strain 5024. We found no evidence that FL-SMN or Δ7SMN transcripts or their ratios were altered from what would be expected from an SMN2 allele (data not shown). We went on to quantify FL-SMN expression from Tg(SMN2)89Ahmb and determine whether the level correlated with the decrease in survival of the Burghes mouse, which has a total of two copies of SMN2, as compared to the Li mouse model (JAX strain 5058) that has a total of 4 copies of SMN2 (2 copies/allele) on a Δ7Smn background. For these experiments we used a real-time PCR assay specific to human FL-SMN transcripts (Heier et al., 2007) and compared the relative FL-SMN expression of each transgene to an exogenous commercially available calibrator, total human reference RNA (THR). Analysis of embryos at E10.5 as well as tissues from PND15 mice that were hemizygous for either transgene on a wild type Smn background demonstrated that Tg(SMN2)2Hung transgene expressed a higher percentage of FL-SMN in whole embryos at E10.5 (7.2±0.8 vs 5.8±0.9) (Fig. 2A). Additionally, the (SMN2)2Hung transgene expressed a higher percentage of FL-SMN transcripts in PND15 spinal cord (8.3±1.8 vs 5.3±1.4), brain (5.0±0.0 vs 2.0±0.0), kidney (4.3±0.7 vs 2.3±0.7) and skeletal muscle (0.4±0.1 vs 0.2±0.0)(Fig. 2B). These results are consistent with the SMN2 copy number of Tg(SMN2)2Hung and Tg(SMN2)89Ahmb transgenic alleles. It also explains that a major contributing difference in survival between the Li and Burghes mouse models is likely due to the total differential expression of their SMN2 transgenes.
Figure 2.

Tg(SMN2)89Ahmb expression analysis and STS content mapping. A) Quantitative real time PCR analysis of FL-SMN in E10.5 mice from (SMN2)2Hung and Tg(SMN2)89Ahmb hemizygous mice. B) Tissue profile of FL-SMN expression for PND 15 (SMN2)2Hung and Tg(SMN2)89Ahmb hemizygous mice. C) STS content map of the SMN2 region in the Tg(SMN2)89Ahmb transgene. Note that the Tg(SMN2)89Ahmb transgene should extend through the 18.1kb region but has a 1.6kb deletion from the 3′ end. Values are mean ±SEM.
Content and integration mapping of Tg(SMN2)89Ahmb
Sequence tagged site (STS) content mapping was used to survey the total content of human sequence present in Tg(SMN2)89Ahmb. PCR primers were designed that spanned the entirety of the 35.5 kb microinjected fragment (Monani et al., 2000). Through this mapping we determined that the total transgene size of Tg(SMN2)89Ahmb is 33.9 kb (Fig. 2C). Only a small fraction of the proximal (<3 kb) and distal ends (1.6 kb) of the microinjected fragment is missing from Tg(SMN2)89Ahmb and that SMN2, including its 3′UTR, appears to be intact.
To identify where in the mouse genome the Tg(SMN2)89Ahmb transgene integrated, we used our STS content mapping data to clone the integration site. Ultimately, we amplified and cloned a fragment containing 423bp of sequence from the transgene and 1,298bp of unknown sequence (clone #1B1B). A blast search of the non-redundant database revealed the sequence to be from intron 4 of the metabotropic glutamate receptor 7(mGluR7) gene (Figs. 3A and 3B). The integration site was subsequently confirmed using a PCR primer from the known 5′ end of Tg(SMN2)89Ahmb to sequence distal to the cloned portion of mGluR7’s intron 4. The addition of a second forward primer corresponding to the uninterrupted mGluR7 locus allowed us to establish a genotyping assay capable of differentiating between wild type, hemizygous and homozygous mice (Fig. 3C).
Figure 3.
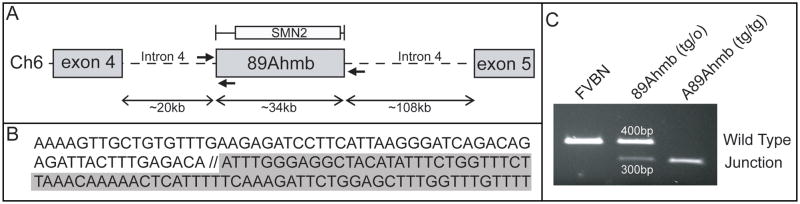
Integration Mapping of the Tg(SMN2)89Ahmb transgene. A) Genome walking was used to clone the integration site of Tg(SMN2)89Ahmb, which was shown to be within intron 4 of the mGluR7 gene. B) Presentation of the sequence at the integration site. Sequence with a white background is from the transgene while sequence in gray is from mGluR7. C) A 3-primer PCR assay of the integration site provides a genotyping assay capable of distinguishing between WT, hemizygous, and homozygous mice.
mGluR7 expression analysis is reduced in Tg(SMN2)89Ahmb mice
During general colony maintenance we and others have noted seizures in mice containing the Tg(SMN2)89Ahmb transgene (JAX strains 5024 and 5025), in some cases resulting in death. Additionally, we have observed a higher frequency of sudden unexpected death within our colony of Tg(SMN2)89Ahmb containing mice. The discovery that Tg(SMN2)89Ahmb integrated within the mGluR7 gene is relevant since mGluR7 loss renders mice susceptible to seizures (Masugi et al., 1999; Sansig et al., 2001). RT-PCR of 6–8 week old mice confirmed a significant 24% (±5%) reduction in mGluR7 cDNA levels (P=0.005, Fig. 4A) and a more modest but consistent 9% (±3%) reduction in mGluR7 protein levels (P=0.05) when Smn was +/+ (Fig. 4B). Interestingly, a 50% reduction in Smn (+/−) appeared to work synergistically with the Tg(SMN2)89Ahmb integration site to further reduce mGluR7 levels by 20% (±7%) of wild type levels (Figure 4B); however, Smn reduction alone (Smn+/+ vs. Smn+/−) did not significantly alter mGluR7 (91% ±7%; P>0.05) in age-matched adult mice (Fig. 4C). We went on to determine if this reduction was present in embryonic and early post natal development by analyzing equivalent genotypes of mice at E12.5, PND1, 5 and 17. We could identify mGluR7 reduced expression in samples that were homozygous for Tg(SMN2)89Ahmb at certain time points and genotypes, but the standard deviation (±14.5% average) due to inter-litter variability and developmental changes of mGluR7 was greater than the original observed reduction. This hampered our ability to consistently and quantitatively determine a diminution in mGluR7 during these early time points (Supplemental Fig. 1). We also evaluated whether the hybrid genetic background of this line influenced our observed mGluR7 adult expression or variability during these early time points by analyzing the fully congenic FVB/N counterpart (JAX strain 7949) but the results were the same in both instances (data not shown).
Figure 4.

mGluR7 expression analysis. A) RT-PCR of mGluR7 exons 3–6. Densitometry quantifies a 24% (±5%) reduction in transcript levels when normalized to Gapdh. B) Western blot analysis of mGluR7 followed by densitometry demonstrates a significant 9±3% reduction in protein levels in (SMN2)89Ahm; Smn +/+ mice and a 20±7% in (SMN2)89Ahmb; Smn +/− mice. C) Smn +/− mice exhibit a minor reduction in mGluR7 levels that fails to reach statistical significance (100±2% vs 92±7%, p=0.14). D) mGluR7 levels are unaltered in mice carrying another commonly used SMN2 transgene, (SMN2)2Hung (100±7% vs 100±2%, p=0.96). Values are mean ±SD.
To definitively determine that our observed decrease in mGluR7 was specifically caused by the integration of Tg(SMN2)89Ahmb, we analyzed mGluR7 in a completely different SMA mouse model, JAX strain 5058 (Hsieh-Li et al., 2000). This SMA model is homozygous for Δ7Smn and its SMN2 transgene, Tg(SMN2)2Hung, is located within an intergenic region on Chrm4 (Gogliotti et al., 2010). Western blot analysis confirmed no significant difference between wild type and Tg(SMN2)2Hung containing mice (Fig. 4D). Taken together, we conclude that the observed mGluR7 decrease in adult homozygous Tg(SMN2)89Ahmb mice is specifically attributed to the integration of this transgene within mGluR7 intron 4.
The integration of Tg(SMN2)89Ahmb into mGluR7 Intron 4 does not affect its localization or cause motor neuron loss
The integration of transgenes within the genome often causes genomic rearrangements at the site of integration that can affect expression of endogenous genes. Given our observed seizures in this line and paucity of information with regards to intronic enhancers/silencers within mGluR7 intron 4 that could have been disrupted, we used immunohistochemistry to localize mGluR7 in adult brains and spinal cords and compared Smn+/+ mice to Tg(SMN2)89Ahm tg/tg; Smn+/+ mice and Tg(SMN2)89Ahmbtg/tg;Smn+/− mice. In all genotypes, mGluR7 was appropriately localized in the cortex, the granule cells of the cerebellum, the blade of the hippocampus, the dorsal spinal cord and in motor neurons (Fig. 5A-E). It made no difference whether the line was a hybrid FVB/N genetic background (strain 5024) or pure FVB/N genetic background (strain 7949).
Figure 5.

Neither transgene integration or Smn levels affect mGluR7 localization. A–B) Consistent with published reports, mGluR7 expression is most prominent in the spinal dorsal horn, with expression in the ventral horn restricted primarily to motor neurons. This pattern remains consistent regardless of genotype. C-E) mGluR7 expression pattern is unaltered in the brain of Tg(SMN2)89Ahmb containing mice with diffuse localization in the cortex, high expression in the blade of the hippocampus and expression restricted to the granule cells of the cerebellum.
One of the cell types within the spinal cord that expresses mGluR7 is motor neurons, but how changes in mGluR7 expression affect motor neuron survival is not known. Because Tg(SMN2)89Ahmb’s primary use is in modeling a motor neuron disease (SMA), we counted toluidine blue stained semi-thin sections of femoral motor axons from 4-month old mice homyzygous for the transgene from both the hybrid and pure congenic FVB/N backgrounds and found no significant difference between mice that were homozygous for the transgene compared to age-matched wild type controls (P>0.05) (Table 1). These results are consistent with previous motor neuron counts between wild type mice and those homozygous for the transgene that were performed in early post natal life (Monani et al., 2000).
Table 1.
Summary of SMN2(Ahmb89) Femoral Motor Axon Counts
| Genotype | JAX Strain | Average Motor Axons (N±SEM) | P-Value |
|---|---|---|---|
| Smn +/+; (FVB/N) | 1800 | 570±18 | |
| SMN2(Ahmb89)+/+; Smn +/+; Hybrid | 5024 | 555±36 | P>0.05 |
| SMN2(Ahmb89)+/+; Smn +/+; Congenic | 7949 | 566±21 | P>0.05 |
Tg(SMN2)89Ahmb containing mice have behavioral deficits in the open field
To investigate the behavioral consequences of Tg(SMN2)89Ahmb’s integration into mGluR7, we studied the exploratory behavior of 11 week old Tg(SMN2)89Ahmbtg/tg, Smn+/+ (N=20); Tg(SMN2)89Ahmbtg/tg, Smn+/− (N=10) and wild type FVB/N (N=20) mice in an open field. While none were observed, it was initially thought that the stress of being placed in an open field might trigger a seizure event. However, mice homozygous for the Tg(SMN2)89Ahmb transgene on a wild type Smn background did explore significantly less than controls, in most cases remaining in the center square for an extended period of time (Fig. 6A). This resulted in a significant increase in the time taken to find a safe wall quadrant (+54s, P= 0.05) as well as a reduction in the number of beams crossed, but this did not reach statistical significance (Fig. 6B).
Interestingly, this behavioral deficit was amplified as Smn dosage was reduced from wild type to heterozygous levels. Tg(SMN2)89Ahmbtg/tg, Smn+/− remained in “danger” squares for an average of 90 seconds longer than controls (+90s, P=0.002) and explored significantly fewer total squares (−37, P=0.007) (Fig. 6B and 6C). As a follow up experiment, we repeated the test with age matched Smn (+/+) and Smn (+/−) FVB/N mice who were wild type at the mGluR7 locus and found no difference in either time to a safe square or total squares explored (Fig. 6D and 6E). This would indicate that if decreased Smn dosage plays a role in open field performance, it does so only in concert with a decrease in mGluR7 expression. Our data suggests that the behavioral abnormalities seen in Tg(SMN2)89Ahmbtg/tg containing mice are associated with a defect in fear response. This is in agreement with the published data on mGluR7 knock-out mice (Callaerts-Vegh et al., 2006; Masugi et al., 1999).
Tg(SMN2)89Ahmb containing mice have increased susceptibility to seizures
mGluR7 knockout mice have been shown to have an increased susceptibility to pentylenetrazol (PTZ) induced seizures (Sansig et al., 2001). As it is relevant to potential SMA therapeutics that might have seizure related side effects, we sought to determine if this is the case in Tg(SMN2)89Ahmb containing mice. Seizure susceptibility analysis of the 3 blinded groups (described in the previous section) was conducted using a sub-threshold 30mg/kg dose of PTZ (≤10% WT seizure frequency). The un-blinded analysis revealed that Tg(SMN2)89Ahmbtg/tg mice demonstrated a higher number (though not significant, P=0.06) and faster onset of myoclonic events, but only if Smn levels were reduced (Fig. 7A and 7B). Additionally, Tg(SMN2)89Ahmbtg/tg; Smn+/+ and Tg(SMN2)89Ahmbtg/tg; Smn+/− mice showed a significantly higher frequency of PTZ induced generalized seizure (0.45, P=0.02 and 0.50, P=0.02, respectively) when compared to wild type FVB/N mice (0.11) (Fig. 7C). No significant difference existed in the time between injection to seizure (Fig. 7D) or in the nature or duration of the generalized seizures, as they all contained myoclonic and tonic elements and lasted ~20s.
Figure 7.

Seizure analysis of Tg(SMN2)89Ahmbtg/tg mice. A) No significant difference existed in the time to the first myoclonic event regardless of Smn dosage. B) Tg(SMN2)89Ahmbtg/tg; Smn+/− mice had a higher number of myoclonic events within a 90% CI. No difference was observed in Tg(SMN2)89Ahmbtg/tg; Smn+/+ mice. C) No significant difference was observed in the time from PTZ injection to generalized seizure between the 3 groups. D) A significantly higher frequency of generalized seizure was observed in mice carrying the transgene relative to wild type FVB/N mice. Values are mean ±SEM.
Discussion
As part of our ongoing effort to re-characterize the SMA mouse models that have been deposited at The Jackson laboratory, we present our characterization of the Tg(SMN2)89Ahmb transgene and carrier SMA mice (JAX Strain 005024) (Monani et al., 2000). This baseline strain and more specifically the SMN2 transgene, is the basis for other SMA model mice that present with varying phenotypes (Le et al., 2005; Monani et al., 2003; Workman et al., 2009). Our survival results of Tg(SMN2)89Ahmbtg/tg; Smn−/− pups indicates a drift to a more severe SMA phenotype resulting in earlier death. We believe this is due to the fact that the mice are now in a more homogenous FVB/N background (66–70%) as compared to what was originally reported (Le et al., 2005; Monani et al., 2000). However, each laboratory will have a slightly different genetic makeup for their 5024 strain depending upon the breeding scheme and fixing of segregating alleles. In addition, environment and nutritional state have been shown to play a role in survival of severe SMA pups (Butchbach et al., 2010; Narver et al., 2008). These additional contributing factors could have impacted our survival results and those obtained by others who utilize this strain.
We have cloned the integration site for Tg(SMN2)89Ahmb and developed a genotyping assay that can differentiate whether this transgene is present in a hemizygous or homozygous state. The assay has been submitted to The Jackson Laboratory, where it is currently used and distributed as the primary mechanism of genotyping mice that contain the Tg(SMN2)89Ahmb transgene (lines TJL#005024 and TJL#005025). Importantly, from our integration site mapping we identified that Tg(SMN2)89Ahmb integrated into intron 4 of the mGluR7 locus. mGluR7 is one of eight members in the metabotropic glutamate receptor family. These are a heterogeneous group of G-protein-coupled receptors (GPCRs) and one of the most important classes of GPCRs in the nervous system. mGluR7 is postulated to be the most important member of this receptor family in regulating CNS function (Conn and Niswender, 2006). It is widely distributed and present at many synapses and localized primarily on the presynaptic terminals where it’s thought to modulate neurotransmitter release (Conn and Niswender, 2006; Kinoshita et al., 1998; Kosinski et al., 1999). mGluR7 knockout mice have been made and homozygotes are viable, but they have defects in taste aversion, depressive behavior, short term memory and most importantly to this study, they have an increased seizure susceptibility (Bushell et al., 2002; Cryan et al., 2003; Holscher et al., 2004; Masugi et al., 1999; Sansig et al., 2001).
Since mGluR7 plays such an important role in CNS function and synapse transmission, we further characterized the consequence of Tg(SMN2)89Ahmb’s integration into this locus and found that its presence created a hypomorphic allele. Normally the small decrease (~10%) in protein reduction would not be expected to have any effect, since mGluR7 heterozygotes (50% reduction) have no abnormalities in fear response and are resistant to chemically induced seizures by PTZ in a C57Bl/6 background (Sansig et al., 2001). However, FVB/N mice are known to be hyper-sensitive to seizures (Schauwecker and Steward, 1997). We postulate that the hypersensitivity of this inbred strain along with the small decrease in mGluR7 levels, caused by the homozygous presence of Tg(SMN2)89Ahmb, are acting synergistically to create the heightened fear response and seizure susceptibility that we have documented. It is interesting to note that decreased Smn dosage when combined with the homozygous Tg(SMN2)89Ahmb transgene amplifies the observed behavioral deficits and reduced mGluR7 dosage, but not on its own. Further research will be required to tease apart this relationship.
There is a paucity of information that addresses motor neuron loss or other motor impairments within mGluR7 knockout mice. Here we found no axonal loss in either the hybrid or pure congenic FVB/N lines of Tg(SMN2)89Ahmb mice. We also did not observe any gross alteration in the localization of mGluR7 or brain and spinal cord morphology, which is consistent with heterozygous mGluR7 mice (Sansig et al., 2001). However, we cannot exclude alterations in microcircuitry, which could contribute to our observed behavioral deficits or potential synaptic transmission defects given mGluR7’s cellular function, the later which we did not address. Since there are known strain specific genetic modifiers, transferring Tg(SMN2)89Ahmb to a non-hypersensitive background would likely eliminate the observed phenotypic abnormalities.
Mice containing the Tg(SMN2)89Ahmb transgene, such as the “delta 7” mouse (stain 5025), are the most widely used model in SMA research (Le et al., 2005). While their use in the characterization of many disease mechanisms should remain unaffected, our results should be viewed as cautionary for those studies that investigate synapse transmission or pre-clinical drug trials. For example, the normal lifespan of the “delta 7” SMA-like mouse is 14 days, however it has recently been demonstrated that TSA, ISSN-1 directed oligonucleotides, and AAV delivered SMN can increase lifespan well into adulthood, thus placing it at higher risk for seizure with increasing age (Avila et al., 2007; Foust et al.,; Narver et al., 2008; Singh et al., 2009).
It has been well documented that seizures trigger a host of inflammatory and immune responses that change the microenvironment in the CNS. We currently lack knowledge in regards to SMA disease mechanisms to ascertain whether this change in microenvironment would be beneficial or detrimental to disease progression; however a seizure induced increase in pro-inflammatory cytokines cannot be dismissed as inconsequential when assessing the phenotypic benefit of a potential therapeutic. Inflammation is meant to serve as a positive response for an area insult but often this can be detrimental to other areas that have been made vulnerable by disease. In fact, a recent study using Lenalidomide to combat the increase in inflammation seen in a mouse model of ALS was able to extend lifespan up to 45%; which might suggest it plays a negative role in some motor neuron diseases (Neymotin et al., 2009).
We maintain that Tg(SMN2)89Ahmb containing mice (strain 5024 and stain 5025) are valuable research tools and have proven useful in many studies addressing disease mechanisms and/or therapeutics. As a compliment to these SMA mice, we previously proposed that the Li SMA mouse model (JAX strain 5058) be used (Gogliotti et al., 2010). We deem the presented data further strengthens that point while contributing to our knowledge of a commonly used SMN2 transgene.
Supplementary Material
mGluR7 expression in Tg(SMN2)89Ahmb containing cannot be accurately quantified during early developmental time points. A) Reduction in mGluR7 expression was seen at most time-points and genotypes, but developmental regulation combined with the inherent variability in the rate at which individual mice and litters of mice develop, created a high average standard deviation (14.5%). This hindered our ability to accurately and reproducibly quantify mGluR7 levels at time points prior to adulthood. (N=5/genotype/time point) Representative samples of N=3 are shown for each time point and genotype. Similar results were obtained with the fully congenic background strain of mice (JAX 7949; N=3/genotype/time point) data not shown.
Acknowledgments
We thank the entire Koh laboratory for their assistance in the open field experiments.
Funding
R.G. is supported by a NIH training grant [T32 AG000260] “Drug Discovery Training in Age-related Disorders”. Research support for these studies was provided by the Families of Spinal Muscular Atrophy DiD0809, the NIH grants #1ROIN5060926 (NINDS), and 1R21HD058311-01A1 (NICHD) to CJD and the SMA Foundation to C.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Avila AM, et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117:659–71. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell TJ, et al. Altered short-term synaptic plasticity in mice lacking the metabotropic glutamate receptor mGlu7. Scientific World Journal. 2002;2:730–7. doi: 10.1100/tsw.2002.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butchbach ME, et al. Effect of diet on the survival and phenotype of a mouse model for spinal muscular atrophy. Biochem Biophys Res Commun. 391:835–40. doi: 10.1016/j.bbrc.2009.11.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaerts-Vegh Z, et al. Concomitant deficits in working memory and fear extinction are functionally dissociated from reduced anxiety in metabotropic glutamate receptor 7-deficient mice. J Neurosci. 2006;26:6573–82. doi: 10.1523/JNEUROSCI.1497-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Niswender CM. mGluR7’s lucky number. Proc Natl Acad Sci U S A. 2006;103:251–2. doi: 10.1073/pnas.0510051103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coovert DD, et al. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet. 1997;6:1205–14. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- Cryan JF, et al. Antidepressant and anxiolytic-like effects in mice lacking the group III metabotropic glutamate receptor mGluR7. Eur J Neurosci. 2003;17:2409–17. doi: 10.1046/j.1460-9568.2003.02667.x. [DOI] [PubMed] [Google Scholar]
- DiDonato CJ, et al. Association between Ag1-CA alleles and severity of autosomal recessive proximal spinal muscular atrophy. Am J Hum Genet. 1994;55:1218–29. [PMC free article] [PubMed] [Google Scholar]
- Foust KD, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. 2010;28:271–4. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gogliotti RG, et al. Molecular and phenotypic reassessment of an infrequently used mouse model for spinal muscular atrophy. Biochem Biophys Res Commun. 2010;391:517–22. doi: 10.1016/j.bbrc.2009.11.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heier CR, DiDonato CJ. Translational read through by the aminoglycoside geneticin (G418) modulates SMN stability in vitro and improves motor function in SMA mice in vivo. Hum Mol Genet. 2009;18:1310–22. doi: 10.1093/hmg/ddp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heier CR, et al. SMN transcript stability: could modulation of messenger RNA degradation provide a novel therapy for spinal muscular atrophy? J Child Neurol. 2007;22:1013–8. doi: 10.1177/0883073807305669. [DOI] [PubMed] [Google Scholar]
- Holscher C, et al. Lack of the metabotropic glutamate receptor subtype 7 selectively impairs short-term working memory but not long-term memory. Behav Brain Res. 2004;154:473–81. doi: 10.1016/j.bbr.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Hsieh-Li HM, et al. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- Kinoshita A, et al. Immunohistochemical localization of metabotropic glutamate receptors, mGluR7a and mGluR7b, in the central nervous system of the adult rat and mouse: a light and electron microscopic study. J Comp Neurol. 1998;393:332–52. [PubMed] [Google Scholar]
- Kosinski CM, et al. Localization of metabotropic glutamate receptor 7 mRNA and mGluR7a protein in the rat basal ganglia. J Comp Neurol. 1999;415:266–84. [PubMed] [Google Scholar]
- Le TT, et al. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14:845–57. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–65. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Masugi M, et al. Metabotropic glutamate receptor subtype 7 ablation causes deficit in fear response and conditioned taste aversion. J Neurosci. 1999;19:955–63. doi: 10.1523/JNEUROSCI.19-03-00955.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melki J, et al. Gene for chronic proximal spinal muscular atrophies maps to chromosome 5q. Nature. 1990;344:767–8. doi: 10.1038/344767a0. [DOI] [PubMed] [Google Scholar]
- Melki J, et al. De novo and inherited deletions of the 5q13 region in spinal muscular atrophies. Science. 1994;264:1474–7. doi: 10.1126/science.7910982. [DOI] [PubMed] [Google Scholar]
- Monani UR, et al. A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J Cell Biol. 2003;160:41–52. doi: 10.1083/jcb.200208079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monani UR, et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9:333–9. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- Narver HL, et al. Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition. Ann Neurol. 2008;64:465–70. doi: 10.1002/ana.21449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neymotin A, et al. Lenalidomide (Revlimid) administration at symptom onset is neuroprotective in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2009;220:191–7. doi: 10.1016/j.expneurol.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellizzoni L, et al. A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell. 1998;95:615–24. doi: 10.1016/s0092-8674(00)81632-3. [DOI] [PubMed] [Google Scholar]
- Sansig G, et al. Increased seizure susceptibility in mice lacking metabotropic glutamate receptor 7. J Neurosci. 2001;21:8734–45. doi: 10.1523/JNEUROSCI.21-22-08734.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci U S A. 1997;94:4103–8. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrank B, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A. 1997;94:9920–5. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NN, et al. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 2009;6:341–50. doi: 10.4161/rna.6.3.8723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman E, et al. A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Hum Mol Genet. 2009;18:2215–29. doi: 10.1093/hmg/ddp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerres K, Rudnik-Schoneborn S. Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol. 1995;52:518–23. doi: 10.1001/archneur.1995.00540290108025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
mGluR7 expression in Tg(SMN2)89Ahmb containing cannot be accurately quantified during early developmental time points. A) Reduction in mGluR7 expression was seen at most time-points and genotypes, but developmental regulation combined with the inherent variability in the rate at which individual mice and litters of mice develop, created a high average standard deviation (14.5%). This hindered our ability to accurately and reproducibly quantify mGluR7 levels at time points prior to adulthood. (N=5/genotype/time point) Representative samples of N=3 are shown for each time point and genotype. Similar results were obtained with the fully congenic background strain of mice (JAX 7949; N=3/genotype/time point) data not shown.