

Abstract
Background
Enteral nutrient-deprivation, via total parenteral nutrition (TPN) administration leads to local mucosal inflammatory responses, but the underlying mechanisms are unknown.
Methods
Wild-type (WT) and MyD88-/- mice underwent jugular vein cannulation. One group received TPN without chow and controls received standard chow. After 7days, we harvested intestinal mucosally-associated bacteria, and isolated small-bowel lamina propria (LP) cells. Bacterial populations were analyzed using 454-pyrosequencing. LP cells were analyzed using quantitative PCR and multi-color flow cytometry.
Results
WT, control mucosally-associated microbiota were Firmicutes-dominant while WT TPN mice were Proteobacteria-domiant. Similar changes were observed in MyD88-/- mice with TPN administration. Unifrac analysis showed divergent small bowel and colonic bacterial communities in controls, merging towards similar microbiota (but distinct from controls) with TPN. The percentage of LP T-regulatory cells significantly decreased with TPN in WT mice. F4/80+CD11b+CD11cdull-neg macrophage derived pro-inflammatory cytokines significantly increased with TPN. These pro-inflammatory immunologic changes were significantly abrogated in MyD88-/- TPN mice.
Conclusions
TPN administration is associated with significant expansion of Proteobacteria within the intestinal microbiota and increased pro-inflammatory LP cytokines. MyD88 signaling blockade abrogated this pro-inflammatory response.
Introduction
Parenteral nutrition (PN), or the removal of enteral nutrition, is commonly used as treatment for many patients, ranging from short-term use in patients with gastrointestinal dysfunction(1), to long-term use in patients with short bowel syndrome(2). While it is life-saving for many, PN use is associated with numerous complications ranging from an increase in enteric-derived infections to a loss of immune reactivity(3, 4).
Previous studies from our laboratory and others have shown that in a mouse model of total PN (TPN) there are a number of significant physical and immunologic changes in the intestinal mucosa(5). Physically, there is atrophy of small bowel villi, an increase in epithelial cell (EC) apoptosis, and a decrease in EC proliferation(6). Immunologically, there is a pro-inflammatory state within the gastrointestinal tract, including increased mucosal and intraepithelial lymphocyte-derived tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), and decreased interleukin (IL)-10(7, 8). However, the mechanisms driving these changes are unknown. Such changes may have a profound impact on the host, including a loss of epithelial barrier function (EBF)(8, 9) and increases in bacterial translocation(10). These findings have been shown in mouse models and in humans(11) on PN.
Several studies(12, 13) have shown a critical role of cross-talk between the EC and lamina propria (LP) compartments. Because of this, we hypothesized that immunologic changes within the LP may be driving the mucosal pro-inflammatory response. A principal function of LP cells is to detect and monitor changes in the intraluminal environment(14-16). A robust body of literature shows the importance of the microbiota in the host's physiology(15, 17, 18). A major pathway through which these microbes interact with the host is via the toll-like receptor (TLR) pathway. Many bacterial components are ligands of TLRs(19), and a major downstream activation pathway for several TLRs is nuclear factor-κB (NFκB) signaling via a myeloid differentiation primary response gene 88 (MyD88) dependent pathway(20). NFκB activation is known to mediate the expression of several pro-inflammatory cytokines including TNF-α(20).
Despite sustaining the host organism with sufficient energy and nutrient needs, TPN puts the intestinal microbiota in an abrupt state of nutrient withdrawal. The consequences to the resident microbial community during this environmental change have not been addressed. The intestinal microbiota is highly sensitive to local environmental changes, and the composition of the population may be rapidly altered in response to such dramatic changes(21). In this study, we show that administration of TPN led to profound shifts in the small intestinal microbiota; moving from a gram-positive Firmicutes-dominant community to a gram-negative Proteobacteria-dominated community. Since the intestinal microbiota interact with the host via the TLR signaling pathway, we hypothesize that blocking the TLR signaling pathway would abrogate the mucosal pro-inflammatory response seen with TPN administration. In particular, we show increased macrophage TLR-4 signaling, a loss of LP regulatory T-cells (Treg) and a pro-inflammatory cytokine response in the LP macrophages in wild-type mice. In MyD88-/- mice, we show a prevention of this loss of Treg cells, and an abrogation of the TPN-associated mucosal pro-inflammatory response. These findings suggest the key role of the TLR and MyD88 signaling pathway on the profound physiologic intestinal changes associated with TPN administration.
Materials and Methods
Animals
All experiments were done in accordance to guidelines set forth by the University of Michigan's University Committee on the Use and Care of Animals. Experimental protocols were approved by this committee (protocol #07703).
Wild-type (WT) male C57BL/6 mice were obtained from the Jackson Laboratory (Bar Harbor, ME). MyD88 knockout (MyD88-/-) mice on a C57BL/6 background(13) were obtained from Dr. Steven Kunkel at University of Michigan, and used with permission from Dr. Shizuo Akira. All were housed and bred in the same room within the University of Michigan Unit for Laboratory Animal Medicine under specific pathogen free conditions. MyD88-/- mice used for experiments were genotyped using primers listed in Supplemental Figure S1a. All mice were at least 2nd generation before being used. All experiments were performed with at least 4 mice in each group.
TPN Model
Mice were used between 10 and 14 weeks of age. WT or MyD88-/- mice underwent internal jugular vein catheterization as previously described(6). Mice were recovered for 2 days after cannulation with full access to chow and water, and 5mL per day of 0.9% normal saline running through the catheter. On day 3, mice in the study group had their chow removed, and started receiving a balanced PN solution as previously described(22) (composition of solution shown in Supplemental Table S1). Control mice continued to have free access to chow, and received normal saline through the catheter. All mice were housed in IVC racks. All mice were killed on day 7 by CO2 asphyxiation.
Bacterial analysis (terminal restriction fragment length polymorphism, T-RFLP; pyrosequencing)
From each mouse, 1cm segments of small intestine and colon were isolated using alcohol-sterilized instruments. Isolation of mucosally-associated bacteria was performed as previously described. (23) Segments were opened, adherent stool rinsed off in sterile media consisting of RPMI 1640 with glutamine (Invitrogen, Carlsbad, CA) and 5% fetal bovine serum (FBS) (Invitrogen), while preserving the mucosally-associated bacteria. This segment was then snap-frozen in liquid nitrogen until analyzed. Bacterial DNA was extracted using established methods(24). T-RFLP was then performed as previously described(25). Raw T-RFLP chromatograms were analyzed using Peak Scanner (Applied Biosystems, Carlsbad, CA) to call the fragment sizes and to build a list of peaks. This process was carried out for every sample, after which all of the peak files were exported as one bulk file. Further analysis was carried out using K9, an in-house designed program for T-RFLP data analysis (freely available at http://www-personal.umich.edu/~jre/Microbiome_Core/K9.html).
The bacterial tag-encoded FLX-Titanium amplicon pyrosequencing method targeting the V1-V3 variable regions of 16S rRNA was used to create amplicon libraries(26). V1-V3 primer sets corresponded to 27F (5’-GAGTTTGATCNTGGCTCAG-3’) and 519R (5’-GTNTTACNGCGGCKGCTG-3’), along with appropriate sample nucleotide bar codes and the Roche A&B primers. Pyrosequencing was performed following established protocols(27) at Research and Testing Laboratories (Lubbock, TX).
Analysis of sequenced data was performed using Mothur, an open-source, community-supported software for describing and comparing microbial communities(28), following the example of Costello Stool Analysis with default software settings.
Isolation of IEC and LP cells
Small intestine underwent removal of Peyer's patches, remaining intestinal segments were split longitudinally, and fecal matter washed out in RPMI 1640 with glutamine on ice. ECs were isolated and purified as previously described(29, 30). The remaining intestinal segments were incubated in a 1mM dithiothreital (Sigma-Aldrich, St. Louis, MO) + 1mM ethylene diaminetetraacetic acid (Sigma) in phosphate buffered solution (PBS, Invitrogen) at 37°C, and stirred at 350 rpm to break off the remaining ECs. This supernatant was discarded, and the remaining segments were incubated in a collagenase mixture (50mL RPMI 1640 with glutamine, 5% FBS, 0.5mM CaCl2, 0.9mM MgCl2, 100-300 U/mL collagenase Invitrogen) for 60 minutes at 37°C. The digested intestinal segments were filtered through a 40 micron filter (BD, San Jose, CA), then run on a 40%/66% Percoll (Sigma) gradient. The cells at the 40% and 66% Percoll interface were washed with RPMI+5% FBS and used for further analysis.
Reverse transcriptase polymerase chain reaction (PCR)
Ribonucleic acid (RNA) from isolated ECs and LP cells was extracted using Trizol reagent (Life Technologies, Inc., MD) according to the manufacturer's directions. Extracted mRNA was reversed transcribed into complementary DNA (cDNA) as previously described(31).
Quantitative real-time PCR
Real-time PCR was performed as previously described(30). Fold changes of target genes were calculated using comparative quantification to β-actin. Primers used are listed in Supplemental Figure S1b.
Immunofluorescence microscopy
Sections of small intestine were taken immediately after killing mice, and fixed in Zinc-formalin as previously described(30). Tissue was then paraffin-embedded, and sectioned to 5μm thickness. Immunofluorescence staining was performed as previously described(6). p65-NFκB (Cell signaling Technology, Beverly, MA) primary antibody with Texas-Red conjugated goat anti-rabbit secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was used. Proliferating cell nuclear antigen (PCNA) staining was performed using PCNA mAb (Cell Signaling) with FITC-conjugated goat anti-rabbit secondary antibody (Santa Cruz). ProLong® Gold antifade reagent with 4’6-diamidino-2-phenylindole (DAPI) (Invitrogen) was added, and slides visualized using a Nikon Ti-E fluorescence microscope.
Flow cytometry
Isolated cells were diluted to ~1-3×106 cells/mL in RPMI 1640 with glutamine, 5% FBS and 10mM HEPES (Invitrogen). GolgiPlug (BD) and GolgiStop (BD) were added according to the manufacturer's instructions, and cells incubated in a 5% CO2 incubator at 37°C for 4-6 hours. Cells were not stimulated with phorbol 12-myristate 13-acetate (PMA) or ionomycin to capture their in-vivo state. After this, cells were washed with a wash buffer consisting of PBS with 1% FBS and 0.1% NaN3, then blocked with Fc blocker (eBioscience, San Diego, CA), stained using a selected cocktail from the antibodies listed in Supplemental Figure S1c, and fixed overnight in Fixation/Permeablization buffer (eBioscience). After fixation/permeablization, the cells were washed with FOXP3 staining buffer (eBioscience), and intracellular markers were stained. An LSR-II (BD) was used to run the cells, and FACSDiva 6.2 (BD) software was used to acquire the data. FlowJo 7.5 (Tree Star, Ashland OR) was used for analysis. Appropriate isotype controls (also listed in Supplemental Figure S1c) were used to set the positive and negative thresholds in gating for analysis.
Physiologic EBF studies
Transepithelial resistance (TER) across full-thickness mouse small intestinal samples was performed using a modified Ussing chamber (Physiologic Instruments, San Diego, CA). Small intestinal tissue was cut along the mesenteric border, and mounted in 0.3cm2 tissue-holders. The tissue was allowed to equilibrate in 37°C Krebs buffer as previously described(9). Three consecutive tissue samples were measured for each mouse. TER was analyzed using instrument-associated software Acquire & Analyze v2.3 (Physiologic Instruments).
Statistical Analysis
All results are expressed as mean±standard deviation unless otherwise specified. Comparison between two groups used the unpaired T-test, and comparison between three or more groups used analysis of variance (ANOVA) with Tukey's posthoc test unless otherwise specified.
Results
Enteral nutrient deprivation with TPN leads to significant changes in the mucosally-associated intestinal microbiota
T-RFLP analysis was performed first to examine if there were significant changes in the small bowel and colonic mucosally-associated bacterial population. A dendrogram was constructed from this data to analyze the relatedness, as measured by Bray-Curtis distances, of small bowel and colon study groups (Figure 1A). We noted that control small bowel and colon samples were more similar to each other than when they were compared with the same portion of the bowel after TPN administration. Figure 1B shows a chromatogram where the differences in the terminal restriction fragments of the mucosally-associated bacteria are easily visualized. These data indicate that significant community shifts occur in the small bowel and colon during TPN administration.
Figure 1.
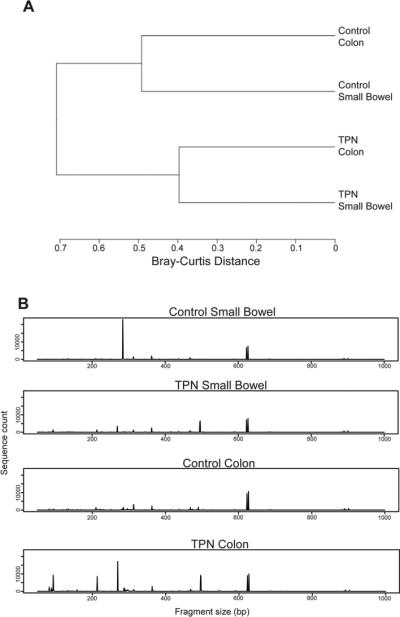
A: Dendrogram representation of small bowel and colonic mucosally-associated bacteria samples in wild type mice using T-RFLP and analysis of similarities (n=6 in each group). Bray-Curtis distances shown by scale at the bottom. P=0.003 between control small bowel and TPN small bowel, and P=0.005 between control colon and TPN colon. B: Chromatogram representation of TRFLP analysis. Restriction fragment size on X-axis, count on Y-axis.
A significant limitation of T-RFLP analysis is the difficulty of identifying bacteria taxonomically. To overcome these limitations, we employed 454-pyrosequencing of the V1-V3 region of bacterial 16S rRNA. A total of 220,656 sequences were obtained from 4 groups of animals (minimum of n=5/group), for an average of 9,593 sequences per animal. Sample sequencing coverage was estimated ranging from 91.4% to 99.7% (mean 97.3%), indicating good, in-depth sampling of the microbial communities. Sequences were annotated using the ribosomal database project classifier(32).
At the phylum level, the vast majority of the mucosally-associated bacteria in the small bowel of the control mice were Firmicutes (Figure 2A). However, in the TPN group, the dominant phyla were Proteobacteria and Bacteroidetes. To examine more specifically which members of the Proteobacteria and Bacteroidetes phyla were expanded in TPN mice, we examined genus level classification. While there were variations between individual mice, as a group, TPN mice had more bacteria in genera Salmonella, Escherichia, Proteus and Bacteroides (Figure 2 B i-iv). These genera are often associated with clinical infections, potentially indicating the development of a pathological state within the intestinal microbial community.
Figure 2.

A: Phylum level analysis after RDP classification of pyrosequenced ileal mucosa-associated bacteria samples. Most common phyla are shown in the graph. There were significantly fewer Firmicutes (2.5±1.9% vs. 93.1±6.2%, P<0.0001), more Proteobacteria (76.2±0.2% vs. 2.6±5.6% P<0.001), more Verrucomicrobia (2.0±1.7% vs. 0.02±0.05%, P<0.01) and more Bacteroidetes (17.9±17.7% vs. 0.2±0.4%, P<0.05) in TPN samples compared to control samples.
Sequence counts of Salmonella (Bi) Escherichia (Bii) Proteus (Biii) and Bacteroides (Biv) genera after genus-level classification of mucosally-associated bacteria samples. While the overall Proteobacteria dominance is apparent, there is variability between individual mice (n=6).
C: Weighted Unifrac principal coordinates analysis of control and TPN small bowel and colon samples. Axis-1 (X) and axis-2 (Y) account for 29.4% and 19.1% of overall differences, respectively.
D: Percentages of Firmicutes, Proteobacteria, Bacteroidetes, Verrucomicrobia and all other phyla in the small bowel of control and TPN mice (both WT and MyD88-/- strains). There is a marked shift from a Firmicutes-dominant flora in control mice to a Proteobacteria dominant flora in TPN mice in both the WT and MyD88-/- mice. Note also an expansion of Verrucomicrobia in the WT mice, and an even larger expansion of this population in MyD88-/- mice (n=4).
Classification-based approaches can be somewhat limited since many of the bacteria present in the intestines are un-culturable. To circumvent this limitation we next analyzed the data using Mothur(28), which bins the data into operational taxonomic units (OTUs) based on percent sequence identity (a 3% sequence identity cutoff was used for these analyses). Figure 2C depicts a principal coordinates analysis (PCoA) using the weighted Unifrac distance metric. In controls, discrete clustering was seen in small bowel and colon communities. However, after TPN, the communities look more similar. This indicates that enteral nutrient deprivation results in a loss of diversity between the large and small intestine.
TLR-signaling pathway is up-regulated in TPN mice
Given these significant shifts in the mucosally-associated bacteria, and the role of the LP in luminal sensing, we hypothesized that there would be associated changes in LP-TLR pathway signaling. As the greatest changes in epithelial barrier function and cytokine changes were seen previously in the small bowel, we focused the remainder of our analysis to the small bowel.
At the mRNA level, the expression of TLR-2, TLR-4 and MyD88 were significantly up-regulated in the LP of WT mice given TPN (Figure 3 A-C). When cell surface TLR-4 expression was examined on LP CD3+ lymphocytes by flow cytometry, only minimal amounts were identified (data not shown). Abundances of TLRs on small bowel epithelial cells failed to show consistent changes between study groups (data not shown). However, when the large cell population of the LP were examined, significant increases were noted in the percentage of F4/80+CD11b+CD11cdull-neg macrophages that expressed TLR-4 in WT TPN mice (Table). CD11chi dendritic cells (DCs) did not show significant differences in cell surface TLR-4 expression (Supplemental Figure S2). These results suggest that the predominant increase in TLR-4 signaling with TPN administration was in LP macrophages.
Figure 3.

Quantitative real time PCR on LP cell mRNA (A. MyD88, B. TLR-2, C. TLR-4, D. TNF-α, E. IFN-γ, F. FOXP3, G. IL-1β, H. IL-2, I. IL-6, J. IL-10, K. IL-17, L. TGF-β1) normalized to β-actin except for panel F where FOXP3 mRNA was normalized to the stable portion of the α-chain of the T-cell receptor to account for the fewer number of LP T-cells in the TPN mice (data not shown). ^P<0.05, †P<0.05 compared to MyD88-/- TPN, *P<0.05 compared to all other samples, #P<0.05 compared to WT control, $P<0.05 compared to both control samples. N=6 for WT mice, n=5 for MyD88-/- mice.
Table.
Flow cytometric analysis of LP cells
| FOXP3 | TLR-4 | IFN-γ | TNF-α | |
|---|---|---|---|---|
| WT control | 13.4±3.0 | 2.7±3.3 | 2.8±1.9 | 3.1±1.5 |
| WT TPN | 6.3±3.2* | 11.6±1.3* | 6.8±3.9* | 6.9±1.1* |
| MyD88-/- control | 11.6±3.5 | 4.1±2.9 | 2.7±1.2 | 2.9±1.5 |
| MyD88-/- TPN | 12.8±3.7 | 2.1±1.0 | 2.6±16 | 3.7±2.5 |
Results shown as mean±SD.
p<0.05 compared to all other groups by ANOVA
FOXP3 column represents the percentage of FOXP3+ cells within CD4+ T-cells by flow cytometry. TLR-4, IFN-γ, TNF-α columns represent percentages of F4/80+CD11b+CD11cdull-neg macrophages expressing each of those proteins.
Pro-inflammatory cytokine signaling is up-regulated in TPN mice
Previous work from our laboratory, and others, have found increased levels of pro-inflammatory cytokines in the mucosa and intraepithelial lymphocyte components of the small bowel of mice receiving TPN(5). In the present work, LP mRNA expression levels of TNF-α, IFN-γ, IL-1β, IL-2, IL-6, IL-10, IL-17 and transforming growth factor-β1 (TGF-β1) were examined. TNF-α and IFN-γ mRNA levels increased significantly in WT TPN mice compared to controls (Figure 3 D and E). IL-1β, IL-2, and IL-6 also increased in WT TPN mice (Figure 3 G, H and I), while TGF-β1 levels decreased significantly with TPN (Figure 3L). However, IL-10 and IL-17 mRNA levels did not significantly change (Figure 3 J and K).
To further define the source of these pro-inflammatory signals, multi-color flow cytometry with intracellular cytokine staining was performed. In TPN mice, there was minimal LP lymphocyte TNF-α and IFN-γ without exogenous PMA and ionomycin stimulation (data not shown). However, even without such exogenous stimulation, there was a significant up-regulation of intracellular TNF-α and IFN-γ in the LP F4/80+CD11b+CD11cdull-neg macrophages (Table), with little change in the CD11chi dendritic cells (Supplemental Figure S2). This line of data also strongly suggests that the pro-inflammatory signals in our TPN model are primarily derived from the LP F4/80+CD11b+ CD11cdull-neg macrophages.
LP Treg population significantly declines in WT TPN mice
As Treg cells are potent anti-inflammatory mediator35, we hypothesized that a loss of the Treg population may be observed in the pro-inflammatory state of the LP. Using flow cytometric analysis, the percentage of FOXP3+ cells within the CD3+/CD4+ lymphocyte population decreased significantly in WT TPN mice (Table). These findings were confirmed at the mRNA level (Figure 3F). Here, we normalized the expression of FOXP3 to the alpha chain of the T-cell receptor to account for differences in the total number of T-cells in the LP between control mice and TPN mice.
MyD88-/- mice baseline characteristics
To examine the role of TLR-signaling in our model, we proceeded to study the mucosal immune response in MyD88-/- mice. This blocked multiple TLR pathways at once. At baseline, the MyD88-/- mice had no gross physical differences versus WT mice. However, baseline intestinal immune cell composition significantly differed. MyD88-/- CD4+ intraepithelial lymphocyte (IEL) percentage was extremely low (Figure 4A) versus WT controls. However, LP CD4+ and CD8+ cell composition in MyD88-/- mice did not differ significantly from the WT controls (Figure 4 B, C and D). Percentage of LP Treg was also unchanged between the WT and MyD88-/- controls (Table).
Figure 4.
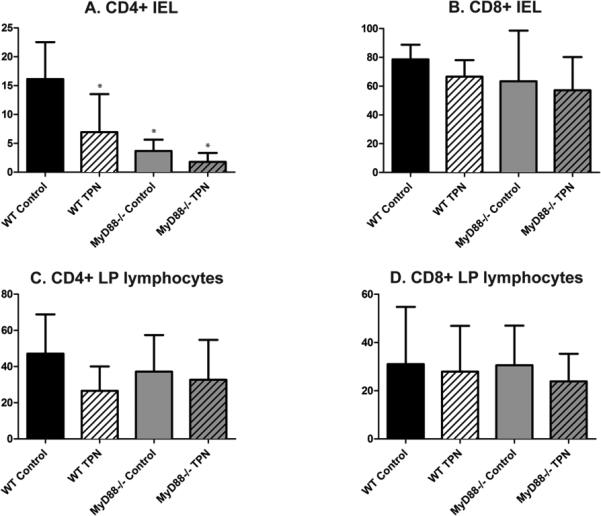
Panels A and B show small bowel CD4+ and CD8+ intraepithelial lymphocyte (IEL) percentages in WT and MyD88-/- control and TPN mice. Panels C and D show small bowel CD4+ and CD8+ lamina propria lymphocyte (LPL) percentages in WT and MyD88-/- control and TPN mice. MyD88-/- mice as well as WT TPN mice had a lower percentage of CD4+ IELs compared to WT control mice, however there were no statistically significant differences between the percentages of other cell populations between study groups. *P<0.05 compared to WT control. N=6 for WT mice, n=5 for MyD88-/- mice.
TPN-associated Macrophage TLR-4 and pro-inflammatory cytokine expression abrogated in MyD88-/- mice
We first examined the effect of MyD88-/- mice on TLR abundance. We found that expression levels of TLR-2 and 4 were similar between the WT control and MyD88-/- control mice (Figure 3 B and C). However, with TPN, TLR-2 and TLR-4 mRNA expression levels that rose in WT mice did not rise in MyD88-/- mice (Figure 3 B and C). Interestingly, the increases in TNF-α and IFN-γ were also abrogated in the MyD88-/- TPN mice (Figure 3 D and E). The up-regulation of cytokines IL-1β, IL-2 and IL-6 observed in the WT mice were also not seen in MyD88-/- mice. However, LP TGF-β1 significantly decreased in MyD88-/- mice (Figure 3L).
At the cellular level, there was minimal TNF-α and IFN-γ in both the CD4+ and CD8+ population, similar to the WT animals (data not shown). However, the up-regulation of intracellular TNF-α and IFN-γ in the LP F4/80+CD11b+ CD11cdull-neg macrophages seen in WT TPN mice was not seen in MyD88-/- TPN mice (Table).
We next asked if this prevention of pro-inflammatory cytokine abundance might be due to an expansion in the Proteobacteria population seen in WT TPN mice. Figure 2D shows phylum level 454 pyrosequencing data (genus level details are seen in Supplemental Table S2). While some differences occurred between WT and MyD88-/- mice, a similar trend of loss of the Firmicutes population and a predominance of Proteobacteria was also seen in the MyD88-/- TPN mouse group. Interestingly, the loss of the MyD88 signaling pathway led to a marked expansion in a much less common population of Verrucomicrobia. Inspection of this population at the genus level showed this to be completely comprised of Akkermansia.
Epithelial barrier function and EC proliferation loss is significantly prevented in MyD88-/- mice
As we have previously reported a loss of jejunal and ileal EBF in our TPN mouse model(8, 9, 33), we next examined if there were any physiologic effects of knocking out the MyD88 signaling pathway. We hypothesized that the failure of MyD88-/- mice to show an increase in pro-inflammatory cytokines with TPN would prevent the loss of EBF. Baseline TER measurements were similar between the WT control mice and MyD88-/- control mice (27.7±4.5 vs. 24.2±5.7 Ω*cm2). WT TPN mice had a significant (P<0.001) drop in TER (12.7±1.6 Ω*cm2). However, TER for MyD88-/- TPN mice was 19.5±3.8 Ω*cm2; while significantly (P<0.05) lower than that in the WT control mice, it was also significantly (P<0.05) higher than that of WT TPN mice (Figure 5A).
Figure 5.

A: TER measured by Ussing chamber, Y-axis units are Ohm*cm2. *P<0.05 between indicated samples. B: Epithelial cell proliferation measured as percentage of PCNA+ cells per crypt. #P<0.05 compared to all other samples. C-F: Representative immunofluorescent images of crypt PCNA staining (C. WT Control, D. WT TPN, E. MyD88-/- Control, F. MyD88-/- TPN). Green–PCNA, blue–DAPI. N=6 for WT mice, n=4 for MyD88-/- mice.
Another consequence of TPN is the loss of EC proliferation. Our laboratory has previously shown that this is due to an imbalance of epidermal growth factor and TNF-α signaling(36). With the prevention of TPN-associated TNF-α increase, we next examined if there would be a preservation of EC proliferation in MyD88-/- mice. As shown by immunofluorescence images (Figure 5 C to F), PCNA expression was maintained in the MyD88-/- TPN mice. The percentage of PNCA+ cells per crypt was maintained in the MyD88-/- mice (Figure 5B), and was not significantly different than WT controls.
Discussion
Our results show a strong association between TPN administration with deprivation of enteral nutrition and profound changes in the intestinal microbiota. Furthermore, we also demonstrated increases in LP TLR signaling and a loss of the Treg population, all of which appear to contribute to a mucosal pro-inflammatory response. These results provide important and novel insights into mechanisms of the local mucosal pro-inflammatory state seen in mice given TPN, and may account for such changes in patients receiving TPN.
Previous work that evaluated mucosal changes in response to TPN showed inflammation at the level of whole mucosal isolates, and the IEL population. Epithelial phenotype changes, such as increased apoptosis and decreased proliferation, as well as loss of epithelial barrier function have also been observed(8, 9). Changes in TLR expression at the whole intestinal level have been described in a mouse TPN model(34). However, the immunologic response within the LP had not been defined, nor was it clear what factors drove these pro-inflammatory changes. Work by Dahan et al(12) showed that epithelial cell phenotype can be markedly affected by LP cells; and this suggests that examining signaling changes in the LP may provide mechanistic insight into known EC changes in our TPN model.
The root cause of the microbiota changes and subsequent inflammatory cascade is most likely due to the lack of enteral nutrition than the TPN solution itself(35). Unfed mice will almost uniformly die within 3 days, and do not describe a clinically relevant physiologic condition. Our model offers a unique method to study the effects of subacute enteral nutrient deprivation while providing sufficient calories for survival. One potential issue was that the experiments were performed using regular rodent chow, and not with a rigorously controlled semi-purified diet. Although we did not see any inconsistencies with the experiments over the time we performed this work, it is possible that this could be a confounding variable that could be addressed in subsequent work.
We hypothesized that deprivation of enteral nutrition reduced nutritional substrate available to luminal bacteria, and promoted the survival of starvation resistant Proteobacteria(36, 37). While the animal model and duration of deprivation of enteral nutrients is very different, a Burmese python model of enteral nutrient deprivation and re-feeding showed significant shifts from Bacteroidetes which were dominant in the fasting state, while Firmicutes established themselves during a fed state(38). In addition to changes in enteral contents from above, goblet cell numbers increase with the administration of TPN(39). This is especially relevant because such EC phenotype changes affect the local environment for the mucosally-associated bacteria, and mucosal glycan foraging ability is directly linked to bacterial fitness within the human gut microbial community(40). These changes lead to altered TLR signaling in antigen presenting cells in the LP, which are known to alter lymphocyte phenotype(14, 16). In MyD88-/- mice, the TLR signaling changes caused by the alteration in luminal bacterial microbiota is not sensed, which leaves the mucosal immune response unchanged from the control state. It is important to note that the shift from a Firmicutes-predominant microbiota in the MyD88-/- control mice to a Proteobacteria predominant microbiota in the MyD88-/- TPN mice was also observed, indicating that similar changes in bacterial signaling are also present in the MyD88-/- mice. While clearly beyond the scope of the present work, we were struck by the marked increase in Akkermansia in the MyD88-/- TPN mice. It is possible that the prevention of Proteobacteria from signaling via TLRs and subsequent loss of pro-inflammatory signaling may have led to a more favorable environment for this relatively rare strain of bacteria. A. muciniphila live off of acidomucins, which have been reported to be increased with TPN administration in other animals41. Further work will need to be done to understand the complex inter-relation between these microbial groups.
We found that in addition to the abrogation of the mucosal pro-inflammatory response, there was also physiologic preservation of EBF in MyD88-/- mice receiving TPN. This was not unexpected, as we saw a similar preservation of barrier function in IFN-γ-/- mice(33), suggesting that the decreased IFN-γ levels in the MyD88-/- mice contributed to the preservation of EBF. An additional physiologic effect we saw was the preservation of EC proliferation. Data from our laboratory has shown that TNF-α receptor-1 and -2 double knockout mice have retained EC proliferation after TPN administration(41), indicating that reduced levels of TNF-α expression in the MyD88-/- mice may be responsible for their preserved proliferative capacity. Whether the prevention of loss of EC proliferation in the MyD88-/- mouse model is due to a single TLR pathway is, though beyond the scope of this manuscript, a very important question. Hsu et al(42) have noted that TLR-4 was critical for effective EC proliferation. Thus, further dissection of these TLRs will be an important line of future investigation.
A major finding in our study is the preservation of the small intestinal Treg population in the MyD88 -/- mice receiving TPN. Atarashi et al(43) have shown that Clostridium strains within the colon promote Treg accumulation, but not within the small intestine. It is known that certain polysaccharide-A producing commensal organisms, such as B. fragilis, have the ability to induce Treg differentiation in the normal gut via a TLR-2 dependent mechanism(44). These findings would seem to argue that there would either be no change in small intestinal LP Tregs, or if anything increase Treg development in our TPN mice (because of increase in TLR-2 levels and increase in the Bacteroides genera in our TPN mice). However, it is unknown if the TPN-microbiota produces polysaccharide-A, and the Treg changes may be driven more by the local TGF-β levels(43) (which were decreased in the TPN model – Figure 3L) than by the existence or lack thereof of Clostridium spp.
Denning et al(45) have shown that LP CD11b+CD11cdull-neg macrophages along with TGF-β1 can lead to Treg differentiation. Furthermore, these macrophages produce IL-10, which results in macrophage anergy, or a loss in the pro-inflammatory response secondary to TLR signaling(46). In our TPN model, the pro-inflammatory phenotype in the LP macrophages is abated when the MyD88 signaling pathway is blocked. There are several possible explanations for this seemingly contradictory finding. One is that the intestinal macrophages in the TPN model may consist of newly recruited macrophages that still possess the ability to respond in a pro-inflammatory manner. Another is that the decreased TGF-β1 levels with TPN may be preventing maintenance of the anergic state, leading to intestinal macrophages with an inflammatory phenotype.
Slack et al(47) have shown protection against increased intestinal permeability with dextran sodium sulfate and NSAIDS in MyD88 and TRIF (Toll/interlukin-1 receptor-domain-containing adapter-inducing interferon-β) double knockout mice. While their model knocks out both Myd88 dependent and independent TLR-signaling, and has different injury mechanisms, the protection of EBF in the knockouts is consistent with our results. A limitation of our study is that it does not directly prove that the changes in the microbiota are responsible for the increased TLR pathway signaling in the TPN mice. However, the near complete loss of a pro-inflammatory response after deleting a major microbial signaling pathway argues strongly for a mechanistic role for the microbiota with TPN administration. Studies with gnotobiotic or single-strain colonized mice (such as in the Atarashi(43) and Slack(47) stuidies), and/or bone marrow chimeras between WT and MyD88-/- mice would be necessary to more conclusively prove this relationship, and to further clarify the role of the LP versus the epithelium in the TPN model, but these are beyond the scope of this manuscript.
In conclusion, enteral nutrient deprivation with TPN administration in mice leads to profound changes in the mucosally-associated microbiota of the small bowel and colon. These micobiota changes are associated with a significant up-regulation in TLR signaling in the LP, and a subsequent increase in LP macrophage pro-inflammatory cytokine expression as well as a loss of the Treg population. This pro-inflammatory response was abrogated upon blockade of the MyD88 signaling pathway. These results suggest a critical role of the intestinal microbiota and TLR signaling pathway in the development of a local inflammatory response in response to deprivation of enteral nutrition. Future strategies to block this signaling pathway in patients receiving TPN, may well provide a beneficial effect and reduce the increased rate of associated infections.
Supplementary Material
Acknowledgements
The authors would like to acknowledge Adam Booth and Keith Bishop for use of their PCR primers; Brian Smereka, Lloyd Mayer, Brian Sheridan, Lynn Puddington and Leo Lefrançois for their guidance in refining the LP cell isolation technique; Weiping Zou, Linda Vatan and Ilona Kryczek for use of the LSR-II and assistance with flow cytometry; Pele Browner for technical assistance with immunofluorescence imaging; Shizuo Akira and Steve Kunkel for usage of the MyD88-/- mice.
Grant Support: T-32HD007505 (to EAM), NIH-R01 AI-44076-11 (to DHT).
Footnotes
Disclosures: No conflicts of interest exist.
REFERENCES
- 1.Braga M, Ljungqvist O, Soeters P, Fearon K, Weimann A, Bozzetti F. ESPEN Guidelines on Parenteral Nutrition: surgery. Clin Nutr. 2009;28:378–386. doi: 10.1016/j.clnu.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Duro D, Kamin D, Duggan C. Overview of pediatric short bowel syndrome. J Pediatr Gastroenterol Nutr. 2008;47(Suppl 1):S33–36. doi: 10.1097/MPG.0b013e3181819007. [DOI] [PubMed] [Google Scholar]
- 3.Gogos CA, Kalfarentzos F. Total parenteral nutrition and immune system activity: a review. Nutrition. 1995;11:339–344. [PubMed] [Google Scholar]
- 4.The Veterans Affairs Total Parenteral Nutrition Cooperative Study Group Perioperative total parenteral nutrition in surgical patients. N Engl J Med. 1991;325:525–532. doi: 10.1056/NEJM199108223250801. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Kudsk KA, Gocinski B, Dent D, Glezer J, Langekamp-Henken B. Effect of parenteral and enteral nutrition on gut-associated lymphoid tissue. Journal of Trauma. 1995;39:44–51. doi: 10.1097/00005373-199507000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Feng Y, Sun X, Yang H, Teitelbaum DH. Dissociation of E-cadherin and beta-catenin in a mouse model of total parenteral nutrition: a mechanism for the loss of epithelial cell proliferation and villus atrophy. J Physiol. 2009;587:641–654. doi: 10.1113/jphysiol.2008.162719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang H, Feng Y, Sun X, Teitelbaum DH. Enteral versus parenteral nutrition: effect on intestinal barrier function. Ann N Y Acad Sci. 2009;1165:338–346. doi: 10.1111/j.1749-6632.2009.04026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun X, Yang H, Nose K, Nose S, Haxhija EQ, Koga H, Feng Y, Teitelbaum DH. Decline in intestinal mucosal IL-10 expression and decreased intestinal barrier function in a mouse model of total parenteral nutrition. Am J Physiol Gastrointest Liver Physiol. 2008;294:G139–147. doi: 10.1152/ajpgi.00386.2007. [DOI] [PubMed] [Google Scholar]
- 9.Yang H, Finaly R, Teitelbaum DH. Alteration in epithelial permeability and ion transport in a mouse model of total parenteral nutrition. Crit Care Med. 2003;31:1118–1125. doi: 10.1097/01.CCM.0000053523.73064.8A. [DOI] [PubMed] [Google Scholar]
- 10.Kudsk KA, Croce MA, Fabian TC, Minard G, Tolley EA, Poret HA, Kuhl MR, Brown RO. Enteral versus parenteral feeding. Effects on septic morbidity after blunt and penetrating abdominal trauma. Ann Surg. 1992;215:503–511. doi: 10.1097/00000658-199205000-00013. discussion 511-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buchman AL, Moukarzel AA, Bhuta S, Belle M, Ament ME, Eckhert CD, Hollander D, Gornbein J, Kopple JD, Vijayaroghavan SR. Parenteral nutrition is associated with intestinal morphologic and functional changes in humans. JPEN J Parenter Enteral Nutr. 1995;19:453–460. doi: 10.1177/0148607195019006453. [DOI] [PubMed] [Google Scholar]
- 12.Dahan S, Roda G, Pinn D, Roth-Walter F, Kamalu O, Martin AP, Mayer L. Epithelial: lamina propria lymphocyte interactions promote epithelial cell differentiation. Gastroenterology. 2008;134:192–203. doi: 10.1053/j.gastro.2007.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asquith MJ, Boulard O, Powrie F, Maloy KJ. Pathogenic and protective roles of MyD88 in leukocytes and epithelial cells in mouse models of inflammatory bowel disease. Gastroenterology. 139:519–529. 529, e511–512. doi: 10.1053/j.gastro.2010.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Novak N, Bieber T. 2. Dendritic cells as regulators of immunity and tolerance. J Allergy Clin Immunol. 2008;121:S370–374. doi: 10.1016/j.jaci.2007.06.001. quiz S413. [DOI] [PubMed] [Google Scholar]
- 15.Chow J, Lee SM, Shen Y, Khosravi A, Mazmanian SK. Host-bacterial symbiosis in health and disease. Adv Immunol. 2010;107:243–274. doi: 10.1016/B978-0-12-381300-8.00008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng SC, Kamm MA, Stagg AJ, Knight SC. Intestinal dendritic cells: their role in bacterial recognition, lymphocyte homing, and intestinal inflammation. Inflamm Bowel Dis. 2010;16:1787–1807. doi: 10.1002/ibd.21247. [DOI] [PubMed] [Google Scholar]
- 17.Chichlowski M, Hale LP. Bacterial-mucosal interactions in inflammatory bowel disease: an alliance gone bad. Am J Physiol Gastrointest Liver Physiol. 2008;295:G1139–1149. doi: 10.1152/ajpgi.90516.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90:859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- 19.Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. 2010;10:131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 20.Karrasch T, Jobin C. NF-kappaB and the intestine: friend or foe? Inflamm Bowel Dis. 2008;14:114–124. doi: 10.1002/ibd.20243. [DOI] [PubMed] [Google Scholar]
- 21.Morowitz MJ, Poroyko V, Caplan M, Alverdy J, Liu DC. Redefining the role of intestinal microbes in the pathogenesis of necrotizing enterocolitis. Pediatrics. 2010;125:777–785. doi: 10.1542/peds.2009-3149. [DOI] [PubMed] [Google Scholar]
- 22.Kiristioglu I, Teitelbaum DH. Alteration of the intestinal intraepithelial lymphocytes during total parenteral nutrition. J Surg Res. 1998;79:91–96. doi: 10.1006/jsre.1998.5408. [DOI] [PubMed] [Google Scholar]
- 23.Gillilland MGI, Erb-Downward JR, Bassis CM, Shen MC, Toews GB, Young VB, Huffnagle GB. Ecological succession of bacterial communities during conventionalization of germ-free mice. Applied and Environmental Microbiology. 2012;78:2359–2366. doi: 10.1128/AEM.05239-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, Young VB, Toews GB, Curtis JL, Sundaram B, Martinez FJ, Huffnagle GB. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One. 2011;6:e16384. doi: 10.1371/journal.pone.0016384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dave M, Johnson LA, Walk ST, Young VB, Stidham RW, Chaudhary MN, Funnell J, Higgins PD. A randomised trial of sheathed versus standard forceps for obtaining uncontaminated biopsy specimens of microbiota from the terminal ileum. Gut. 2011 doi: 10.1136/gut.2010.224337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dowd SE, Callaway TR, Wolcott RD, Sun Y, McKeehan T, Hagevoort RG, Edrington TS. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol. 2008;8:125. doi: 10.1186/1471-2180-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bailey MT, Dowd SE, Parry NM, Galley JD, Schauer DB, Lyte M. Stressor exposure disrupts commensal microbial populations in the intestines and leads to increased colonization by Citrobacter rodentium. Infect Immun. 78:1509–1519. doi: 10.1128/IAI.00862-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grossman J, Maxson J, Whitacre C, Orosz D, Berger N, Fiocchi C, Levine A. New isolation technique to study apoptosis in human intestinal epithelial cells. American Journal of Pathology. 1998:53–62. doi: 10.1016/S0002-9440(10)65545-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng Y, McDunn JE, Teitelbaum DH. Decreased phospho-Akt signaling in a mouse model of total parenteral nutrition: a potential mechanism for the development of intestinal mucosal atrophy. Am J Physiol Gastrointest Liver Physiol. 2010;298:G833–841. doi: 10.1152/ajpgi.00030.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang H, Gumucio DL, Teitelbaum DH. Intestinal specific overexpression of interleukin-7 attenuates the alternation of intestinal intraepithelial lymphocytes after total parenteral nutrition administration. Ann Surg. 2008;248:849–856. doi: 10.1097/SLA.0b013e31818a1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang H, Kiristioglu I, Fan Y, Forbush B, Bishop DK, Antony PA, Zhou H, Teitelbaum DH. Interferon-gamma expression by intraepithelial lymphocytes results in a loss of epithelial barrier function in a mouse model of total parenteral nutrition. Ann Surg. 2002;236:226–234. doi: 10.1097/00000658-200208000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikeda T, Hiromatsu K, Hotokezaka M, Chijiiwa K. Up-regulation of intestinal Toll-Like receptors and cytokines expressions change after TPN administration and a lack of enteral feeding. J Surg Res. 2010;160:244–252. doi: 10.1016/j.jss.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 35.Wildhaber BE, Yang H, Spencer AU, Drongowski RA, Teitelbaum DH. Lack of enteral nutrition--effects on the intestinal immune system. J Surg Res. 2005;123:8–16. doi: 10.1016/j.jss.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 36.Sinclair JL, Alexander M. Role of resistance to starvation in bacterial survival in sewage and lake water. Appl Environ Microbiol. 1984;48:410–415. doi: 10.1128/aem.48.2.410-415.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Durand L, Zbinden M, Cueff-Gauchard V, Duperron S, Roussel EG, Shillito B, Cambon-Bonavita MA. Microbial diversity associated with the hydrothermal shrimp Rimicaris exoculata gut and occurrence of a resident microbial community. FEMS Microbiol Ecol. 2009;71:291–303. doi: 10.1111/j.1574-6941.2009.00806.x. [DOI] [PubMed] [Google Scholar]
- 38.Costello EK, Gordon JI, Secor SM, Knight R. Postprandial remodeling of the gut microbiota in Burmese pythons. ISME J. 2010;4:1375–1385. doi: 10.1038/ismej.2010.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conour JE, Ganessunker D, Tappenden KA, Donovan SM, Gaskins HR. Acidomucin goblet cell expansion induced by parenteral nutrition in the small intestine of piglets. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1185–1196. doi: 10.1152/ajpgi.00097.2002. [DOI] [PubMed] [Google Scholar]
- 40.Martens EC, Chiang HC, Gordon JI. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. 2008;4:447–457. doi: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng Y, Teitelbaum DH. Modulation of TNF-α/Epidermal Growth Factor (EGF) Signaling Leads to Loss of Epithelial Barrier Function (Ebf) and Intestinal Atrophy in a Mouse Model of Total Parenteral Nutrition (TPN). Gastroenterology. 2010;138:S-272–273. [Google Scholar]
- 42.Hsu D, Fukata M, Hernandez YG, Sotolongo JP, Goo T, Maki J, Hayes LA, Ungaro RC, Chen A, Breglio KJ, Xu R, Abreu MT. Toll-like receptor 4 differentially regulates epidermal growth factor-related growth factors in response to intestinal mucosal injury. Laboratory Investigations. 2010;90:1295–1305. doi: 10.1038/labinvest.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov II, Umesaki Y, Itoh K, Honda K. Induction of Colonic Regulatory T Cells by Indigenous Clostridium Species. Science. 2011;331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107:12204–12209. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8:1086–1094. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 46.Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, Orenstein JM, Smith PD. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest. 2005;115:66–75. doi: 10.1172/JCI19229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slack E, Hapfelmeier S, Stecher B, Velykoredko Y, Stoel M, Lawson MAE, Geuking MB, Beutler B, TEdder TF, Hardt W, Bercik P, Verdu EF, McCoy KD, Macpherson AJ. Innate and Adaptive Immunity Cooperate Flexibly to Maintain Host-Microbiota Mutualism. Science. 2009;325:617–620. doi: 10.1126/science.1172747. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.