

Abstract
The gastrointestinal tract is the largest mucosal surface in our body. It houses diverse microorganisms that collectively form the commensal microbial community. The security of this community is kept by host-microbial interactions and is violated by foreign pathogens that induce local as well as systemic pathology. In most cases, gastrointestinal infections are caused by Gram-negative enteropathogens, which trigger host immune responses through the TLR4 signaling pathways. Although TRIF is one of the major pathways downstream of TLR4, very little is known about how the TRIF pathway contributes to intestinal defense against pathogenic infection. Recently, we reported a unique role of TRIF signaling in host response to an enterophathogen Yersinia enterocolitica, which consisted of IFN-β induction from regional macrophages followed by activation of NK cells in the mesenteric lymph nodes. In this addendum, we show distinct roles for TRIF-dependent host response in intestinal vs. systemic infection with Gram-negative enterophathogens.
Keywords: bacteria, host defense mechanism, host-pathogen interactions, infection, innate immunity, intestine, macrophages, mucosal immunology
Introduction
Bacterial infections occur locally or systemically, while most infections are established through the internal surface of our body called mucosa. The infecting bacterial pathogen colonizes at the mucosa or associated lymphoid tissues, where antigen-presenting cells (APC) mobilize host immune defenses. A systemic infection occurs when the bacterial pathogen overwhelms the local host defense. It also depends on the virulence factors of the pathogen by which the pathogen can invade tissues, evade immune defenses, or survive within immune cells.1-3 The gastrointestinal tract possesses the largest mucosal surface in our body and thus it may receive a variety of pathogenic infections. However, it naturally houses myriad microorganisms in which bacteria are the major components. Therefore, host defense mechanism in the gastrointestinal tract may be unique as it needs to induce strong immune response against invading pathogens while maintaining tolerance to the normal flora. However, how the host immune system differently responds to intestinal pathogens between intestinal vs. systemic infections is still obscure.
In the face of enteric mucosal infection, innate immunity plays major roles in establishing primary host defense against the responsible pathogen in intestinal interface. The innate immune response is rapid and antigen-nonspecific, which can provide immediate and broad defense against diverse pathogens. This response is initiated by recognizing pathogen patterns by pattern-recognition receptors (PRRs) of APCs. Multiple molecular patterns of the pathogen can be sensed by several PRRs of a single APC, e.g., a bacterial pathogen can be recognized by toll-like receptors (TLRs) and nucleotide binding oligomerization domain (NOD)-like receptors (NLR)s in the plasma membrane or endosomes and the cytoplasm, respectively. Activated PRRs induce individual intracellular signaling pathways that result in the release of anti-microbial peptides, induction of phagocytosis, secretion of cytokines and chemokines, initiation of adaptive immunity, and finally cellular apoptosis. NOD signaling activate mitogen-activated protein kinases (MAPK)s and NF-κB, and another NLRs lead to activation of caspase-1 that release IL-1β and IL-18 and contribute to the cell apoptosis. Most TLRs induce the MyD88 (myeloid differentiation primary response gene 88) pathway except for TLR3 that exclusively induces the TRIF (TIR-domain-containing adaptor protein inducing IFN-β) pathway. The MyD88 pathway mainly leads to a strong activation of NF-κB, while the TRIF pathway induces type I interferons and slower NF-κB activation; both pathways also induce MAPKs_. Only TLR4, which recognizes lipopolysaccharide (LPS) a molecular pattern of Gram-negative bacteria, has the ability to induce both the MyD88 and TRIF pathways, suggesting a specific role of the TRIF pathway in host response to Gram-negative bacteria.
Several reports have demonstrated the important roles of TLR4 signaling, both through the MyD88 and TRIF pathways, in mucosal defense against Gram-negative bacterial pathogens in the lungs, cornea, urinary tract, and female reproductive tract.4-10 However, specific contribution of the individual pathways to intestinal host defense during infection with Gram-negative pathogens remains elusive, despite the fact that most bacterial pathogens that infect through the gastrointestinal mucosa are Gram-negative species. Furthermore, it has been totally unclear whether the roles played by individual pathways of TLR4 differ during host response to intestinal vs. systemic infection with Gram-negative pathogens. Recently, we reported a unique role of the TRIF pathway on establishing intestinal protective immunity during infection with Gram-negative Y. enterocolitica.11 In this addendum, we reconfirmed intestinal specificity of TRIF-mediated immune defense by challenging TRIF-deficient mice with systemic infection with Y. enterocolitica (WA-314 serotype O:8).
The role of Individual TLR4 Signaling Pathways in Systemic Vs. Mucosal Infection with Gram-negative Pathogens
C3H/HeJ mice, which have defective TLR4 signaling due to a natural mutation of TLR4 gene, are known to survive longer in polymicrobial sepsis than TLR4 sufficient counterpart.12 However, C3H/HeJ mice are more susceptible to systemic (intraperitoneal) infection with Gram-negative Salmonella typhimurium.12-14 A more detailed study has demonstrated that TLR4-deficient (TLR4−/−) mice are impaired in control of S. typhimurium growth in the liver in initial (phagocytic killing within 24 h) as well as the plateau phases (cytokine mediated killing at later time point) of intravenous infection.15 In this systemic S. typhimurium infection model, both the MyD88 and TRIF pathways are required for the initial and plateau phases of bacterial elimination, as both MyD88−/− and TRIF−/− mice showed similar growth pattern of S. typhimurium in the liver as TLR4−/− mice.15 These findings suggest that TLR4 signaling is required for the protection of host from systemic infection with Gram-negative pathogens, but it may work as a detrimental signaling during polymicrobial sepsis. In this regard, the MyD88 pathway but not the TRIF pathway has shown to be responsible for induction of cardiac dysfunction during polymicrobial sepsis.16 By contrast, the TRIF pathway may be beneficial because induction of CXCL10, which is largely regulated by TRIF signaling is important for prevention of disease progression in this model.17 Therefore, the roles of TLR4-mediated MyD88 or TRIF pathway in host defense mechanism differ in each organ during systemic infection with Gram-negative pathogens.
The host defense mechanism induced by the individual pathways of MyD88 and TRIF have been examined during infection with Gram-negative pathogens in several mucosal organs.4-10 Both MyD88−/− mice and TRIF−/− mice have demonstrated increased bacterial burden and greater mortality than WT mice in the lung infection with Klebsiella pneumoniae due to impaired neutrophil influx and local cytokine responses.6 Similarly, these mice have shown a defective bacterial clearance during Pseudomonas aeruginosa pneumonia.5,18 Requirement of both pathways of the MyD88 and TRIF in neutrophil influx has also been described in Escherichia coli pneumonia, suggesting the impaired neutrophil influx is a common phenotype of these mice in the lung infection with Gram-negative pathogens.7 However, it is unlikely that both pathways of the MyD88 and TRIF are always required for pulmonary defense against Gram-negative pathogens, because TRIF−/− mice have shown similar resistance to WT mice in the lung infection with other Gram-negative pathogens such as Burkholderia pseudomallei and Hemophilus influenzae.19,20 Selection of the dominant pathway either MyD88 or TRIF to induce local inflammation in response to individual Gram-negative pathogens appears to depend on the structure of their LPS.21
In the intestine, there are two important pathologies during mucosal infection with Gram-negative pathogens; mucosal inflammation and systemic dissemination of the pathogen. Both manifestations are consequential because intestinal mucosal inflammation leads to deregulation of body fluid-electrolyte balance and bacteremia causes systemic inflammatory response syndrome. In this context, mucosal TLR4 signaling may block bacterial dissemination but contribute to induction of mucosal inflammation. TLR4−/− mice have shown to experience an increased bacterial burden in the Peyer’s patches (PPs) and the mesenteric lymph nodes (MLNs) compared with WT mice after oral infection with S. typhimurium, especially during the early phase of infection.22 This increase of bacterial burden is more profound in MyD88−/− mice suggesting the involvement of other upstream signaling that transduce the MyD88 pathway during host defense against enteric S. typhimurium infection.22 Similarly, TLR4-deficient C3H/HeJ mice have demonstrated higher Y. enterocolitica colonization in the liver and the spleen compared with TLR4-sufficient counterparts after oral infection.23 On the other hand, TLR4-deficient mice show reduced intestinal inflammation and morbidity in enteric Citrobacter rodentium infection.24 MyD88 deficiency reduced intestinal inflammation in Helicobacter hepaticus infection in RAG2−/− mice compared with MyD88 sufficient RAG2−/− counterparts.25 Similarly, streptomycin-pretreated MyD88−/− mice have shown a reduced severity of cecal inflammation compared with WT mice after oral S. __typhimurium infection.26 These findings indicate that TLR4 signaling through the MyD88 pathway is responsible for the induction of mucosal inflammation in response to enteric infection with Gram-negative pathogens, which is required for the host defense against bacterial dissemination to the peripheral organs. Contribution of the TRIF pathway to the induction of intestinal mucosal inflammation has also been suggested in oral Campylobacter jejuni model of gnotobiotic mice.27 However, very little is known about the role of the TRIF pathway in intestinal defense against bacterial dissemination during oral infection with Gram-negative pathogens.
TRIF-mediated Intestinal Defense Mechanism Against Gram-Negative Enteropathogens
In order to understand the unique role of the TRIF pathway in host defense against dissemination of Gram-negative enterophathogens, we orogastrically infected TRIF-deficient (TrifLPS2) mice with Y. enterocolitica.11 We found that TrifLPS2 mice are very susceptible to intestinal infection with Y. enterocolitica compared with WT mice. Almost half of the TrifLPS2 mice died within 3 d due to massive bacterial dissemination to the spleen and the liver, suggesting an impaired mucosal defense against invading Y. enterocolitica. Since most enteropathogens first colonize in the PPs then spread systemically through the MLNs, we examined what happens to the PPs and MLNs 48 h post infection.28-30 In the PPs, TrifLPS2 mice had an impaired neutrophil influx along with defective induction of CXCL10. In addition, there were a reduced number of macrophages that were associated with Y. enterocolitica in the PPs. Consistently, peritoneal macrophages from TrifLPS2 mice exhibited an impaired ability in phagocytosis and intracellular bacterial killing during in vitro infection with Y. enterocolitica. In the MLNs, TrifLPS2 mice had defective inductions of IFN-β from macrophages and IFN-γ from NK cells 48 h post infection. IFN-β supplementation restored in vitro bactericidal function of TrifLPS2 macrophages and in vivo resistance of TrifLPS2 mice against oral Y. enterocolitica infection. Therefore, TRIF-dependent intestinal defense against Gram-negative Y. enterocolitica infection comprises of macrophage activation and neutrophil recruitment in the PPs, and sequential induction of IFNs in the MLNs.
TRIF-mediated Protective Immunity: Is This Intestinal Specific?
Next we questioned whether the observed TRIF-mediated defense mechanism also applies to systemic infection with Gram-negative pathogens. To answer this question, we infected WT and TrifLPS2 mice intravenously with 2 x 105 CFU of Y. enterocolitica.11 This infection dose has been reported as LD50 during seven weeks period of intravenous Y. enterocolitica infection.31 Interestingly, both WT and TrifLPS2 mice survived and showed similar Y. enterocolitica burdens in the spleen during five days post infection (Fig. 1A). It is possible that the infection dose of 2 x 105 CFU was too low or five days observation period was too short to examine primary host response to systemic Y. enterocolitica infection. However, when we increased the infection dose to 2 x 107 CFU, the same dose as we observed greater mortality and bacterial burden in TrifLPS2 mice than WT mice during oral infection, WT and TrifLPS2 mice still showed similar Y. enterocolitica burden in the liver and spleen during seven days of observations (Fig. 1B and C).
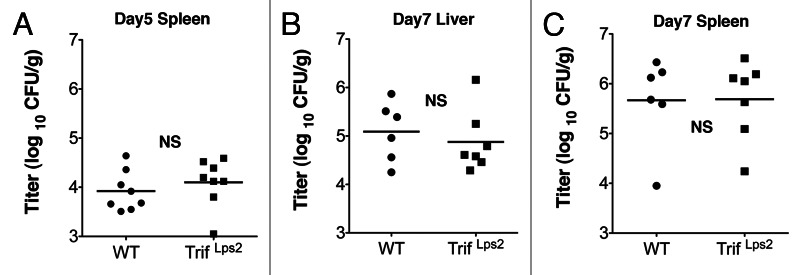
Figure 1. TrifLPS2 mice have similar Y. enterocolitica burden in the spleen and liver as WT mice after intravenous infection. (A) Y. enterocolitica titers in the spleen of infected WT and TrifLps2 mice taken five days post intravenous infection (2 x 105 CFU, error bars; s.e.m. NS = not significant). (B) Y. enterocolitica titers in the liver and spleen of infected WT and TrifLps2 mice taken seven days post intravenous infection (2 x 107 CFU, error bars; s.e.m. NS = not significant).
In contract to these findings in systemic Y. enterocolitica infection, a previous report has shown increased bacterial burden in the liver of TRIF−/− mice compared with WT mice after intravenous infection with other Gram-negative pathogen, S. typhimurium.15 One of the major components of innate immune defense is induction of acute inflammatory cell influx to the infection site. TRIF−/− mice have shown defective neutrophil influx in the liver seven days post intravenous infection with S. typhimurium, and we found an impaired neutrophil recruitment to the PPs in TrifLPS2 mice after oral infection with Y. enterocolitica.11,15 However, acute inflammatory cell influx in the liver was similar between WT mice and TrifLPS2 mice seven days post intravenous infection with Y. enterocolitica (Fig. 2). Since we have observed greater expression of splenic TNF-α in survived TrifLPS2 mice compared with WT mice 72 h post oral Y. enterocolitica infection, we concluded that this TNF-α induction would be a compensatory defense mechanism by which TrifLPS2 mice could survive systemic Y. enterocolitica dissemination.11 Similarly, the enhanced IFN-γ production by splenocytes was suggested as a reason for preserved resistance of TRIF−/− mice against intravenous S. typhimurium infection, although we did not see such IFN-γ enhancement in TrifLPS2 mice in our oral Y. enterocolitica infection system.11,15 At day seven of intravenous Y. enterocolitica infection (2 x 107 CFU), some of the TrifLPS2 mice showed higher expression of TNF-α or IFN-γ mRNA in the spleen compared with WT mice but the mean differences did not reach statistical significance (Fig. 3A, B).
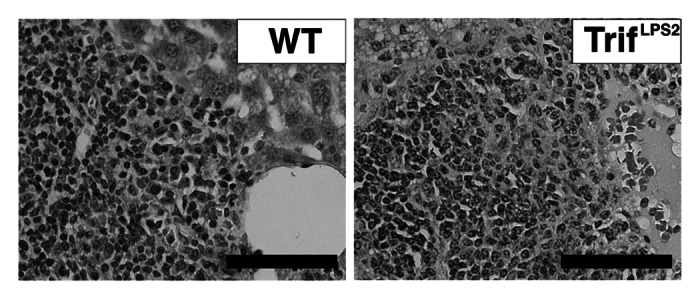
Figure 2. TrifLPS2 mice have similar liver histology as WT mice seven days post intravenous infection. H&E staining of the liver taken seven days post infection (2 x 107 CFU). Representative pictures from 6 WT mice and 7 TrifLPS2 mice. Original magnification, x40, Bars: 50µm.
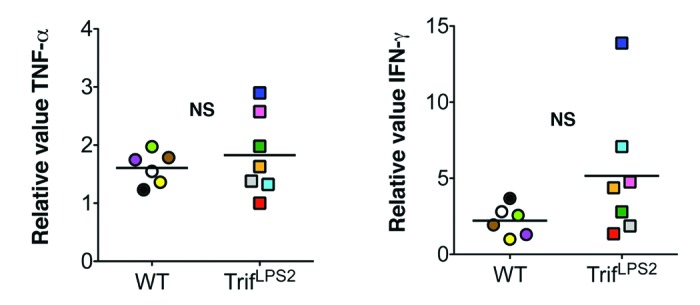
Figure 3. Splenic pro-inflammatory cytokine responses are similar between WT mice and TrifLPS2 mice after intravenous Y. enterocolitica infection. Left panel: Real-time PCR analysis of TNF-α mRNA expression in the spleen. Right panel: Real-time PCR analysis of IFN-γ mRNA expression in the spleen. TrifLPS2 mice n = 7, WT n = 6, p = 0.13, NS: not significant, error bars, s.e.m. Each color on graph indicates the value of individual mice.
These data indicate that TRIF signaling is responsible for a particular aspect of intestinal defense mechanism against Gram-negative enteropathogens but there are multiple compensatory mechanisms that can replace the TRIF-mediated host defense during systemic dissemination of the enteropathogen. It is puzzling why TrifLPS2 mice succumb to systemic dissemination with Y. enterocolitica during intestinal infection but survive intravenous infection with Y. enterocolitica. Many factors may be involved in the mechanism for the difference in resistance of TrifLPS2 mice to systemic Y. enterocolitica dissemination between intestinal and intravenous infection. A specific feature of intestinal infection is that enteropathogens colonize the intestine and proliferate locally before disseminate to other organs.28,32 Systemic dissemination can occur through lymphatics as well as the blood stream, and locally grown colonies may serve as a continuous feeder source of bacteremia.28-30 Therefore, systemic Y. enterocolitica dissemination from intestine may be different from intravenous infection in dose, route, and frequency, which depend on the phase of infection. A time course study comparing intestinal and intravenous infection in WT and TrifLPS2 mice will provide important clues as to whether local colonization control is the key to the TRIF-dependent host defense during the intestinal and systemic phases of Y. enterocolitica infection.
In summary, TLR4-mediated host response is indispensable for mounting full defense mechanism during mucosal as well as systemic infection with Gram-negative bacterial pathogens. However, individual roles of the MyD88 and TRIF pathways differ between mucosal and systemic infection. Both pathways equally contribute to acute inflammatory cell influx in mucosal interface. Although it is associated with regional pathology of the site of infection, adequate inflammation is necessary to prevent systemic dissemination of pathogens. During systemic infection, both MyD88 and TRIF signaling contribute to the induction of pro-inflammatory cytokines in the spleen, which is an important defense mechanism but the overproduction of these cytokines may lead to multiple organ dysfunction. However, unlike the local response, the systemic cytokine response may be compensated by other signaling pathways. In the intestine, the TRIF pathway plays a unique role in establishing local immune defense against infection with Gram-negative pathogens. TRIF-mediated intestinal protective immunity is mainly mediated by macrophages, which orchestrate bacterial elimination process in the MLN by attracting NK cells through IFN signaling. Unlike MyD88, TRIF has limited upstream inputs. Therefore, TRIF can be a potent target for the prevention and treatment of enteric infection with Gram-negative pathogens.
Acknowledgments
This study was supported by National Institute of Allergy and Infectious Diseases (R56 AI095255–01) for M.F. The authors have no conflicting financial interests.
Footnotes
Previously published online: www.landesbioscience.com/journals/gutmicrobes/article/20873
References
- 1.Griffin AJ, McSorley SJ. Development of protective immunity to Salmonella, a mucosal pathogen with a systemic agenda. Mucosal Immunol. 2011;4:371–82. doi: 10.1038/mi.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sotolongo J, Ruiz J, Fukata M. The role of innate immunity in the host defense against intestinal bacterial pathogens. Curr Infect Dis Rep. 2012;14:15–23. doi: 10.1007/s11908-011-0234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnold R, Jehl A, Rattei T. Targeting effectors: the molecular recognition of Type III secreted proteins. Microbes Infect. 2010;12:346–58. doi: 10.1016/j.micinf.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Mijares LA, Wangdi T, Sokol C, Homer R, Medzhitov R, Kazmierczak BI. Airway epithelial MyD88 restores control of Pseudomonas aeruginosa murine infection via an IL-1-dependent pathway. J Immunol. 2011;186:7080–8. doi: 10.4049/jimmunol.1003687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skerrett SJ, Liggitt HD, Hajjar AM, Wilson CB. Cutting edge: myeloid differentiation factor 88 is essential for pulmonary host defense against Pseudomonas aeruginosa but not Staphylococcus aureus. J Immunol. 2004;172:3377–81. doi: 10.4049/jimmunol.172.6.3377. [DOI] [PubMed] [Google Scholar]
- 6.Cai S, Batra S, Shen L, Wakamatsu N, Jeyaseelan S. Both TRIF- and MyD88-dependent signaling contribute to host defense against pulmonary Klebsiella infection. J Immunol. 2009;183:6629–38. doi: 10.4049/jimmunol.0901033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeyaseelan S, Young SK, Fessler MB, Liu Y, Malcolm KC, Yamamoto M, et al. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF)-mediated signaling contributes to innate immune responses in the lung during Escherichia coli pneumonia. J Immunol. 2007;178:3153–60. doi: 10.4049/jimmunol.178.5.3153. [DOI] [PubMed] [Google Scholar]
- 8.Sun Y, Karmakar M, Roy S, Ramadan RT, Williams SR, Howell S, et al. TLR4 and TLR5 on corneal macrophages regulate Pseudomonas aeruginosa keratitis by signaling through MyD88-dependent and -independent pathways. J Immunol. 2010;185:4272–83. doi: 10.4049/jimmunol.1000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Packiam M, Wu H, Veit SJ, Mavrogiorgos N, Jerse AE, Ingalls RR. Protective role of Toll-like receptor 4 in experimental gonococcal infection of female mice. Mucosal Immunol. 2012;5:19–29. doi: 10.1038/mi.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer H, Yamamoto M, Akira S, Beutler B, Svanborg C. Mechanism of pathogen-specific TLR4 activation in the mucosa: fimbriae, recognition receptors and adaptor protein selection. Eur J Immunol. 2006;36:267–77. doi: 10.1002/eji.200535149. [DOI] [PubMed] [Google Scholar]
- 11.Sotolongo J, España C, Echeverry A, Siefker D, Altman N, Zaias J, et al. Host innate recognition of an intestinal bacterial pathogen induces TRIF-dependent protective immunity. J Exp Med. 2011;208:2705–16. doi: 10.1084/jem.20110547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alves-Filho JC, de Freitas A, Russo M, Cunha FQ. Toll-like receptor 4 signaling leads to neutrophil migration impairment in polymicrobial sepsis. Crit Care Med. 2006;34:461–70. doi: 10.1097/01.CCM.0000198527.71819.E1. [DOI] [PubMed] [Google Scholar]
- 13.O’Brien AD, Rosenstreich DL. Genetic control of the susceptibility of C3HeB/FeJ mice to Salmonella typhimurium is regulated by a locus distinct from known salmonella response genes. J Immunol. 1983;131:2613–5. [PubMed] [Google Scholar]
- 14.Vazquez-Torres A, Vallance BA, Bergman MA, Finlay BB, Cookson BT, Jones-Carson J, et al. Toll-like receptor 4 dependence of innate and adaptive immunity to Salmonella: importance of the Kupffer cell network. J Immunol. 2004;172:6202–8. doi: 10.4049/jimmunol.172.10.6202. [DOI] [PubMed] [Google Scholar]
- 15.Talbot S, Tötemeyer S, Yamamoto M, Akira S, Hughes K, Gray D, et al. Toll-like receptor 4 signalling through MyD88 is essential to control Salmonella enterica serovar typhimurium infection, but not for the initiation of bacterial clearance. Immunology. 2009;128:472–83. doi: 10.1111/j.1365-2567.2009.03146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng D, Chen H, Yang KX, Fang F. Pre-peritoneal lipoleiomyoma with hyperoestrogenism in a postmenopausal woman. J Obstet Gynaecol. 2011;31:556–7. doi: 10.3109/01443615.2011.578777. [DOI] [PubMed] [Google Scholar]
- 17.Kelly-Scumpia KM, Scumpia PO, Delano MJ, Weinstein JS, Cuenca AG, Wynn JL, et al. Type I interferon signaling in hematopoietic cells is required for survival in mouse polymicrobial sepsis by regulating CXCL10. J Exp Med. 2010;207:319–26. doi: 10.1084/jem.20091959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Power MR, Li B, Yamamoto M, Akira S, Lin TJ. A role of Toll-IL-1 receptor domain-containing adaptor-inducing IFN-beta in the host response to Pseudomonas aeruginosa lung infection in mice. J Immunol. 2007;178:3170–6. doi: 10.4049/jimmunol.178.5.3170. [DOI] [PubMed] [Google Scholar]
- 19.Wiersinga WJ, Wieland CW, Roelofs JJ, van der Poll T. MyD88 dependent signaling contributes to protective host defense against Burkholderia pseudomallei. PLoS One. 2008;3:e3494. doi: 10.1371/journal.pone.0003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wieland CW, Florquin S, Maris NA, Hoebe K, Beutler B, Takeda K, et al. The MyD88-dependent, but not the MyD88-independent, pathway of TLR4 signaling is important in clearing nontypeable haemophilus influenzae from the mouse lung. J Immunol. 2005;175:6042–9. doi: 10.4049/jimmunol.175.9.6042. [DOI] [PubMed] [Google Scholar]
- 21.Bowen WS, Minns LA, Johnson DA, Mitchell TC, Hutton MM, Evans JT. Selective TRIF-dependent signaling by a synthetic toll-like receptor 4 agonist. Sci Signal. 2012;5:ra13. doi: 10.1126/scisignal.2001963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiss DS, Raupach B, Takeda K, Akira S, Zychlinsky A. Toll-like receptors are temporally involved in host defense. J Immunol. 2004;172:4463–9. doi: 10.4049/jimmunol.172.7.4463. [DOI] [PubMed] [Google Scholar]
- 23.Sing A, Tvardovskaia N, Rost D, Kirschning CJ, Wagner H, Heesemann J. Contribution of toll-like receptors 2 and 4 in an oral Yersinia enterocolitica mouse infection model. Int J Med Microbiol. 2003;293:341–8. doi: 10.1078/1438-4221-00277. [DOI] [PubMed] [Google Scholar]
- 24.Khan MA, Ma C, Knodler LA, Valdez Y, Rosenberger CM, Deng W, et al. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect Immun. 2006;74:2522–36. doi: 10.1128/IAI.74.5.2522-2536.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Asquith MJ, Boulard O, Powrie F, Maloy KJ. Pathogenic and protective roles of MyD88 in leukocytes and epithelial cells in mouse models of inflammatory bowel disease. Gastroenterology 2010; 139:519-29, 29 e1-2. [DOI] [PMC free article] [PubMed]
- 26.Keestra AM, Godinez I, Xavier MN, Winter MG, Winter SE, Tsolis RM, et al. Early MyD88-dependent induction of interleukin-17A expression during Salmonella colitis. Infect Immun. 2011;79:3131–40. doi: 10.1128/IAI.00018-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bereswill S, Fischer A, Plickert R, Haag LM, Otto B, Kühl AA, et al. Novel murine infection models provide deep insights into the “ménage à trois” of Campylobacter jejuni, microbiota and host innate immunity. PLoS One. 2011;6:e20953. doi: 10.1371/journal.pone.0020953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Westphal S, Lügering A, von Wedel J, von Eiff C, Maaser C, Spahn T, et al. Resistance of chemokine receptor 6-deficient mice to Yersinia enterocolitica infection: evidence of defective M-cell formation in vivo. Am J Pathol. 2008;172:671–80. doi: 10.2353/ajpath.2008.070393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oellerich MF, Jacobi CA, Freund S, Niedung K, Bach A, Heesemann J, et al. Yersinia enterocolitica infection of mice reveals clonal invasion and abscess formation. Infect Immun. 2007;75:3802–11. doi: 10.1128/IAI.00419-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jepson MA, Clark MA. The role of M cells in Salmonella infection. Microbes Infect. 2001;3:1183–90. doi: 10.1016/S1286-4579(01)01478-2. [DOI] [PubMed] [Google Scholar]
- 31.Handley SA, Dube PH, Revell PA, Miller VL. Characterization of oral Yersinia enterocolitica infection in three different strains of inbred mice. Infect Immun. 2004;72:1645–56. doi: 10.1128/IAI.72.3.1645-1656.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monack DM, Hersh D, Ghori N, Bouley D, Zychlinsky A, Falkow S. Salmonella exploits caspase-1 to colonize Peyer’s patches in a murine typhoid model. J Exp Med. 2000;192:249–58. doi: 10.1084/jem.192.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]