

Abstract
Autophagy is activated in response to a variety of cellular stresses including metabolic stress. While elegant genetic studies in yeast have identified the core autophagy machinery, the signaling pathways that regulate this process are less understood. AMPK is an energy sensing kinase and several studies have suggested that AMPK is required for autophagy. The biochemical connections between AMPK and autophagy, however, have not been elucidated. In this report, we identify a biochemical connection between a critical regulator of autophagy, ULK1, and the energy sensing kinase, AMPK. ULK1 forms a complex with AMPK, and AMPK activation results in ULK1 phosphorylation. Moreover, we demonstrate that the immediate effect of AMPK-dependent phosphorylation of ULK1 results in enhanced binding of the adaptor protein YWHAZ/14-3-3ζ; and this binding alters ULK1 phosphorylation in vitro. Finally, we provide evidence that both AMPK and ULK1 regulate localization of a critical component of the phagophore, ATG9, and that some of the AMPK phosphorylation sites on ULK1 are important for regulating ATG9 localization. Taken together these data identify an ULK1-AMPK signaling cassette involved in regulation of the autophagy machinery.
Keywords: 14-3-3 proteins, AMP-activated protein kinase, Atg9, autophagy, energy metabolism, metabolic stress, phosphorylation, Unc-51-like kinase 1
Introduction
Macroautophagy is an evolutionarily conserved cellular mechanism for turnover of long-lived proteins and organelles. Commonly referred to as autophagy, this process is upregulated in response to various intra- and extracellular stressors such as organelle damage, nutrient deprivation, energy stress and pathogens. Moreover, misregulation of autophagy has been implicated in a number of diseases including cancer and neurodegenerative disorders.1 Thus, understanding how this process is regulated is crucial.
Elegant genetic studies in yeast have identified a number of genes (termed Atg) encoding the core autophagy machinery, and homologs of many of these genes have been identified in other organisms including mammals.2,3 While the various steps at which these genes function have been defined, the precise signal transduction mechanisms regulating autophagy are still poorly understood.
Studies originally done in yeast have identified the serine/threonine kinase Atg1 as a key initiator and the nutrient sensing serine/threonine kinase target of rapamycin (Tor) as a negative regulator of this process.4-7 In mammals, Atg1 has at least two homologs, Unc51-like kinase (ULK)1 and ULK2; and both have been shown to regulate autophagy.8-10 There are three additional proteins which share homology with ULK1/2 in the kinase domain, ULK3, ULK4 and STK36/Fused, but only ULK3 has been linked to autophagy.11,12
Functionally, Atg1/ULK1 is important for recruitment and recycling of proteins to and from the phagophore assembly site (PAS) and for autophagosome formation. For example, in yeast, Atg9 cannot be retrieved from the PAS in the absence of Atg1.13 In mammalian cells, regulation of both, recruitment of proteins to the PAS and autophagosome formation by ULK1/2 is dependent on their kinase activity, but substrates of ULK1/2 that may regulate these and other functions are only beginning to be identified.10,14-17 In yeast, a high-throughput proteomic screen for targets of Atg1 identified more than 150 candidate substrates although they have not been validated.18 Previous work by our lab identified the focal adhesion protein PXN/paxillin, which is also required for autophagy, as an in vitro substrate of ULK1; and AMBRA1, a component of the class III phosphatidylinositol 3-kinase-BECN1 complex required for autophagosome nucleation, is also a substrate of ULK1.19,20
Upon induction of autophagy in yeast, Atg1 forms a complex with additional Atg-proteins, including Atg17 and Atg13, which modulate its kinase activity.5,21,22 Mammalian counterparts for Atg13 (ATG13) and Atg17 [focal adhesion kinase interacting protein of 200 kDa (RB1CC1/FIP200)] have been identified based on functional similarity.10,14,15,17,23 Furthermore, ULK-ATG13-RB1CC1 complexes provide a molecular link between both the autophagy machinery and the nutrient- and growth factor-activated mechanistic target of rapamycin complex 1 (MTORC1) signaling pathway. ULKs and ATG13 are directly phosphorylated by MTORC1, and the ULK1 and the MTORC1 complexes interact with each other.14-17,24 However, ULK1 has been shown to be required for autophagy under conditions where MTOR is not required.25-27 These data suggest that regulators of ULK1 other than MTORC1 might exist.
Adenosine monophosphate-activated protein kinase (AMPK) has been identified as a regulator of autophagy, yet its role in the process remains controversial and is not fully understood.28-33 While two regulators of MTORC1, TSC2 and RPTOR, are well established as AMPK substrates, a more direct connection of this kinase to the autophagy machinery in mammalian cells has not been demonstrated.34-36
Here we report the identification of the energy sensing kinase AMPK as an ULK1-interacting protein. We showed that AMPK can phosphorylate ULK1 on at least three specific sites and that activation of AMPK results in increased binding of regulatory proteins of the YWHA/14-3-3-family to ULK1. Furthermore, we found that AMPK, like ULK1, is required for correct intracellular localization of ATG9 and provide evidence that AMPK-dependent phosphorylation of ULK1 is important for ULK1 regulation of ATG9-trafficking. Taken together, our data support a direct connection between the cell’s energy sensing system and autophagy and suggest that one step modulated by the AMPK-ULK1 signaling cassette is trafficking of ATG9.
Results
Several kinases are predicted to regulate ULK1
ULK1/2 regulate autophagy but how these kinases themselves are regulated is poorly understood. To begin to address this issue, we stably expressed myc-tagged ULK1 (myc-ULK1) in mouse embryonic fibroblasts (MEFs) and used a combination of microcapillary liquid chromatography/tandem mass spectrometry (LC/MS/MS) and bioinformatics to identify and predict phosphorylation sites on ULK1 (Table S1). LC/MS/MS analyses on myc-ULK1 immunopurified from MEFs grown under nutrient-rich or nutrient-depleted conditions identified six sites that appear to be constitutively phosphorylated and 24 sites whose phosphorylation was nutrient status-dependent. Three of the constitutively phosphorylated sites and 14 of the sites that were detected under a subset of conditions have not been reported in previous studies.37-46 Analysis with the bioinformatics tool Scansite predicted various kinases as regulators of these sites, as well as additional candidate sites (Table S1).47 Interestingly, among the kinases predicted to phosphorylate ULK1 is the energy sensing kinase AMPK, which has been implicated in autophagy.28,48,49
AMPK associates with ULK1
Given these data, we investigated a potential functional relationship between AMPK and ULK1. We first tested whether AMPK and ULK1 can associate by performing a series of co-immunoprecipitation experiments involving exogenous and endogenous proteins. Myc-tagged ULK1 co-immunoprecipitated with FLAG-tagged PRKAA1/AMPKα1 when both proteins were transiently overexpressed in HEK293T cells (Fig. 1A). Similarly, YFP-tagged ULK2 co-immunoprecipitated with FLAG-PRKAA1. Thus, both ULK1 and ULK2 can interact with AMPK (Fig. 1B). This did not require overexpression of AMPK since endogenous AMPK also co-immunoprecipitated with myc-ULK1 (Fig. 1C). In addition, antibodies against either the α1 or the α2 isoform of endogenous PRKAA co-immunoprecipitated myc-ULK1 in COS7 cells (Fig. 1D and E). Finally, a small amount of endogenous ULK1 was co-immunoprecipitated with endogenous PRKAA in mouse embryonic fibroblasts (MEFs). Taken together, these data support the existence of an ULK1-AMPK-complex.

Figure 1. ULK1 and ULK2 associate with AMPK. (A) FLAG-AMPKα1 can interact with myc-ULK1. HEK293T cells were cotransfected with the indicated plasmids and FLAG-AMPKα1 was immunoprecipitated from the cell lysates using anti-FLAG agarose beads. Cell lysates and immunoprecipitates were analyzed by western blot with the antibodies indicated. (B) YFP-ULK2 can interact with FLAG-AMPKα1. HEK293T cells were cotransfected and processed as described in (A). (C) Endogeneous AMPKα1 can be detected in myc-ULK1 immunoprecipitates of myc-ULK1 COS7 cell lysates. Lysates from COS7 cells infected with control virus or virus encoding myc-ULK1 were incubated with the indicated antibodies and processed as described in Materials and Methods. Western blots shown were probed with the indicated antibodies. (D) Endogeneous AMPKα1 can be detected in myc-ULK1 immunoprecipitates from myc-ULK1 infected 293T cells. 293T cells were infected with myc-ULK1 encoding virus and lysates from these cells were incubated with the indicated antibodies and immunoprecipitates analyzed by western blot with the indicated antibodies. (E) Endogeneous AMPKα2 can be co-immunoprecipitated with myc-ULK1. Myc-ULK1 293T cells were generated and processed as described in (D). Western blots were probed with the antibodies indicated. (F) Endogeneous AMPKα1 and ULK1 can be co-immunoprecipitated. MEFs were lysed and endogenous AMPKα1 was immunoprecipitated as in (D). Cell lysates and immunoprecipitates were analyzed by western blot with the antibodies indicated. The asterisk in (A, C, D and E) denotes IgG, the asterisk in (B) denotes a nonspecific band.
Kinase activity is not required for the ULK1-AMPK association
To determine whether the kinase activity of ULK1 or AMPK is important for the interaction, we tested the ability of kinase dead ULK1 to associate with wild-type AMPK and of kinase dead AMPK to associate with wild-type ULK1. Myc-kinase dead ULK1 co-immunoprecipitated with endogenous AMPK (Fig. 2A). Similarly, myc-ULK1 co-immunoprecipitated with FLAG-kinase dead PRKAA1 (Fig. 2B). These results demonstrated that the interaction between ULK1 and AMPK does not require the kinase activity of either enzyme.

Figure 2. Association of ULK1 and AMPK is independent of their kinase activities and requires the ULK1 spacer region and the AMPKα subunit. (A) The kinase activity of ULK1 is not required for its association with AMPK. COS7 cells infected with wild-type (wt) or kinase dead (kd) myc-ULK1 were lysed and subjected to immunoprecipitation using an AMPKα2-specific rabbit antibody or non-specific rabbit-anti-mouse IgG (RαM) as a control. Western blot analysis was performed with the antibodies indicated. (B) The kinase domain of AMPK can associate with ULK1 and AMPK kinase activity is not required. HEK293T cells were cotransfected with myc-ULK1 and wild-type, kinase dead or constitutively active (ca) versions of FLAG-AMPKα1 or an empty vector. FLAG-AMPKα1 proteins were immunoprecipitated using anti-FLAG agarose beads. Western blot analysis was performed with the antibodies indicated. (C) The ULK1 spacer region can co-immunoprecipitate AMPK. HEK293T cells were transfected with empty vector or FLAG-tagged ULK1 fragments containing various combinations of the three ULK1 domains depicted in the schematic drawing. ULK1-FLAG proteins were immunoprecipitated using anti-FLAG agarose beads and cell lysates and immunoprecipitates were analyzed by western blot with the antibodies indicated.
The catalytic domain of AMPK and spacer region of ULK1 are required for formation of the ULK1-AMPK complex
AMPK is composed of three subunits, a catalytic subunit (α) and two regulatory subunits (β and γ). To determine if all three subunits are important for formation of the ULK1-AMPK-complex, we examined various mutants of AMPK. Both wild-type and catalytically inactive α subunits will associate with the β and γ subunits. In contrast, the constitutively active AMPK mutant does not associate with either regulatory subunit because it lacks the C-terminal β/γ-binding region.50 As shown in Figure 2B, the kinase domain of PRKAA is sufficient for the association with ULK1, as FLAG-tagged, constitutively active PRKAA1 co-immunoprecipitated myc-ULK1.
ULK1/2 are defined by three regions, (1) the N-terminal kinase domain (aa 1–278), (2) a serine-proline rich spacer region (aa 279–828), and (3) a C-terminal domain (CTD; aa 829–1051) (Fig. 2C). To define the region of ULK1 that is required for the association with AMPK, ULK1-FLAG variants were transiently expressed in HEK293T cells and tested for their ability to co-immunoprecipitate AMPK (Fig. 2C). Deletion of the kinase domain or CTD of ULK1 did not disrupt the ability of ULK1-FLAG to form a complex with AMPK. Similarly, neither the FLAG-tagged kinase domain nor the CTD alone were able to co-immunoprecipitate AMPK. In contrast, AMPK was able to co-immunoprecipitate with the spacer region. These data suggest that the spacer region is important for mediating the interaction between ULK1 and AMPK.
Nutrient and energy stress differentially affect the ULK1-AMPK interaction
Previous studies in yeast have identified Tor, Atg13 and Atg17 as regulators of Atg1, and recent work in mammalian cells has found that homologs of these proteins can interact with ULK1.5,10,14-17,22,51 ULK1 forms a constitutive complex with ATG13 and the ATG17 counterpart, RB1CC1. In contrast, its association with MTORC1 is only seen under nutrient-rich conditions. To determine if the association between ULK1 and AMPK is constitutive or regulated by energy stress, we treated myc-ULK1 COS7 cells with various activators of AMPK. Stimulation of these cells with the direct AMPK activator A-769662 resulted in AMPK activation but had little to no effect on the ULK1-AMPK interaction (Fig. 3A). Similarly, other activators of AMPK [AICAR, 2-deoxyglucose (2DG) or phenformin], also had little to no effect on the ULK1-AMPK complex (data not shown).
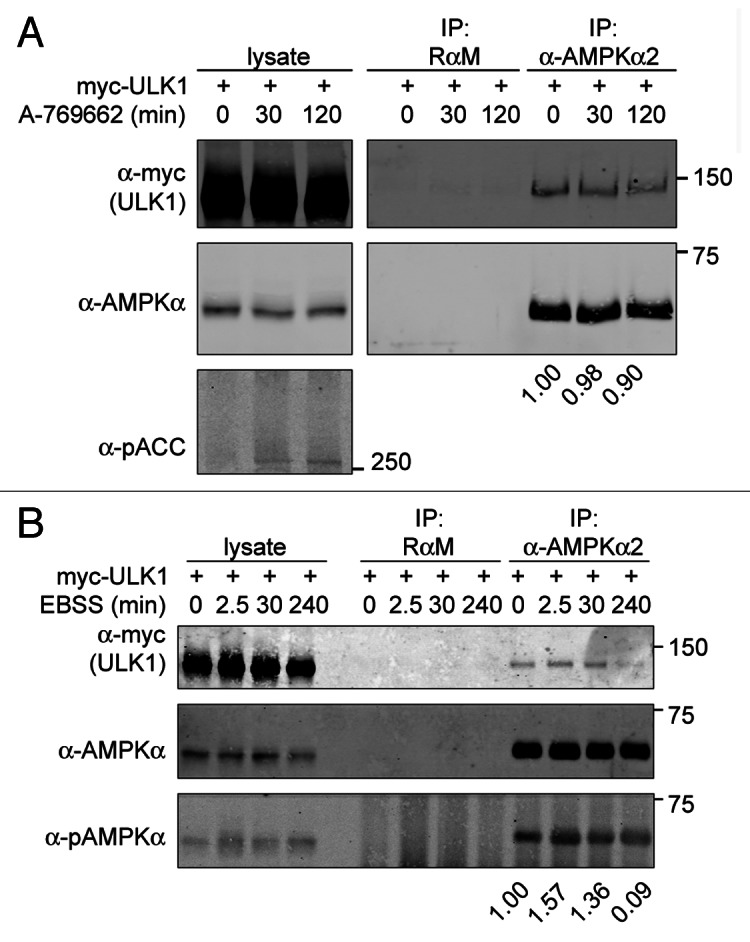
Figure 3. Stability of the ULK1-AMPK-complex is largely independent of AMPK-activation but sensitive to prolonged nutrient deprivation. (A) AMPK activation with A-769662 does not alter the ULK1-AMPK complex. Myc-ULK1-infected COS7 cells were treated with vehicle (0 min) or 100 µM A-769662 for the times indicated and AMPK-activation was monitored by examining phosphorylation of the AMPK substrate Acetyl-CoA carboxylase (pACC). Cell lysates were subjected to immunoprecipitation (IP) with the indicated antibodies. Antibodies used for western analysis are as labeled. Western blot signals were quantified. The numbers indicate the ratio of myc-ULK1 to AMPK in the AMPK IP at each time point normalized to the ratio at the 0 min time point. The result shown is representative of three independent experiments. (B) Nutrient deprivation has some effect on the amount of ULK1 co-immunoprecipitating with AMPK. COS7 cells stably overexpressing myc-ULK1 were starved with EBSS for the times indicated and immunoprecipitation was performed as in (A). Lysates and immunoprecipitates were analyzed by western blot with the antibodies indicated. Western blot signals were quantified and data normalized as in (A). The result shown is representative for four independent experiments.
To determine whether this complex was regulated by nutrient stress, myc-ULK1-COS7 cells were placed under nutrient-deprived conditions by incubation in Earl’s Balanced Salt Solution (EBSS). AMPK activation was seen within 5 min and subsequently declined to baseline levels (Fig. S2A). Interestingly, in myc-ULK1 COS7 cells, ULK1 association with AMPK showed a small but consistent increase at 2.5 min, but when these cells were incubated in EBSS over longer time periods (greater than 2 h) the amount of ULK1 precipitating with AMPK decreased over time (Fig. 3B). Some of these starvation-induced changes in the ULK1-AMPK association and AMPK activation, however, may be cell-type dependent (Fig. S1 and S2). Taken together, these data suggest that the ULK1-AMPK complex is present under a variety of conditions and the extent of its regulation by energy and nutrient stress differs between cell types.
AMPK phosphorylates ULK1
The association between AMPK and ULK1 is consistent with the possibility that AMPK may phosphorylate ULK1. We first examined whether AMPK could phosphorylate ULK1 in vitro. As shown in Figure 4A, AMPK was able to phosphorylate kinase dead ULK1. Bioinformatics analyses revealed that most of the potential AMPK phosphorylation sites lie in the ULK1 spacer region, although there are also two sites predicted outside of this domain (Fig. 4B). We therefore tested different fragments of ULK1 to determine if they could be phosphorylated by AMPK. The spacer region but not the CTD was phosphorylated by AMPK, suggesting that most of the AMPK phosphorylation sites lie in the spacer region (Fig. 4A).

Figure 4. AMPK phosphorylates ULK1. (A) Kinase dead (kd) myc-ULK1 or FLAG-tagged ULK1 fragments comprising the kinase domain and spacer region (1–828), the kinase domain alone (1–278), the spacer region and CTD (279–1051), the spacer region alone (279–828) or the CTD alone (829–1051) were transiently overexpressed in HEK293T cells, immunoprecipitated from the cell lysates with antibodies against the affinity tag, and used as substrates for in vitro kinase assays with purified AMPK. For each substrate, one reaction was performed without AMPK and AMP to control for phosphorylation of ULK1 by coprecipitating kinases and ULK1-autophosphorylation for mutants that contained an active kinase domain. Autoradiographs are shown. (B) Predicted AMPK phosphorylation sites in murine ULK1. The optimal AMPK-phosphorylation motif is shown for reference.35 AMPK phosphorylation sites were predicted using Scansite. Phosphorylation sites detected by mass spectrometry are marked by asterisks (compare Tables S1–S3). (C) Identification of phosphorylation sites on ULK1 induced with 2DG-mediated AMPK activation. Myc-tagged ULK1 was immunoprecipitated from lysates of COS7-cells treated with vehicle or 25 mM 2DG for 15 min and analyzed by LC/MS/MS. Signals obtained for the peptide TPpSSQNLLTLLAR containing residue S637 of ULK1 were quantified.
As discussed above, our initial mass spectrometry data identified potential AMPK phosphorylation sites (Fig. 4B; Table S1); yet subsequent examination of AMPK activation during EBSS-starvation revealed that this is only seen at very early and at very late time points (Fig. S2). In addition, other groups have reported candidate AMPK sites on ULK1 under normal growth conditions (Tables S1 and S3). Therefore, we repeated the mass spectrometry studies to examine phosphorylation of ULK1 under conditions where AMPK was active. When cells were treated with 2DG or short-term EBSS-starvation, two of the predicted AMPK sites, S494 and S637 were identified by mass spectrometry (Table S2). Quantification of the mass spectrometry data on S637 revealed a 3.76-fold increase in phosphorylation of this site upon AMPK activation (Fig. 4C). Additional candidate AMPK phosphorylation sites that were detected by mass spectrometry in our previous analyses or by other groups include S467, S555 and T659 (Fig. 4B; Tables S1–S3).
AMPK phosphorylates ULK1 in vitro at S555, S637 and T659
Next, we examined whether the candidate AMPK phosphorylation sites in ULK1 predicted by Scansite and identified by quantitative mass spectrometry analyses by us (S494, S555, S637, T659, Table S1) or other groups (S467, T574, Table S3), were phosphorylated by AMPK in vitro. Various ULK1 mutants that were catalytically inactive and had mutations in one or more AMPK phosphorylation sites were transiently expressed in HEK293T cells and the immunopurified ULK1 mutants were subjected to in vitro kinase assays in the presence or absence of AMPK (Fig. 5A and B). While AMPK efficiently phosphorylated wild-type and kinase dead myc-ULK1, mutation of the 2DG-induced site detected by mass spectrometry, S637 (see above), to alanine strongly reduced phosphorylation of the ULK1-protein. A smaller decrease was seen when two other sites, S555 and T659, which not only closely match the AMPK phosphorylation motif but also conform to the optimal YWHA/14-3-3-binding motif (see below), were mutated. Mutation of all three sites resulted in decreased phosphorylation of myc-ULK1 in comparison to that observed for the S637A single mutant. Moreover, the extent of residual phosphorylation of this S555A-S637A-T659A triple mutant was similar to that detected for an ULK1–6A-mutant, in which these three residues along with S467, S494 and T574 were replaced with alanine (Figs. 4B and 5A). Taken together, these data suggest that the major residues in ULK1 phosphorylated by AMPK are S555, S637 and T659, with S637 being the predominant site in vitro and in vivo.
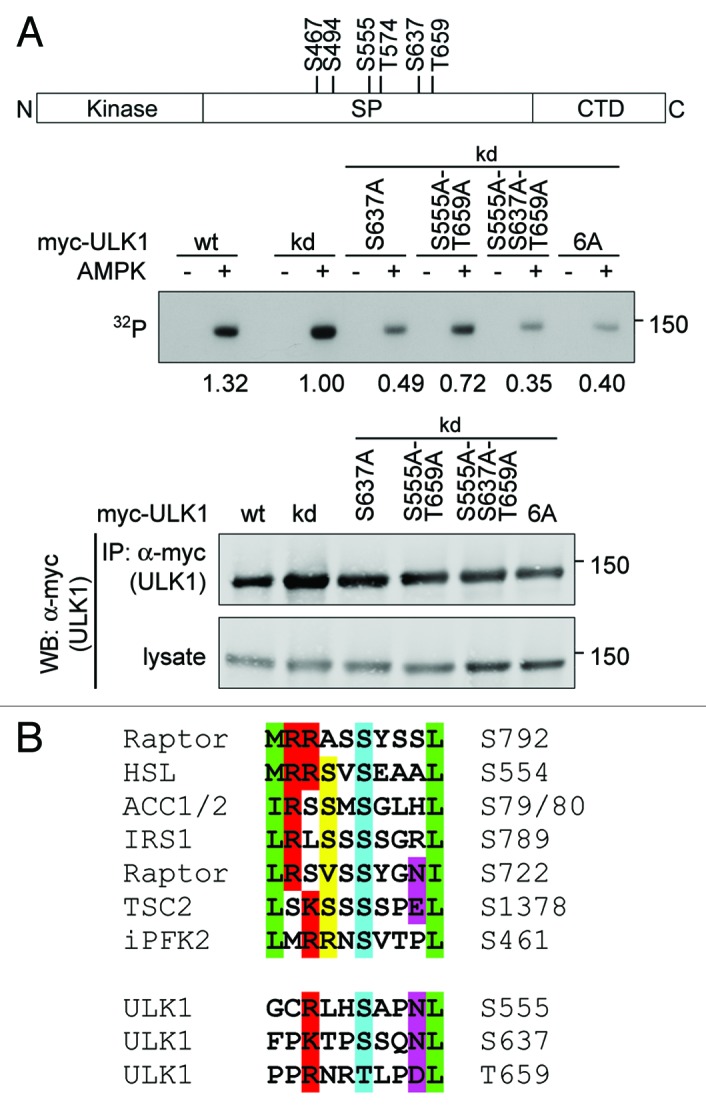
Figure 5. ULK1 S555, S637 and T659 are phosphorylated by AMPK in vitro. (A) Myc-tagged wild-type (wt) ULK1, kinase dead ULK1 (kd) and kinase dead mutants S637A, S555A-T659A, S555A-S637A-T659A and S467A-S494A-S555A-T574A-S637A-T659A (6A) were transiently expressed in HEK293T cells, immunoprecipitated from the cell lysates with anti-myc-antibody and used as substrates in in vitro kinase assays with purified AMPK in the presence of its allosteric activator AMP or analyzed by western blotting. For each substrate, one reaction was performed in the absence of AMPK and AMP to control for phosphorylation by coprecipitating kinases. Autoradiographs are shown. The numbers represent the ratio of the signal in the in vitro kinase assay to the signal in the western blot of the anti-myc immunoprecipitation. The schematic drawing depicts the location of the candidate AMPK phosphorylation sites examined in this experiment. (B) Comparison of the AMPK phosphorylation sites in ULK1 validated in this study with known AMPK phosphorylation sites in other proteins. Residues defined by the AMPK consensus motif are highlighted as in Figure 4B.
AMPK-activation increases YWHA/14-3-3 binding to ULK1
Previous reports have identified AMPK as a kinase that prefers a basic residue in the -3 position relative to the phosphoacceptor.35,52,53 Interestingly, the YWHA/14-3-3 protein family recognizes phosphoserine/phosphothreonine proteins that carry the motif, RXXpSXP.54 Consistent with this data, YWHA proteins can bind to some AMPK substrates.35 Thus, we investigated the hypothesis that the immediate effect of AMPK-dependent phosphorylation of ULK1 is enhanced binding of YWHA proteins. Myc-ULK1-overexpressing COS7-cells were treated with control vehicle or an AMPK activator and lysates were incubated with GST-YWHAZ. GST-YWHAZ was able to coprecipitate a small amount of myc-ULK1 under normal growth conditions, but AMPK activation resulted in increased coprecipitation of myc-ULK1 and GST-YWHAZ (Fig. 6A–C). This was not dependent on ULK1 overexpression as endogenous ULK1 was also found in GST-YWHAZ precipitates, and the amount of ULK1 detected increased with AMPK activation (Fig. 6D). Moreover, upon AMPK activation, increasing amounts of myc-ULK1 could be co-immunoprecipitated with endogenous 14-3-3 (Fig. 6E).

Figure 6. AMPK regulates YWHAZ binding to ULK1. COS7 cells infected with myc-ULK1 were treated with (A) 25 mM 2DG; (B) 1 mM AICAR; or (C) EBSS for the times indicated. The 0 min time point indicates treatment with vehicle for the longest timepoint. Cell lysates were incubated with GST-YWHAZ bound to glutathione sepharose beads or plain glutathione sepharose beads as a control. Immunoblots were probed with the antibodies indicated. (D) Wild-type MEFs and MEFs lacking the genes for both (PRKAA1−/−,PRKAA2−/−) or one of the PRKAA isoforms (PRKAA1−/− and PRKAA2−/−) were treated with vehicle (0 min time point) or 25 mM 2DG for 15 min and endogenous ULK1 was pulled down with GST-YWHAZ. The GST-tag alone served as a negative control. Western blot analysis was performed with the antibodies indicated. (E) MEFs stably overexpressing myc-ULK were treated with 25 mM 2DG as in (D) and endogenous YWHAZ was immunoprecipitated using a pan-14–3-3 antibody or nonspecific mouse IgG1 as a negative control. Western blot analysis was performed with the antibodies indicated. (F) Wild-type or nonphosphorylatable serine to alanine mutants of myc-ULK1 at four potential YWHAZ-binding sites, S467A, S494A, S555A and T659A as well as the S555A-T659A double mutant were stably overexpressed in COS7 cells were treated with 25 mM 2DG as in (D). Following lysis, myc-ULK1 variants were pulled down with GST or GST-YWHAZ. Western blot analysis was performed with the antibodies indicated.
To determine if YWHAZ binding was dependent on AMPK, we incubated GST-YWHAZ beads with lysates prepared from untreated or 2DG-treated wild-type MEFs and from MEFs lacking the genes for either one or both isoforms of the catalytic AMPKα subunit (Fig. 6D). While the amount of ULK1 detected in GST-YWHAZ precipitates increased upon AMPK activation in wild-type cells, there was no increase in binding in the PRKAA1−/−;PRKAA2−/− MEFs. Analysis of single knockout cells indicated that loss of the major isoform in MEFs, PRKAA1, also caused a dramatic decrease in the amount of ULK1 detected in GST-YWHAZ precipitates. Taken together, these results suggest that ULK1 may be phosphorylated by AMPK under normal growth conditions and that AMPK activation results in increased YWHAZ binding to ULK1. It should be noted that in the absence of AMPK, we consistently observed a small but detectable decrease in ULK1 protein levels, suggesting that AMPK may regulate ULK1 expression or stability (Fig. S3).
S555 and T659 mediate AMPK phosphorylation-dependent YWHAZ-binding to ULK1
Having established that AMPK activation leads to increased binding of YWHAZ to ULK1, we next sought to determine which sites are important for this enhanced association. Candidate AMPK phosphorylation sites and YWHAZ binding motifs in ULK1 were mutated to alanine and tested for their ability to complex with GST-YWHAZ (Tables S1–S3; Fig. 6F; Fig. S4A). Mutation of S467 or S494 did not alter the 2DG-induced increase in ULK1 binding to GST-YWHAZ (Fig. 6F). In contrast, the S555A-mutant showed little to no increase in GST-YWHAZ binding after 2DG treatment. Mutation of T659 also altered binding of GST-YWHAZ to ULK1 but this was more apparent in the S555A-T659A double mutant. In an alternative approach, we examined a set of ULK1 mutants in which the AMPK-site confirmed by mass spectrometry, S637, and three out of the four strong candidate AMPK/YWHAZ-binding-sites, S467, S494, S555 and T659, were mutated to alanines, leaving only one site available for phosphorylation (Fig. S4A). In line with our previous result, quantification of the immunoblots revealed that increased YWHAZ binding to ULK1 upon AMPK activation required intact S555 or T659 (Fig. S4B). Thus S555 and T659 are important for enhanced binding of ULK1 to GST-YWHAZ upon AMPK activation.
AMPK phosphorylation and ULK1 kinase activity
Next, we sought to determine the consequence of AMPK-phosphorylation and AMPK-phosphorylation-dependent YWHAZ-binding on ULK1 function, focusing on ULK1 kinase activity. To investigate whether AMPK regulated ULK1 kinase activity, we performed in vitro kinase assays on myc-tagged ULK1 isolated from wild-type MEFs that were untreated or treated for 15 min with 2 DG (Fig. 7A). In addition, to block the activity of the coprecipitating AMPK, we added the AMPK inhibitor, compound C, to the in vitro kinase reactions.55 Although we consistently saw an increase in AMPK activation with 2DG-treatment, we did not see a consistent increase in ULK1 phosphorylation upon AMPK activation. This is likely due to phosphorylation of ULK1 in vivo reducing the number of sites available for phosphorylation in vitro. Given these issues, we compared phosphorylation of myc-ULK1 in wild-type and AMPK PRKAA1−/−;PRKAA2−/− MEFs in the presence of compound C. In the presence of compound C, phosphorylation of myc-ULK1 from PRKAA1−/−;PRKAA2−/− MEFs was still considerably lower than phosphorylation of myc-ULK1 from wild-type MEFs. This is consistent with AMPK promoting ULK1 autophosphorylation. Alternatively, AMPK may regulate the activity of a coprecipitating kinase.

Figure 7. AMPK regulates phosphorylation of ULK1 by other kinases. (A) Wild-type MEFs or MEFs lacking the genes for both AMPKα isoforms (α1−/−,α2−/−) stably overexpressing myc-tagged wild-type ULK1 were treated with vehicle (0 min time point) or 25 mM 2DG for 15 min to activate AMPK in wild-type MEFs. Myc-ULK1 was immunoprecipitated from the cell lysates using anti-myc-antibody and immunoprecipitates were subjected to analysis by in vitro kinase assays and western blotting. 50 µM of the AMPK inhibitor compound C or vehicle (DMSO) were added to the kinase assays (k.a.) as indicated, to control for phosphorylation by AMPK coprecipitating with myc-ULK1 in lysates from wild-type MEFs. Phosphorylation of myc-ULK1 was detected by autoradiography. Signals were quantified and normalized to the corresponding western blot signals. The numbers indicate in vitro phosphorylation in the presence of compound C of myc-ULK1 from the two cell types, relative to the phosphorylation of myc-ULK1 from control-treated (0 min time point) wild-type MEFs. (B) ULK1−/− MEFs stably overexpressing myc-tagged wild-type ULK1 or nonphosphorylatable mutants on previously established AMPK phosphorylation sites were subjected to control- or 2DG treatment as in (A). Immunoprecipitation and analysis of myc-ULK1 constructs by in vitro kinase assays and western blotting was performed as in (A). Phosphorylation of myc-ULK1 was detected by autoradiography and signals were quantified and normalized to the corresponding western blot signals as in (A). The numbers indicate in vitro phosphorylation in the presence of compound C of the various myc-ULK1-constructs, relative to phosphorylation of wild-type myc-ULK1 from control-treated (0 min time point) cells.
Given the observation that AMPK is required for increased ULK1 in vitro phosphorylation, we examined whether the three previously identified AMPK-phosphorylation sites in ULK1—S637 and the two YWHAZ-binding sites S555 and T659—were important for this regulation. Myc-tagged ULK1 wild-type or non-phosphorylatable mutants on these AMPK target sites were stably overexpressed in ULK1−/− MEFs, isolated from cells following treatment with 2DG for 0 min or 15 min to activate AMPK, and subjected to in vitro kinase assays (Fig. 7B). Similar to the experiments shown in 7A, compound C was added to the kinase reaction to block the in vitro kinase activity of coprecipitating AMPK. When ULK1-mutants were isolated from cells in which AMPK was activated by 2DG, we observed increased phosphorylation of the S555A-T659A and the S555A-S637A-T659A-mutant, but not of the S637A-mutant. This implies that the AMPK-dependent YWHAZ-binding to ULK1 inhibits phosphorylation of ULK1 at other sites. Whether this is due to YWHAZ affecting the ability of ULK1 to phosphorylate itself at other sites or whether it is due to regulation of an associated kinase remains to be determined.
AMPK promotes efficient degradation of SQSTM1/p62 in early to intermediate stages of starvation-induced autophagy
Given contradictory reports in the literature on the requirement for AMPK in autophagy of mammalian cells, we re-examined its role in autophagy in EBSS-starved MEFs using GFP-LC3 puncta formation and SQSTM1 degradation as markers for autophagy.28,33 In agreement with a previous report, we found that after 3–4 h of starvation, there was little to no difference in GFP-LC3 puncta in PRKAA1−/−;PRKAA2−/− MEFs compared with wild-type MEFs (data not shown).33 Thus, AMPK appears to be dispensable for GFP-LC3 puncta formation and upregulation of autophagy upon nutrient deprivation. Yet, under nutrient-rich conditions, PRKAA1−/−;PRKAA2−/− MEFs displayed a small but significant increase in GFP-LC3-puncta. Given that the GFP-LC3-puncta formation assay relies on ectopic expression of a reporter construct, we further sought to clarify the requirement for AMPK in autophagy by monitoring autophagic degradation of endogenous SQSTM1-protein. PRKAA1−/−;PRKAA2−/− MEFs showed an increase in SQSTM1 relative to wild-type MEFs (Fig. S5A and S5C, and data not shown). Furthermore, when we examined the fold change in SQSTM1 levels, degradation of SQSTM1 appeared to be delayed in early-intermediate stages of EBSS starvation (3–6 h) in AMPK null MEFs compared with wild-type MEFs (Fig. S5A and S5B, and data not shown). Yet, at late time points (12 h), the fold change in SQSTM1 levels was similar in AMPK-null MEFs and wild-type MEFs (Fig. S5A and S5B). When starvation was continued for more than 12 h, we observed massive cell death in AMPK-null MEFs but not in wild-type MEFs, as reported previously (data not shown).33,56 The discrepancy of the GFP-LC3-marker and the SQSTM1 marker at early to intermediate starvation times in PRKAA1−/−;PRKAA2−/− MEFs is reminiscent of previous studies in HEK293T cells where siRNA depletion of ATG9 did not impair starvation-induced GFP-LC3 puncta formation, but did impair long-lived protein degradation.57 Therefore, we concluded that AMPK—in addition to its role during long-term starvation, where it promotes cell survival—increases autophagy efficiency in early to intermediate stages of the process.
AMPK-dependent phosphorylation of ULK1 is important for correct intracellular localization of ATG9
In yeast and mammalian cells, Atg1/ULK1 is important for cycling of Atg9/ATG9, a key regulator of the phagophore assembly site.13,58,59 In HEK293T cells, ATG9 accumulates in perinuclear clusters colocalizing with markers of the trans Golgi network under nutrient-rich conditions, while under conditions of nutrient stress, it is found in peripheral endosomal compartments. Knockdown of ULK1 blocks this relocalization and ATG9 remains in perinuclear clusters.57 Given the dependence of ATG9 cycling on ULK1, the biochemical connection between ULK1 and AMPK, and the observation that AMPK as well as ATG9 appear to differentially affect the autophagic markers SQSTM1 and GFP-LC3, we investigated whether AMPK was also required for correct ATG9 localization.
We first sought to confirm whether ATG9 localization was altered in MEFs and whether this was dependent upon ULK1. Littermate matched wild-type MEFs or ULK1−/− MEFs were cultured in full media or subjected to starvation with EBSS, and localization of endogenous ATG9 was analyzed by immunofluorescence microscopy. While we observed the previously reported localization pattern in wild-type MEFs (perinuclear ATG9 clusters under nutrient-rich conditions that are lost upon starvation), we unexpectedly found that the intensity of these clusters was markedly decreased in ULK1−/− MEFs, even when cultured under nutrient-rich conditions. No difference in ATG9 localization was seen between ULK1−/− MEFs and wild-type MEFs under starvation conditions (Fig. 8). This was not due to differences in ATG9 levels between wild-type and ULK1−/− MEFs (Fig. S6). Importantly, re-expression of wild-type myc-ULK1 was able to largely rescue the defect in perinuclear ATG9 cluster formation seen under nutrient-rich conditions (Fig. 8A and B). Thus, our data suggests that ULK1 is important for localization of ATG9 to perinuclear clusters under nutrient-rich conditions, or for recycling of ATG9 from peripheral pools to perinuclear clusters. Similarly, reduced accumulation in perinuclear clusters was also seen for ATG9 in fully fed PRKAA1−/−;PRKAA2−/− MEFs compared with littermate matched wild-type MEFs (Fig. 8C and D). These results indicate that AMPK, like ULK1, regulates ATG9 accumulation in perinuclear clusters under nutrient-rich conditions or the cycling of ATG9 between the peripheral compartments and perinuclear clusters.

Figure 8. ULK1 and AMPK regulate the intracellular localization of ATG9. (A) Wild-type MEFs carrying an empty vector or ULK1−/− MEFs expressing either an empty vector (V) or myc-tagged wild-type (wt) ULK1 or the ULK1 S555A-S637A-T659A (“3A”) mutant were cultured in regular growth media (“full media”) or EBSS (“starvation”) for 3 h, and intracellular localization of endogenous ATG9 (red staining) was examined by immunoflourescence. Nuclei were counterstained with DAPI (blue). (B) Quantitation of ATG9-staining in the experiment shown in (A). Analysis was performed using Cellprofiler Image analysis software. Each data point represents one cell, horizontal bars and error bars indicate means with 95% confidence intervals. A minimum of 20 cells per cell line and treatment condition was analyzed. Statistical significance was determined by unpaired t-tests with Welch’s correction. *** indicates a nominal p-value < 0.001, n.s. indicates a nonsignificant nominal p-value > 0.05. The result shown are representative for 3 independent experiments. (C) Wild-type MEFs and MEFs lacking the genes for both catalytic PRKAA-subunits (PRKAA1−/−;PRKAA2−/−) were treated and examined for intracellular ATG9-localization as in (A). (D) Quantitation of ATG9-staining in the experiment shown in (C). Image analysis and statistics were performed as in (B). Nominal p-values of p < 0.001, p < 0.05 and p > 0.05 are denoted as ***, *, and n.s. (nonsignificant), respectively.
Given these data, we next tested whether the major AMPK phosphorylation sites we identified on ULK1 are important for regulating ATG9 localization. Expression of the myc-ULK1–3A mutant (S555A-T659A-S637A) in ULK1−/− MEFs was unable to rescue the defect in ATG9 accumulation in perinuclear clusters (Fig. 8A and B). Taken together, these data suggest that both AMPK and ULK1 are important for localization of ATG9 to perinuclear clusters and that this localization is regulated at least in part by AMPK phosphorylation of ULK1 at specific sites.
Discussion
Induction of autophagy is a common cellular response to a variety of stress conditions. The signaling networks regulating autophagy, however, are only beginning to be defined. In the present study, we identified AMPK as an interaction partner of ULK1/2 and demonstrated that ULK1 is a substrate of AMPK. Similar results have been recently reported by other groups.60-64 We identified three sites in ULK1 that are phosphorylated by AMPK, S555, S637 and T659. Phosphorylation of two of these sites, S555 and T659, results in an AMPK-dependent increase in binding of the adaptor protein YWHAZ to ULK1. Moreover, our data indicates that this AMPK-dependent phosphorylation of ULK1 contributes to efficient shuttling of the putative autophagosomal membrane carrier protein ATG9.
Changes in ULK1 phoshorylation in response to nutrient deprivation
We identified six constitutive and 24 nutrient-sensitive phosphorylation sites in ULK1, and 17 of these sites have not been previously reported (Tables S1–S3). Bioinformatic analysis using Scansite predicts candidate regulators of ULK1 and potentially autophagy. Consistent with this idea, at least two kinases known to regulate autophagy, CDK5 and MAPK14/p38, are predicted to phosphorylate ULK1.65-67 Determining whether these different proteins modulate ULK1 function and/or autophagy will provide further insight on the ULK1 regulatory networks.
Several of the phosphorylation sites we found to be induced by starvation have been detected under nutrient-rich conditions in previous reports. This difference may be due to variations in the cell types used or technical difficulties in eliminating influences from stress-signaling pathways that are activated when cells are lysed. In any case, our data suggest that ULK1 phosphorylation may vary at different stages of autophagy; this is consistent with ULK1’s predicted role in regulating autophagy at multiple steps as well as its role in other cellular processes distinct from autophagy.68
Signaling pathways regulating the ULK1-AMPK-interaction
We and others have identified an interaction between ULK1 and AMPK.60-64 This interaction does not require kinase activity since catalytically inactive ULK1 can associate with AMPK and catalytically inactive AMPK can associate with ULK1 (Fig. 2). In addition, the ULK1-AMPK complex can be detected under normal growth conditions where the negative regulator of autophagy, MTORC1, is active and AMPK activity is minimal, and under a variety of conditions where AMPK is activated and MTORC1 activity reduced (Fig. 3A and B). Interestingly, in the cell lines we examined, there was a decrease in the ULK1-AMPK complex detected after extended nutrient deprivation (Fig. 3B; Fig. S1), and Shang et al. have reported similar results.64 Taken together, these data preclude a simple model in which this complex only forms and functions during autophagy and in which MTORC1 regulates formation of this complex. Our results and those of Shang et al. are in contrast with those recently reported by Kim et al. who found that constitutive activation of MTOR (via overexpression of RHEB) resulted in a decrease in the ULK1-AMPK complex detected while inhibition of MTOR (via rapamycin treatment of RHEB-overexpressing cells) resulted in increased binding of ULK1 to AMPK.62,64 Given that the sensitivity of the ULK1-AMPK complex to various activators of AMPK and MTOR shows some cell-type dependence, regulation of the ULK1-AMPK cassette is likely to be more complicated and suggests that it may be dependent on additional proteins or other factors that are differentially expressed or activated in these various cell lines.
AMPK and autophagy
Similarly, a simple model of how AMPK regulates autophagy is not apparent. In S. cerevisiae, the AMPK-homolog Snf1 was found to function upstream of Atg1 and Atg13 in autophagy.69 In mammalian cells, expression of dominant negative AMPK or knockdown of AMPK blocks autophagy induced by nutrient stress as well as a variety of other agents.28,48,49 In contrast, AMPK knockout cells show higher levels of autophagy under low glucose conditions, although they are more susceptible to apoptotic cell death upon prolonged and exacerbated stress.33 We also observed massive cell death in AMPK null MEFs but not in wild-type MEFs upon extended EBSS-starvation (> 12 h, data not shown), which may reflect the fact that AMPK is required for entry into autophagy instead of apoptosis during prolonged glucose deprivation.56 In addition, using SQSTM1-degradation as a read-out for autophagic activity, we find that in MEFs, AMPK is not absolutely essential for autophagy at early and intermediate stages of EBSS-starvation, but it enhances the efficiency of the process (Fig. S5). In contrast, starvation-induced GFP-LC3 puncta formation was unaffected by the absence of AMPK (data not shown). A similar discrepancy between regulation of GFP-LC3 puncta formation and long-lived protein degradation has been reported previously for another mammalian autophagy protein, ATG9.57 This is consistent with the notion that AMPK and ATG9 proteins may function at a common step in autophagy, and indeed, we find that AMPK is important for correct intracellular localization of ATG9 (see below).
Mechanistically, AMPK may regulate autophagy via regulation of the MTORC1 pathway as AMPK can phosphorylate a regulator of MTORC1, TSC2, and one of its components, RPTOR.34-36 Our work and other recent reports suggest a more direct link between AMPK and the autophagy machinery, consisting of its interaction with and phosphorylation of ULK1. While the exact position of the ULK1 phosphorylation sites is not strictly conserved across species, AMPK phosphorylation sites can be found in the spacer region of several ULK1 homologs. Therefore, ULK1 phosphorylation by AMPK is likely to constitute a conserved mechanism for regulation of ULK1 function.
ULK1, however, may not be the only autophagy target of AMPK. Bioinformatic analysis predicts additional ATG proteins as potential substrates of AMPK including ATG9 (see below), ATG12, ATG16L1, ATG18 and BECN1. Furthermore, these predicted AMPK substrates also contain Atg1-consensus motifs, raising the possibility that the two kinases function as a signaling cassette to phosphorylate common substrates.70 Such coordinated regulation could serve to make regulation of autophagy more efficient but also more selective. Intriguingly, the ULK1-AMPK complex could also be involved in comodulating signals for other cellular processes. Searching protein databases for phosphorylation motifs of Atg1/ULK1 and AMPK identified proteins implicated in processes such as metabolism, cell migration and vesicular trafficking (data not shown). For example, CLIP1/CLIP170, which regulates cell migration and is a known AMPK substrate has predicted ULK1 phosphorylation sites and both AMPK and ULK1 have been implicated in cell migration and polarity.71-73 Similarly, we identified the focal adhesion protein paxillin as an ULK1 substrate, and this protein, which is important for cell migration and autophagy, also contains predicted AMPK phosphorylation sites. In principle, such coordinated action is facilitated by the fact that the ULK1-AMPK cassette is detected under physiological growth conditions and also relatively nutrient- and energy stress-insensitive, at least in certain cell types and upon short-term exposure to stressors.
Role of AMPK phosphorylation and 14-3-3 binding
In addition to AMPK, we also found that the adaptor protein YWHAZ co-immunoprecipitated with ULK1. Moreover, AMPK-activation increases YWHAZ binding to ULK1, which has also been reported by Egan et al.61 We identified two AMPK phosphorylation sites in ULK1 that mediate this effect: S555 and T659. While these two sites are important for AMPK-activation-induced YWHAZ-binding, there are additional YWHAZ sites predicted in ULK1 that may account for the association of these two proteins under basal conditions. In general, the finding that these two proteins interact per se suggests that YWHAZ could be a regulator of autophagy. With regard to regulation of ULK1, our data clearly demonstrate that YWHAZ-binding to AMPK-phosphorylation sites blocks in vitro phosphorylation of other sites in ULK1. Whether this is due to YWHAZ regulating the ability of ULK1 to phosphorylate these other sites, or whether it is due to regulation of an associated kinase, remains to be determined. Our preliminary results indicate that there is at least one additional kinase associated with ULK1 whose activity may be regulated by AMPK activation (data not shown).
In line with our results (Fig. 5E63), Lee et al. observed increased amounts of endogenous YWHAZ coprecipitating with exogenous ULK1 upon AMPK activation.63 It is unclear whether YWHAZ binds directly to ULK1 or to one of its interacting proteins, in particular to RPTOR, a component of the MTORC1 complex.16 RPTOR also binds YWHAZ and this interaction is enhanced by AMPK activation.35 While this raises the possibility that ULK1 may indirectly interact with YWHAZ via RPTOR, the lysis conditions we used for our studies are unlikely to preserve the ULK1-RPTOR complex.16,74 Of note, a recent comprehensive proteomic study that used lysis conditions similar to ours did not detect interaction of ULK1 and RPTOR while ULK1 was found to interact with YWHAZ.60
Regulation of ATG9-localization by ULK1 and AMPK
Our study describes a particular step of autophagy jointly regulated by ULK1 and AMPK, namely the intracellular trafficking of ATG9, whose likely function is the delivery of membranes to the expanding autophagosome.75,76 Previous studies in S. cerevisiae and HEK293T cells found that starvation-induced Atg9/ATG9 cycling from perinuclear clusters to peripheral compartments is Atg1/ULK1 dependent.13,57 In contrast, using ULK1−/− MEFs, we find that ATG9 localization to these perinuclear clusters also is ULK1-dependent. Further studies are required to elucidate whether this reflects an adaption to chronic ULK1 deficiency or whether there is a general difference in the role of ULK1 in ATG9 localization among various cell types.
Importantly, both AMPK null and ULK1−/− MEFs showed similar defects in ATG9 localization under nutrient-rich conditions. Moreover, re-expression of an ULK1-mutant that lacks the major AMPK phosphorylation sites S555, S637 and T659 failed to restore ATG9 clustering in ULK1−/− MEFs. This indicates that AMPK and ULK1 both promote localization of ATG9 to perinuclear clusters and that the mechanistic basis for this includes AMPK-dependent phosphorylation of ULK1. Given that enhanced binding of YWHAZ is defective in this mutant and that the 3A mutant also shows increased phosphorylation in vitro, one can speculate that YWHAZ binding may be important for downregulating ULK1 phosphorylation and moving ATG9 from peripheral compartments to perinuclear clusters.
At first glance, it may be surprising that AMPK phosphorylates ULK1 and regulates ATG9-localization under nutrient-rich conditions but, in our hands, does not seem to affect ATG9-localization under starvation conditions. Under nutrient-rich conditions, AMPK activity would be expected to be lower than under starvation conditions. However, consistent with a previous report we found that EBSS-starvation did not robustly increase AMPK-activity (Fig. S3).64 Moreover, prolonged EBSS-starvation, which we applied in our ATG9-localization studies, causes destabilization of the ULK1-AMPK-complex (Fig. 3; Fig. S1), and this destabilization is also observed in MEFs, i.e., the cell type we use in the ATG9-localization studies (data not shown). Thus, we propose a model in which ATG9-localization is regulated by the fraction of ULK1 that is associated with and phosphorylated by AMPK. As we show, ATG9-localization is regulated by AMPK-dependent phosphorylation of ULK1 under nutrient-rich conditions, which are the conditions under which interaction of ULK1 and AMPK is maximal. Hence, low basal activity of AMPK is likely to be sufficient to phosphorylate the ULK1-molecules directly bound to it. We note that close proximity of ULK1 and AMPK also is central in the model for ULK1-regulation by AMPK suggested by other authors.64
Although we provide evidence that AMPK contributes to ATG9 localization by phosphorylating ULK1, it is interesting to note that there are additional players in the ATG9 pathway that are candidate or established substrates of AMPK. For example, ATG9 contains AMPK- as well as ULK1-consensus sites. In addition, the Drosophila non-muscle myosin regulatory light chain, Sqh, is an AMPK-substrate and also plays a role in Atg9 localization.77,78 Interestingly, Sqh is regulated by a recently identified substrate of ULK1, DAPK3/ZIPK.78 Clearly, additional studies are needed to dissect the precise mechanism of ATG9 trafficking and the contributions of ULK1 and AMPK to this process.
In addition to mislocalization of ATG9, loss of AMPK also caused inhibition of SQSTM1-degradation at the beginning of starvation-induced autophagy, suggesting that AMPK is required for maximum efficiency of this process (Fig. S5). How could AMPK-dependent regulation of ATG9-localization increase autophagy efficiency? A recent comprehensive proteomic study of the autophagy network implies that the autophagy machinery to a large extent is already pre-assembled under nutrient-rich conditions. This situation, in which the cell is “ready to go” as soon as an autophagy-inducing stimulus arrives, is impaired when ATG9 is mislocalized, and therefore, it is reasonable to speculate that upregulation of autophagy takes more time in this setting. In addition, it is possible that the cell has several mechanisms to form autophagosomes, only some of which are dependent on ATG9. This hypothesis is supported by the fact that autophagosomal membranes may be derived from multiple different sources including the plasma membrane, outer mitochondrial membrane, endoplasmic reticulum and Golgi.79-82 In yeast, Atg9 has been suggested to contribute Golgi membranes to autophagosome formation via its cycling and in mammalian cells, ATG9 has been postulated to contribute membranes from the trans-Golgi network or endosomes.57,75 Clearly, a deeper understanding of ATG9 function is required to precisely define the mechanisms by which ATG9, and ultimately AMPK, potentially via phosphorylation of ULK1, contributes to efficient autophagy.
Taken together, our data identify AMPK as an ULK1 kinase that contributes to its function in intracellular trafficking of the putative autophagosomal membrane carrier protein, ATG9, and thereby provide further mechanistic insights into how these two kinases regulate autophagy.
Materials and Methods
Plasmids
pGEX-4T1-14-3-3ζ GST was described previously.54 pCX-myc-ULK1 was provided by Dr. Mary Hatten (Rockefeller University) and p3xFLAG-CMV14-ULK1 variants were generously provided by Dr. Noboru Mizushima (Tokyo Medical and Dental University, Tokyo, Japan).16,83 ULK1 mutants were generated using the QuickChange II XL site-directed mutagenesis kit (Agilent Technologies, 200522) and verified by sequencing. pEX-EF1-YFP-ULK2 was obtained from ATCC/AfCs (1037973). The pcDNA3-FLAG-AMPKα1 plasmid was constructed by cloning the human AMPKα1 cDNA into the EcoRI/XhoI-sites of pcDNA3-FLAG.84
Antibodies and reagents
Anti-phospho-AMPKα Thr172 (2535), anti-AMPKα (2603 and 2793) and anti-phospho-Acetyl-CoA carboxylase Ser79 (3661) antibodies were from Cell Signaling Technology, anti-AMPKα1 (04-323) and anti-AMPKα2 (07-363) antibodies from Millipore, and anti-β-Actin (A1978), anti-FLAG (F3165) and anti-ULK1 (A7481) antibodies from Sigma. Anti-pan-14-3-3 antibody used for immunoprecipitation was from Thermo Fisher (MS-1504-PABX) and anti-14-3-3 used for western blot from Santa Cruz Biotechnology (sc-629). For immunoprecipitation and western blot detection of myc-tagged proteins, mouse monoclonal antibody 9E10 was used. The monoclonal anti-ATG9A antibody (clone 14F28B1) used for immunofluorescence and the polyclonal anti-ATG9A antibody used for western blot (ab71795 and ab54500) were purchased from Abcam. 2-deoxyglucose was purchased from Sigma (D8375), A-769662 from Tocris (3336) and AICAR from Toronto Research Chemicals (A611700).
Cell culture and transfection
Mouse embryonic fibroblasts (MEFs) deficient in the genes encoding one or both isoforms of PRKAA and corresponding wild-type MEFs were provided by Dr. B. Viollet (INSERM U567; Paris, France) and described previously.85-87 ULK1−/− MEFs and corresponding wild-type MEFs were provided by Dr. Mondira Kundu (St. Jude Children’s Hospital, Memphis, TN/USA).88 All cells were cultured in Dulbecco’s modified Eagle medium (DMEM, Cellgro, 10–013), supplemented with 10% fetal bovine serum (Atlanta Biologicals, S11550) and 100 µg/ml penicillin/streptomycin (Invitrogen, 15140-122) at 37°C, 10% CO2. For starvation treatments, cells were washed twice with phosphate-buffered saline (PBS) and incubated in Earl’s balanced salt solution (EBSS, Invitrogen, E2888) for the times indicated. HEK293T and COS7 cells were transfected using polyethylenimine (PEI; Polysciences Inc., 23966-2) at a 3:1 and 5:1 ratio to DNA, respectively.
Production of retrovirus and retroviral infection
For production of retrovirus, target plasmids were cotransfected with packaging plasmids pCMV-Tat2, pJK3 and pVSVG into HEK293T cells using PEI. Supernatant was collected 24, 36 and 48 h post transfection, pooled and centrifuged to eliminate cells. For infections, cells were incubated in viral supernatant containing 8 µg/ml polybrene (Sigma, H9268) for 16 h.
Immunoprecipitation and western blotting
For co-immunoprecipitation of myc-ULK1 with endogenous PRKAA, cells were lysed in Triton lysis buffer (20 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1% Triton X-100). For co-immunoprecipitation of myc-ULK1 and FLAG-AMPKα1 lysis was performed using CHAPS lysis buffer (40 mM HEPES, pH 7.5, 120 mM NaCl, 1 mM EDTA, 0.3% CHAPS). For immunoprecipitation of endogenous AMPK and ULK1, CHAPS lysis buffer without salt was used. For co-immunoprecipitation of endogenous YWHAZ and myc-ULK1, cell lysates were prepared in NP-40 lysis buffer (0.75% NP-40 in PBS). All lysis buffers contained protease inhibitors (7.7 U/ml aprotinin, 1 µM leupeptin, 1 mM PMSF) and phosphatase inhibitors (2.5 mM β-glycerophosphate, 2.5 mM sodium fluoride, 1 mM sodium pyrophosphate, 200 µM pervanadate). Lysates were cleared by centrifugation and incubated with Pansorbin cells (Calbiochem, 507861) for 2 × 15 min. For immunoprecipitation of endogenous PRKAA-isoforms, lysates were incubated with the desired antibody over night at 4°C, followed by incubation with Protein A-agarose beads (Repligen, 10-1003). For immunoprecipitation of myc-ULK1 or endogenous YWHAZ, lysates were incubated with the antibody at 4°C for 2 h followed by incubation with Protein G-sepharose-beads (Invitrogen, 10-1242). For immunoprecipitation of FLAG-tagged proteins, anti-FLAG M2 affinity gel (Sigma, A2220) was added to the lysates for 45 min. Precipitates were washed four times with lysis buffer and eluted with Laemmli buffer. Samples were separated by SDS-PAGE and transferred to PVDF membranes (Bio-Rad, 162–0177 or Millipore, IPFL00010). Membranes were incubated with primary antibodies at 1:1,000 dilution (exceptions: α-Atg9A 1:2,000; α-β-Actin:1:5,000; α-myc 1:7,000; anti-ULK1 1:500) followed by secondary antibody conjugates to HRP (α-Mouse-IgG, Bio-Rad, 172–1011; α-Rabbit-IgG, Cell Signaling Technology, 7074) or fluorescent dyes (α-Mouse-IgG Alexa Flour 680 conjugate, Invitrogen, A21057; α-Rabbit-IgG IRDye® 800 conjugate, Rockland, 611-132-122) and subsequently detected with enhanced chemiluminescence (Millipore, WBKLS0500) on X-ray films (Denville, E3018) or on a molecular imaging system (Carestream, GL 4000 Pro) with fluorescence on a Odyssey® infrared imager (LI-COR® Biosciences).
GST-YWHAZ pulldown
Recombinant GST-YWHAZ/14-3-3ζ was expressed in E. coli BL21 and purified with glutathione sepharose (GE Healthcare, 17-0756-01). For pulldown experiments, cell lysates were prepared in modified RIPA (10 mM Tris, pH 7.4, 158 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate) with protease and phosphatase inhibitors as described above and, after clearing by centrifugation, incubated with GST-YWHAZ bound to glutathione sepharose beads at 4°C for 4 h. Samples were washed, eluted, separated by SDS-PAGE and analyzed by western blot as described above.
Kinase assays
For use as substrates in AMPK kinase assays, various tagged ULK1 constructs were transiently overexpressed in HEK293T cells and immunoprecipitated from the cell lysates as described above with addition of a high salt buffer wash (10 mM Tris, pH 7.4, 500 mM NaCl) before the low salt buffer wash of the precipitates. Lysis was performed in modified RIPA buffer (10 mM Tris, pH 7.4, 158 mM NaCl, 0.5 mM EDTA pH 8.0, 1% v/v Triton X-100, 1% w/v deoxycholate) containing protease and phosphatase inhibitors (compare above). At the final wash step the sample was split into three, with one sample being subjected to SDS-PAGE followed by western blotting and the two remaining to in vitro kinase assays by incubation either in kinase buffer mix [1 mM ATP and 10 mCi g32P]-ATP in 1× kinase buffer (20 mM HEPES, pH 7.0, 0.4 mM DTT, 0.01% Brij, and 10 mM MgCl2)] alone or in kinase buffer mix with AMPK (Millipore, 14-305) and 300 mM AMP. Reactions were incubated at 30°C for 15 min and stopped by addition of 2X Laemmli buffer. For ULK1 kinase assays, myc-tagged ULK1-constructs were stably overexpressed in MEFs and immunoprecipitated as described above. Cells were lysed in Triton lysis buffer containing protease and phosphatase inhibitors (compare above). Precipitates were washed as described above, with addition of a second wash with low salt buffer at which samples were split into 2–3 samples, one for western blot analysis and 1–2 for in vitro kinase assays. Kinase assays were performed as for AMPK, with addition of 50 µM compound C (Sigma, P5499) or vehicle to the kinase buffer. Reaction time was 20 min at 30°C.
Immunofluorescence
Immunofluorescence was essentially done as previously described.89 Briefly, cells were grown on Fibronectin coated glass coverslips, fixed with 3.7% paraformaldehyde, permeabilized with 0.2% Triton X-100 and blocked with 5% milk powder in PBS. Coverslips were incubated overnight with anti-Atg9A-antibody (1:100) at 4°C and subsequently with DyLight 549-conjugated secondary antibody (1:500; Jackson Immunoresearch, 127-505-099) for 1 h at room temperature. Nuclei were counterstained with 4',6-diamidino-2-phenylindole (DAPI; Sigma, D9542) added at 0.5 µg/ml. Coverslips were mounted using Mowiol. Slides were visualized under a 63×/1.4 NA apochromat oil objective on a confocal laser scanning microscope (Carl Zeiss, LSM510). Image analysis was performed with CellProfiler cell image analysis software and the pipeline given in the supplementary materials available online.90
LC/MS/MS tandem mass spectrometry
For all mass spectrometry (MS) experiments, myc-ULK1 immunoprecipitates were separated using SDS-PAGE, the gel was stained with Coomassie blue, and the myc-ULK1 band was excised. Samples were subjected to reduction with dithiothreitol, alkylation with iodoacetamide, and in-gel digestion with trypsin or chymotrypsin overnight at pH 8.3, followed by reversed-phase microcapillary/tandem mass spectrometry (LC/MS/MS). LC/MS/MS was performed using an EASY-nLC nanoflow HPLC (Proxeon Biosciences/ Thermo Fisher Scientific, Waltham, MA/USA) with a self-packed 75 μm id × 15 cm C18 column connected to a hybrid LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) in the data-dependent acquisition and positive ion mode at 300 nL/min. A LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific) was also used for some experiments. MS/MS spectra collected via collision-induced dissociation in the ion trap were searched against the concatenated target and decoy (reversed) single entry ULK1 and full Swiss-Prot protein databases using Sequest (Proteomics Browser Software, Thermo Fisher Scientific) with differential modifications for Ser/Thr/Tyr phosphorylation (+79.97) and the sample processing artifacts Met oxidation (+15.99), deamidation of Asn and Gln (+0.984) and Cys alkylation (+57.02). Phosphorylated and unphosphorylated peptide sequences were identified if they initially passed the following Sequest scoring thresholds against the target database: 1+ ions, Xcorr ≥ 2.0 Sf ≥ 0.4, p ≥ 5; 2+ ions, Xcorr ≥ 2.0, Sf ≥ 0.4, p ≥ 5; 3+ ions, Xcorr ≥ 2.60, Sf ≥ 0.4, p ≥ 5 against the target protein database. Passing MS/MS spectra were manually inspected to be sure that all b- and y-fragment ions aligned with the assigned sequence and modification sites. Determination of the exact sites of phosphorylation was aided using FuzzyIons and GraphMod and phosphorylation site maps were created using ProteinReport software (Proteomics Browser Software suite, Thermo Fisher Scientific). False discovery rates (FDR) of peptide hits (phosphorylated and unphosphorylated) were estimated below 1.5% based on reversed database hits.
Supplementary Material
Acknowledgments
The authors thank Dr. Noboru Mizushima (Tokyo Medical and Dental University) for providing ULK1-FLAG-constructs, Dr. Benoit Viollet (INSERM U567) for the complete set of AMPK-knockout-MEFs, Dr. Mondira Kundu (St Jude Children’s Research Hospital) for ULK1−/− MEFs, and Dr. Karl Munger (Brigham and Women’s Hospital) for helpful scientific discussions and support. H.I.D.M would like to thank Dr. Karl-Friedrich Becker (Technische Universität München) for support and S.M.T. would like to thank Dr. Marti Ni for fruitful discussions. This work was funded by graduate fellowships from the Bavarian State Ministry of Sciences, Research and the Arts, the German Academic Exchange Service and the German National Academic Foundation (H.I.D.M), American Cancer Society grant RSG041751 (S.M.T) and NIH R00CA133245 (B.Z.).
Glossary
Abbreviations:
- 2DG
2-deoxyglucose
- ACC
acetyl-CoA carboxylase
- AICAR
5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside
- AMPK
adenosine monophosphate- activated protein kinase
- Atg1
autophagy-related protein 1
- CTD
C-terminal domain
- EBSS
Earle’s balanced salt solution
- IP
immunoprecipitation
- MEF
mouse embryonic fibroblast
- LC/MS/MS
liquid chromatography/tandem mass spectrometry
- MTOR
mechanistic target of rapamycin
- MTORC1
mechanistic target of rapamycin complex 1
- PRKAA1
protein kinase, AMP-activated, alpha 1 catalytic subunit
- RB1CC1/FIP200
focal adhesion kinase family interacting protein of 200 kDa
- TSC2
tuberous sclerosis complex protein 2
- ULK1
Unc-51-like kinase 1
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials can be found at: www.landesbioscience.com/journals/autophagy/article/20586
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/20586
References
- 1.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thumm M, Egner R, Koch B, Schlumpberger M, Straub M, Veenhuis M, et al. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994;349:275–80. doi: 10.1016/0014-5793(94)00672-5. [DOI] [PubMed] [Google Scholar]
- 3.Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333:169–74. doi: 10.1016/0014-5793(93)80398-E. [DOI] [PubMed] [Google Scholar]
- 4.Matsuura A, Tsukada M, Wada Y, Ohsumi Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene. 1997;192:245–50. doi: 10.1016/S0378-1119(97)00084-X. [DOI] [PubMed] [Google Scholar]
- 5.Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150:1507–13. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Díaz-Troya S, Pérez-Pérez ME, Florencio FJ, Crespo JL. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy. 2008;4:851–65. doi: 10.4161/auto.6555. [DOI] [PubMed] [Google Scholar]
- 7.Neufeld TP. TOR-dependent control of autophagy: biting the hand that feeds. Curr Opin Cell Biol. 2010;22:157–68. doi: 10.1016/j.ceb.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomoda T, Bhatt RS, Kuroyanagi H, Shirasawa T, Hatten ME. A mouse serine/threonine kinase homologous to C. elegans UNC51 functions in parallel fiber formation of cerebellar granule neurons. Neuron. 1999;24:833–46. doi: 10.1016/S0896-6273(00)81031-4. [DOI] [PubMed] [Google Scholar]
- 9.Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. 2007;282:25464–74. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- 10.Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. 2009;29:157–71. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 12.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reggiori F, Tucker KA, Stromhaug PE, Klionsky DJ. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell. 2004;6:79–90. doi: 10.1016/S1534-5807(03)00402-7. [DOI] [PubMed] [Google Scholar]
- 14.Ganley IG, Lam H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ptacek J, Devgan G, Michaud G, Zhu H, Zhu X, Fasolo J, et al. Global analysis of protein phosphorylation in yeast. Nature. 2005;438:679–84. doi: 10.1038/nature04187. [DOI] [PubMed] [Google Scholar]
- 19.Chen GC, Lee JY, Tang HW, Debnath J, Thomas SM, Settleman J. Genetic interactions between Drosophila melanogaster Atg1 and paxillin reveal a role for paxillin in autophagosome formation. Autophagy. 2008;4:37–45. doi: 10.4161/auto.5141. [DOI] [PubMed] [Google Scholar]
- 20.Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol. 2010;191:155–68. doi: 10.1083/jcb.201002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheong H, Yorimitsu T, Reggiori F, Legakis JE, Wang CW, Klionsky DJ. Atg17 regulates the magnitude of the autophagic response. Mol Biol Cell. 2005;16:3438–53. doi: 10.1091/mbc.E04-10-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell. 2005;16:2544–53. doi: 10.1091/mbc.E04-08-0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meijer WH, van der Klei IJ, Veenhuis M, Kiel JA. ATG genes involved in non-selective autophagy are conserved from yeast to man, but the selective Cvt and pexophagy pathways also require organism-specific genes. Autophagy. 2007;3:106–16. doi: 10.4161/auto.3595. [DOI] [PubMed] [Google Scholar]
- 24.Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal. 2009;2:pe51. doi: 10.1126/scisignal.284pe51. [DOI] [PubMed] [Google Scholar]
- 25.Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170:1101–11. doi: 10.1083/jcb.200504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A. 2007;104:19023–8. doi: 10.1073/pnas.0709695104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, et al. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281:34870–9. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 29.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–9. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–87. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–52. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 32.Grotemeier A, Alers S, Pfisterer SG, Paasch F, Daubrawa M, Dieterle A, et al. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal. 2010;22:914–25. doi: 10.1016/j.cellsig.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 33.Williams T, Forsberg LJ, Viollet B, Brenman JE. Basal autophagy induction without AMP-activated protein kinase under low glucose conditions. Autophagy. 2009;5:1155–65. doi: 10.4161/auto.5.8.10090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004;18:1533–8. doi: 10.1101/gad.1199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/S0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 37.Dorsey FC, Rose KL, Coenen S, Prater SM, Cavett V, Cleveland JL, et al. Mapping the phosphorylation sites of Ulk1. J Proteome Res. 2009;8:5253–63. doi: 10.1021/pr900583m. [DOI] [PubMed] [Google Scholar]
- 38.Oppermann FS, Gnad F, Olsen JV, Hornberger R, Greff Z, Kéri G, et al. Large-scale proteomics analysis of the human kinome. Mol Cell Proteomics. 2009;8:1751–64. doi: 10.1074/mcp.M800588-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daub H, Olsen JV, Bairlein M, Gnad F, Oppermann FS, Körner R, et al. Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol Cell. 2008;31:438–48. doi: 10.1016/j.molcel.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 40.Beausoleil SA, Villén J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–92. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 41.Pan C, Gnad F, Olsen JV, Mann M. Quantitative phosphoproteome analysis of a mouse liver cell line reveals specificity of phosphatase inhibitors. Proteomics. 2008;8:4534–46. doi: 10.1002/pmic.200800105. [DOI] [PubMed] [Google Scholar]
- 42.Chen RQ, Yang QK, Lu BW, Yi W, Cantin G, Chen YL, et al. CDC25B mediates rapamycin-induced oncogenic responses in cancer cells. Cancer Res. 2009;69:2663–8. doi: 10.1158/0008-5472.CAN-08-3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trinidad JC, Thalhammer A, Specht CG, Lynn AJ, Baker PR, Schoepfer R, et al. Quantitative analysis of synaptic phosphorylation and protein expression. Mol Cell Proteomics. 2008;7:684–96. doi: 10.1074/mcp.M700170-MCP200. [DOI] [PubMed] [Google Scholar]
- 44.Wissing J, Jänsch L, Nimtz M, Dieterich G, Hornberger R, Kéri G, et al. Proteomics analysis of protein kinases by target class-selective prefractionation and tandem mass spectrometry. Mol Cell Proteomics. 2007;6:537–47. doi: 10.1074/mcp.T600062-MCP200. [DOI] [PubMed] [Google Scholar]
- 45.Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, et al. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105:10762–7. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cantin GT, Yi W, Lu B, Park SK, Xu T, Lee JD, et al. Combining protein-based IMAC, peptide-based IMAC, and MudPIT for efficient phosphoproteomic analysis. J Proteome Res. 2008;7:1346–51. doi: 10.1021/pr0705441. [DOI] [PubMed] [Google Scholar]
- 47.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–41. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herrero-Martín G, Høyer-Hansen M, García-García C, Fumarola C, Farkas T, López-Rivas A, et al. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J. 2009;28:677–85. doi: 10.1038/emboj.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 50.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–93. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 51.Kamada Y, Yoshino K, Kondo C, Kawamata T, Oshiro N, Yonezawa K, et al. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol Cell Biol. 2010;30:1049–58. doi: 10.1128/MCB.01344-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Michell BJ, Stapleton D, Mitchelhill KI, House CM, Katsis F, Witters LA, et al. Isoform-specific purification and substrate specificity of the 5′-AMP-activated protein kinase. J Biol Chem. 1996;271:28445–50. doi: 10.1074/jbc.271.45.28445. [DOI] [PubMed] [Google Scholar]
- 53.Stapleton D, Mitchelhill KI, Gao G, Widmer J, Michell BJ, Teh T, et al. Mammalian AMP-activated protein kinase subfamily. J Biol Chem. 1996;271:611–4. doi: 10.1074/jbc.271.2.611. [DOI] [PubMed] [Google Scholar]
- 54.Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, et al. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 1997;91:961–71. doi: 10.1016/S0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 55.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–24. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 57.Young AR, Chan EY, Hu XW, Köchl R, Crawshaw SG, High S, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006;119:3888–900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 58.Webber JL, Tooze SA. New insights into the function of Atg9. FEBS Lett. 2010;584:1319–26. doi: 10.1016/j.febslet.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 59.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010;5:e15394. doi: 10.1371/journal.pone.0015394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A. 2011;108:4788–93. doi: 10.1073/pnas.1100844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Furuya T, Kim M, Lipinski M, Li J, Kim D, Lu T, et al. Negative regulation of Vps34 by Cdk mediated phosphorylation. Mol Cell. 2010;38:500–11. doi: 10.1016/j.molcel.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prick T, Thumm M, Köhrer K, Häussinger D, Vom Dahl S. In yeast, loss of Hog1 leads to osmosensitivity of autophagy. Biochem J. 2006;394:153–61. doi: 10.1042/BJ20051243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.vom Dahl S, Dombrowski F, Schmitt M, Schliess F, Pfeifer U, Häussinger D. Cell hydration controls autophagosome formation in rat liver in a microtubule-dependent way downstream from p38MAPK activation. Biochem J. 2001;354:31–6. doi: 10.1042/0264-6021:3540031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–9. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 69.Wang Z, Wilson WA, Fujino MA, Roach PJ. Antagonistic controls of autophagy and glycogen accumulation by Snf1p, the yeast homolog of AMP-activated protein kinase, and the cyclin-dependent kinase Pho85p. Mol Cell Biol. 2001;21:5742–52. doi: 10.1128/MCB.21.17.5742-5752.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mok J, Kim PM, Lam HY, Piccirillo S, Zhou X, Jeschke GR, et al. Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci Signal. 2010;3:ra12. doi: 10.1126/scisignal.2000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng B, Cantley LC. Regulation of epithelial tight junction assembly and disassembly by AMP-activated protein kinase. Proc Natl Acad Sci U S A. 2007;104:819–22. doi: 10.1073/pnas.0610157104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang L, Li J, Young LH, Caplan MJ. AMP-activated protein kinase regulates the assembly of epithelial tight junctions. Proc Natl Acad Sci U S A. 2006;103:17272–7. doi: 10.1073/pnas.0608531103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nakano A, Kato H, Watanabe T, Min KD, Yamazaki S, Asano Y, et al. AMPK controls the speed of microtubule polymerization and directional cell migration through CLIP-170 phosphorylation. Nat Cell Biol. 2010;12:583–90. doi: 10.1038/ncb2060. [DOI] [PubMed] [Google Scholar]
- 74.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–75. doi: 10.1016/S0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 75.Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, Reggiori F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol. 2010;190:1005–22. doi: 10.1083/jcb.200912089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reggiori F, Shintani T, Nair U, Klionsky DJ. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy. 2005;1:101–9. doi: 10.4161/auto.1.2.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee JH, Koh H, Kim M, Kim Y, Lee SY, Karess RE, et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature. 2007;447:1017–20. doi: 10.1038/nature05828. [DOI] [PubMed] [Google Scholar]
- 78.Tang HW, Wang YB, Wang SL, Wu MH, Lin SY, Chen GC. Atg1-mediated myosin II activation regulates autophagosome formation during starvation-induced autophagy. EMBO J. 2011;30:636–51. doi: 10.1038/emboj.2010.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–67. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–7. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 81.Mari M, Reggiori F. Atg9 reservoirs, a new organelle of the yeast endomembrane system? Autophagy. 2010;6:1221–3. doi: 10.4161/auto.6.8.13792. [DOI] [PubMed] [Google Scholar]
- 82.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12:747–57. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tomoda T, Kim JH, Zhan C, Hatten ME. Role of Unc51.1 and its binding partners in CNS axon outgrowth. Genes Dev. 2004;18:541–58. doi: 10.1101/gad.1151204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–35. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jørgensen SB, Viollet B, Andreelli F, Frøsig C, Birk JB, Schjerling P, et al. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2004;279:1070–9. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- 86.Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, et al. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26:5336–47. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Viollet B, Andreelli F, Jørgensen SB, Perrin C, Geloen A, Flamez D, et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–8. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112:1493–502. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thomas SM, Soriano P, Imamoto A. Specific and redundant roles of Src and Fyn in organizing the cytoskeleton. Nature. 1995;376:267–71. doi: 10.1038/376267a0. [DOI] [PubMed] [Google Scholar]
- 90.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.