

Abstract
Adoptive cell therapy with tumor infiltrating lymphocytes (TILs) represents an effective treatment for patients with metastatic melanoma. However, most of the antigen targets recognized by effective melanoma reactive TILs remain elusive. In this study, patient 2369 experienced a complete response, including regressions of bulky liver tumor masses ongoing beyond seven years following adoptive TILs transfer. The screening of a cDNA library generated from the autologous melanoma cell line resulted in the isolation of a mutated PPP1R3B (protein phosphatase 1, regulatory (inhibitor) subunit 3B) gene product. The mutated PPP1R3B peptide represents the immunodominant epitope recognized by tumor reactive T cells in TIL 2369. Five years following adoptive transfer, peripheral blood T lymphocytes obtained from patient 2369 recognized the mutated PPP1R3B epitope. These results demonstrate that adoptive T cell therapy targeting a tumor-specific antigen can mediate long-term survival for a patient with metastatic melanoma. This study also provides an impetus to develop personalized immunotherapy targeting tumor-specific, mutated antigens.
Introduction
Patients with metastatic melanoma have a poor prognosis, as the five-year survival rate in this population is approximately 5% (1). Other than the conventional chemotherapy, the available treatments include interleukin-2 (IL-2), anti-cytotoxic T lymphocyte antigen 4 (CTLA-4) antibody ipilimumab, BRAF V600E inhibitor vemurafenib, and adoptive cell therapy. Among these treatments, adoptive cell therapy can be an effective salvage treatment, after patients have progressed after other therapies (2).
Adoptive cell therapy involves the transfer of autologous T cells with antitumor activity to the cancer-bearing patient. Tumor infiltrating lymphocytes (TILs) within surgically resected melanoma deposits can be grown to large numbers in culture medium containing IL-2, while retaining reactivity against autologous tumor. On three sequential clinical trials, patients were treated with the adoptive transfer of autologous TILs after ex vivo expansion in conjunction with high-dose IL-2 following a lymphodepleting preparative regimen (3). Adoptive TIL transfer mediated the objective regression of metastatic melanoma in up to 72% of patients, including the induction of up to 36% of complete durable responses ongoing beyond five years (4).
The results of studies have indicated that immunosuppressive factors present in the tumor microenvironment may restrain the in vivo activity of TILs (5, 6). The ex vivo culture of TILs with the stimulation of IL-2 can reverse this inhibitory state, resulting in their activation and clonal expansion. In spite of the strong anti-tumor activities of TILs ex vivo, the majority of patients receiving adoptive TIL transfer have not experienced durable regressions. The results from these clinical trials imply that some of the TILs from these patients may not recognize critical antigens, as the tumor may simply lose expression of antigens generated from non-essential genes and escape from immunosurveillance (7). Therefore, TILs targeting antigens derived from essential gene products may mediate long-term complete regression more effectively than TILs targeting antigens derived from non-essential gene products, such as MART-1 and gp100. However, most of the antigens recognized by adoptive transferred TILs that mediated long-term complete regressions remain elusive (8). To further examine this hypothesis, we identified the immunodominant target of a TIL product, which was administered to a patient with metastatic melanoma who experienced a durable complete regression without tumor recurrence.
Materials and Methods
Patient materials and cell lines
All patient materials were obtained in the course of a National Cancer Institute Institutional Review Board approved clinical trial. Patient 2369 was enrolled on a clinical trial (Trial registration ID: NCT00096382 at www.clinicaltrials.gov) that has been described in detail previously (9). The patient underwent a resection from which both a TIL line and a tumor cell line were established. TILs used for this study were generated by methods described previously (10). Briefly, tumor fragments were excised and cultured in media containing IL-2. TIL cultures that expanded were screened for recognition of autologous or HLA-matched tumor, and reactive TILs were expanded using a rapid expansion protocol (REP) with IL-2, anti-CD3 antibody and irradiated feeder cells to large numbers for patient infusion (11). A small portion of TILs underwent a second REP for the experiments shown in this report. For co-culture assays, T cells and tumor cells were cultured at 1:1 ratio in a 96-well plate with 200 μL medium (AIM-V medium supplemented with 5% human serum) for 16 hr. In antibody blocking experiments, melanoma cells were pre-incubated with HLA-B,C (B1.23.1) or HLA-A,B,C (W6/32) blocking antibodies (40 μg/mL) for 3 hr, followed by co-cultured with T cells. The concentration of IFN-γ in the supernatant was determined by ELISA (Thermo Scientific).
cDNA library construction, PCR amplification and TCR analysis
A cDNA library was generated from Mel 2369 mRNA using the SMARTer RACE cDNA Amplification Kit (Clontech), and the cDNA library was cloned into pCMV6 vector using the In-Fusion Advantage PCR Cloning Kit (Clontech), according to the manufacture's instruction. The following primer sets were used for PCR amplification: 5'-CAT GAT GGC TGT GGA CAT CGA GTA C-3' and 5'- GTG AGC AGA GCT AGG CTT GTC TGT G-3' for the amplification of PPP1R3B ORF in Exon 2 from genomic DNA. 5'- TGC CCT CGG GAC TTA TGA GCT GAA C-3' and 5'- GGC TTC CGA ACT GGT CAA AGG ATA T-3' for the amplification of PPP1R3B transcription variant 1 from melanoma cDNA. 5'- GCG GCC CAA AAG CCT GTT CAT CTA G-3' and 5'- GGC TTC CGA ACT GGT CAA AGG ATA T-3' for the amplification of PPP1R3B transcription variant 2 from melanoma cDNA. For PPP1R3B transcription variant 1 quantitative PCR, the following primer set and probe were used: 5'-CCT CGG GAC TTA TGA GCT GAA-3', 5'-GAG CCA TGC AGT TGT ATC TGT ACT C-3', probe 5'- ATC TAG CCC CAT GAT GGC TGT GGA CAT-3'. For PPP1R3B transcription variant 2 quantitative PCR, the following primer set and probe were used: 5'- CGG CCC AAA AGC CTG TT-3', 5'-GAG CCA TGC AGT TGT ATC TGT ACT C-3', probe 5'- ATC TAG CCC CAT GAT GGC TGT GGA CAT-3'. The method for T cell receptor (TCR) clonotype analysis has been described previously (12).
Transfection of tumor cell lines
For siRNA knockdown assays, melanoma cells were transfected with siRNA and Lipofectamine RNAiMAX (Life Technologies) for 48 hr, according to the manufacturer's instruction. ON-TARGETplus siRNAs and non-targeting siRNA control were purchased from Dharmacon (Lafayette, CO). The following siRNA were used: HLA-A1 (CUG UGU UCG UGU AGG CAU A). PPP1R3B (1) (GCA GAU UAC UUA GAC UUU A). PPP1R3B (2) (GGC AAG AAC UAU AGG AUC A). PPP1R3B (3) (GGA CAC UUA UGC CGG UUC A). Expression plasmids were introduced into melanoma cells using the Neon transfection system (Life Technologies), according to the manufacturer's instruction.
Flow cytometric analyses
Analysis of intracellular IFN-γ staining was carried out by pulsing 293-A1 cells with 10 μM peptide for 2 hr, followed by a co-culture with T cells for 4 hr in the presence of GolgiPlug (BD Biosciences, San Jose, CA). Cells were then fixed, permeabilized and stained, according to the manufacturer's instructions (eBioscience Inc., San Diego, CA). The following antibodies were used for flow cytometry analysis: Anti-IFN-γ antibody (BD Biosciences, San Jose, CA), Anti-TCR Vβ antibodies (Beckman Coulter, Inc., Brea, CA), Anti-HLA-A1, A26 antibody (One Lambda, Canoga Park, CA).
Chromium-release assays
To measure cytotoxic T lymphocytes killing activity, a standard method was used as described previously (13). Briefly, target cells were pulsed with 51Cr solution (0.1 mCi/mL) with or without peptides (10 μM) for 2 hr. After washing, target cells were co-cultured with effector cells at various ratios for 4 hr, followed by the measurement of 51Cr release.
HLA binding assays
A standard protocol was followed to measure the binding affinity of short peptides to HLA molecules (14). Briefly, autologous EBV-transformed B cells were treated with acid to remove HLA-bound peptides, followed by an incubation for 24 hr at 4°C with a fluorescent reference peptide Fl-A1 , together with various concentrations of test peptides. The cells were then subjected to flow cytometric analysis and the percentage of inhibition of fluorescent peptide was calculated. The following peptides were synthesized from a commercial source: Fl-A1 [YLEPAC(Fl)AKY], A*01 consensus sequence Ctrl-A1 (YLEPAIAKY) (IC50 = 200 nM), PPP1R3B 172wt (YTDFPCQYVK), and PPP1R3B 172mut (YTDFHCQYVK) (Atlantic Peptides, ME).
Evaluation of peptide binding was also carried out using the REVEAL MHC binding assay, performed by ProImmune (Oxford, United Kingdom). Briefly, the assay measures the relative affinity of binding between a peptide to an HLA molecule by its ability to stabilize a peptide-MHC complex. Renatured peptide-MHC complexes were detected using an anti-MHC antibody that binds to native MHC molecules.
Results
Patient 2369 with metastatic melanoma experienced a long-term complete regression following adoptive cell therapy
Patient 2369 had metastatic melanoma with a primary lesion in the neck, and metastatic lesions in the liver, brain and periportal lymph node. In September 2004, he underwent a left lateral segmentectomy of the liver (segment 2 and 3), and TILs were grown from the liver metastasis (Fig. 1A). He received high-dose IL-2 therapy but developed a new brain metastasis which was resected. In addition, the largest lesion, present in segment 4 of the liver also continued to progress up until July 2005 (Fig. 1B), when the patient was treated with adoptive cell transfer with TILs plus high dose IL-2 following a lymphodepleting regimen. The patient then went on to experience a complete regression of this lesion, and a stable periportal lymph node that was resected 7 months after the adoptive cell therapy contained no viable tumor on pathological examination. This patient was declared a complete responder and remains free of disease seven years following the treatment. None of the CT and MRI scans showed abnormalities consistent with melanoma in the last five years (Fig. 1C).
FIGURE 1.

Response of patient 2369 after adoptive cell therapy. Computed tomography (CT) and magnetic resonance imaging (MRI) scans showed the disease status after the hepatic resection for TIL harvest (A), prior to the treatment (B) and seven years after the adoptive cell therapy (C). White arrows indicate the lesions.
TIL 2369 T cells recognize HLA-A1-restricted mutated PPP1R3B gene product
In order to obtain sufficient cells for experiments in this study, the infused TIL 2369 T cells were further expanded in vitro using a REP. To examine the TCR repertoire before and after the REP, TCR clonotype analysis was performed with deep sequencing. As shown in Supplemental Table 1, the majority of TCR clonotypes, including the most highly represented BV27 clonotype (Vβ14), were comparable in the original and in vitro expanded TIL samples. Experiments were then carried out to identify the predominant antigen target recognized by TIL 2369. The HLA loci of patient 2369 are A*01, A*26, B*07, B*14, Cw07, Cw08. The ability of autologous Mel 2369 tumor cells to stimulate IFN-γ release from TIL 2369 T cells was not inhibited by incubation with a blocking antibody that binds to all HLA-B and C loci, indicating that TIL 2369 predominantly recognized the autologous melanoma in the context of HLA-A*01 or A*26 (Fig. 2A). Mel 2369 cells transfected with an siRNA targeting the HLA-A*01 3'-untranslated region (3'-UTR) stimulated lower levels of IFN-γ release from TIL 2369 T cells than the cells transfected with a non-targeting siRNA (Fig. 2B and 2C). In addition, transfection of tumor cells with an HLA-A*01 cDNA lacking the natural 3'-UTR interfered with the ability of the HLA-A*01-specific siRNA to inhibit the recognition of the Mel 2369 by autologous TIL (Fig. 2B and 2C). These results indicate that TIL 2369 predominantly recognized the autologous tumor cells in the context of HLA-A*01.
FIGURE 2.

Identification of dominant HLA-restriction element of TIL 2369 T cells. (A) Mel 2369 cells were pre-incubated with HLA-B,C (B1.23.1) or HLA-A,B,C (W6/32) blocking antibodies for 3 hr, followed by co-cultured with TIL 2369 T cells, Mage-A3 TCR-transduced or Mage-A12 TCR-transduced T cells overnight. (B) Mel 2369 cells were transfected with HLA-A*01 siRNA and cDNA, and the expression of HLA-A1 and A26 was determined by flow cytometry (NT, non-targeting). (C) Mel 2369 cells were transfected with HLA-A*01 siRNA and cDNA, and then co-cultured with autologous TIL 2369 T cells. The secretion of IFN-γ by T cells was determined by ELISA.
TIL 2369 T cells do not recognize any shared melanoma antigens (Supplemental Fig. 1). Identification of the antigen recognized by TIL 2369 T cells was carried out by the transient transfection of HLA-A*01-expressing HEK293 cells (293-A1 cells) with pools of 50 cDNA clones generated from the autologous melanoma 2369. These transfected cells were co-cultured with TIL 2369 T cells overnight and the secretion of IFN-γ was detected by ELISA (Fig. 3A). After screening 1000 cDNA library pools, a single positive pool was identified and confirmed. All the positive clones isolated from this pool corresponded to the transcript of PPP1R3B gene. According to the GenBank database, alternate splicing of PPP1R3B results in two transcript variants, which encode the same protein. The sequence of the PPP1R3B coding region was identical to sequences in GenBank database, with a single C to A transversion at 527 b.p., which resulted in a substitution of histidine for proline at position 176 of the wild-type (WT) PPP1R3B protein (Fig. 3B). The Exon 2 of PPP1R3B, which encodes the entire protein, was amplified from genomic DNA isolated from both the Mel 2369 and the peripheral blood mononuclear cells (PBMC) from patient 2369 (Fig. 3C). Genomic DNA and cDNA amplified from Mel 2369 cells contained both the mutated and WT nucleotide at position 527 in the PPP1R3B coding region, whereas DNA isolated from the PBMC of patient 2369 corresponded to the WT sequence, indicating that this represented a somatic mutation in Mel 2369 cells. In addition, the PPP1R3B coding region from both transcript variants was also amplified from Mel 2369 cDNA. The results indicate that both transcript variants contained both the mutated and WT nucleotide at position 527 (Fig. 3D).
FIGURE 3.

Identification of mutated PPP1R3B as the potential antigen. (A) cDNA library screening of the potential antigen. A single colony containing a mutated PPP1R3B cDNA fragment was isolated from a pool of 50 colonies. (B) Gene structure of PPP1R3B. Both transcripts encode the same protein. (C) DNA sequence chromatogram results for PPP1R3B genomic DNA obtained from autologous PBMC and Mel 2369 cells. (D) DNA sequence chromatogram results for PPP1R3B cDNA obtained from autologous Mel 2369 cells.
Exomic sequence analysis of DNA isolated from 14 additional melanoma cell lines revealed that while this residue was not mutated in other melanoma cells, one additional melanoma cell line contained a non-synonymous mutation in the PPP1R3B gene (15). The DNA from Mel 2167 cell line contained a C to T transition resulting in a substitution of Phe for the Ser residue at amino acid position 16 of the WT PPP1R3B protein.
Protein phosphatase 1 (PP1) represents a eukaryotic protein serine/threonine phosphatase that is comprised of one catalytic subunit (PP1c) and one of the over fifty regulatory subunits. The regulatory subunits, which include the PPP1R3B, define the substrate specificity, subcellular location and functional diversity of the PP1 complex (16). Previous studies have demonstrated that PP1c, together with the regulatory subunit PPP1R3 family, plays an important role in glycogen metabolism (16). For instance, the expression of PPP1R3B in liver cells is up-regulated in response to insulin stimulation. The newly formed protein phosphatase 1 protein complex (PP1c-PPP1R3B) dephosphorylates glycogen synthase. As a result, the activity of glycogen synthase is increased through allosteric regulation (17–19). PPP1R3B transcription variant 1 was highly expressed in human liver, consistent with the previous reports (Supplemental Fig. 2A) (18, 20). However, PPP1R3B transcript variant 2 was expressed at high levels in a variety of human tissues, including placenta, leukocytes, prostate and spleen (Supplemental Fig. 2A), which have not been reported previously. Both transcript variants of PPP1R3B were also expressed at a high level in various melanoma cell lines (Supplemental Fig. 2B).
The relationship between glycogen metabolism and carcinogenesis remains largely unclear. Studies showed that the glycogen level was higher in tumor comparing to the adjacent or normal tissue, and the glycogen level was negatively correlated with the tumor cell proliferation rate (21–23). Recent studies also demonstrated that hypoxia could induce the accumulation of glycogen, but the detailed molecular mechanism is still unknown (24, 25). In this study, the mutation of PPP1R3B found in Mel 2369 (Fig. 3B) was located within the proposed glycogen binding domain of PPP1R3B protein (26). Even though the non-synonymous PPP1R3B mutation was rare in melanoma, many non-synonymous mutations have been identified among PPP1R3 family. These findings imply that members of the PPP1R3 gene family may play a role in carcinogenesis (15, 27, 28).
To further confirm the recognition of PPP1R3B by TIL 2369 T cells, transfection of Mel 2369 cells with PPP1R3B siRNAs reduced the transcription of both PPP1R3B transcript variants (Fig. 4A). Transfection of Mel 2369 cells with PPP1R3B siRNAs significantly inhibited the ability to stimulate IFN-γ release from TIL 2369, but did not alter the responses of control HLA-A*01-restricted MAGE-A3-specific T cells (Fig. 4B). In addition, COS-7 cells were transfected with either WT or mutated PPP1R3B cDNA, together with HLA-A*01, HLA-A*02 or HLA-A*26 cDNA. TIL 2369 T cells only recognized cells with the mutated but not WT PPP1R3B gene product, in an HLA-A*01 restriction manner (Fig. 4C). Taken together, these results indicated that a mutated product of the PPP1R3B gene represented an immunodominant target of TIL 2369 T cells.
FIGURE 4.

TIL 2369 T cells recognize mutated, but not wild-type PPP1R3B gene product. (A) PPP1R3B mRNA expression in melanoma cell lines after the transfection of Mel 2369 cells with PPP1R3B siRNAs. The copy numbers of PPP1R3B transcript variant 1 and variant 2 from Mel 2369 cells after siRNA transfection were determined by quantitative PCR (NT, non-targeting). (B) Mel 2369 cells were transfected with PPP1R3B siRNAs, and then co-cultured with TIL 2369 T cells or HLA-A*01-restricted Mage-A3 TCR-transduced T cells. (C) COS-7 cells were transfected with HLA cDNA constructs, together with wild-type or mutated PPP1R3B cDNA construct. These transfected cells were co-culture with TIL 2369 T cells overnight. The secretion of IFN-γ was determined by ELISA.
The antigenic region of mutated PPP1R3B was determined by transfection of 293-A1 cells with constructs encoding truncations of the carboxy terminus of the mutated PPP1R3B protein (Fig. 5A), followed by the co-culture with TIL 2369 T cells. The result indicates that the T cell epitope mapped to the region between amino acids 175 and 186 (cDNA 525 ~ 558 b.p.), which included the mutated amino acid (176 a.a.) of PPP1R3B (Fig. 5B). An HLA peptide binding prediction program was used to identify candidate HLA-A*01-binding peptides encompassed by residues 175 to 186, which were then synthesized and tested for their ability to be recognized by TIL 2369 (Fig. 5C) (29). 293-A1 cells were pulsed with this set of peptides and then co-cultured with TIL 2369 T cells. The results demonstrate that cells pulsed with a decamer corresponding to residues 172–181 (172mut) stimulated the release of high levels of IFN-γ from TIL 2369 T cells, and the peptide was recognized at a minimum concentration of 0.1 μM. In contrast, the corresponding WT peptide did not induce significant IFN-γ release at a concentration as high as 10 μM (Fig. 5D). Consistent with IFN-γ ELISA results, TIL 2369 T cells specifically lysed Mel 2369 cells, as well as 293-A1 cells that were pulsed with 172mut peptide (Fig. 5E).
FIGURE 5.

Identification of the PPP1R3B epitope. (A) Constructs encoding the truncations of the carboxy terminus of the mutated PPP1R3B (ORF, open reading frame). (B) 293-A1 cells were transfected with these constructs, followed by co-cultured with TIL 2369 T cells. The secretion of IFN-γ was determined by ELISA. (C) Multiple peptides covered the region of 175 ~ 186 a.a. were synthesized. (D) 293-A1 cells were pulsed with these peptides, followed by co-cultured with TIL 2369 T cells. (E) Target cells, including melanoma cells or 293-A1 cells pulsed with 172wt or 172mut peptide, were incubated with TIL 2369 T cells in a 51Cr-release assay. HLA-A*01+ Mel 2556 cells lack the PPP1R3B mutation.
Assays were then carried out to evaluate the relative HLA-A*01 binding affinities of 172wt and 172mut peptides. In a competition-based cellular peptide binding assay for HLA-A*01, both 172wt and 172mut inhibited binding of the fluorescent peptides at a 2~3-fold lower concentration than the un-labeled control peptide (Supplementary Fig. 3A). Additionally, the results of a REVEAL MHC-peptide binding assay suggest that both 172wt and 172mut are good binders (Supplementary Fig. 3B). These results demonstrate that both of the PPP1R3B 172wt and 172mut peptides represent relatively strong binders that have similar affinities for HLA-A*01. These results indicate that the mutated residue at position 5 in the 172mut peptide does not play a major role in MHC binding, since the anchor residues at positions two and three as well as the carboxy terminus were not altered.
The mutated PPP1R3B is the immunodominant epitope recognized by TIL 2369 T cells
The antigen reactivity of TIL 2369 T cells was then further evaluated by intracellular IFN-γ staining after co-culture with the target cells. As shown in Fig. 6A, approximately half of the TIL 2369 T cells up-regulated IFN-γ in response to Mel 2369 cells. This result indicates that around half of the TIL 2369 T cells recognized Mel 2369 cells, but the rest of the T cells either were inactive or did not recognized autologous melanoma cells. A similar percents of TIL 2369 T cells recognized 293-A1 cells pulsed with PPP1R3B 172mut peptide, suggesting that 172mut is the immunodominant epitope recognized by TIL 2369. Furthermore, the majority of the IFN-γ+ cells also expressed Vβ14 (Fig. 6B). Cells expressing Vβ14 beta chain variable region represented the dominant T cell population present in TIL 2369, based on the antibody staining and TCR clonotype analysis (Supplemental Table 1). The cDNA encoding the TCR from this dominant T cell population was isolated. Activated T cells obtained from the peripheral blood of healthy donors were transduced with a recombinant retroviral construct encoding this dominant TCR. These T cells recognized Mel 2369 cells, as well as 293-A1 cells pulsed with PPP1R3B 172mut peptide but not the corresponding WT peptide (Fig. 6C). In addition, TCR-transduced T cells could kill Mel 2369 cells but not Mel 2556 cells, an HLA-A*01+ melanoma that lacks the PPP1R3B mutation (Fig. 6D). Taken together, these results suggest that the majority of autologous tumor-reactive T cells in TIL 2369 recognize the mutated PPP1R3B antigen.
FIGURE 6.

Mutated PPP1R3B-reactive T cells are responsible for tumor recognition. Intracellular IFN-γ staining (A) and TCR Vβ14 co-staining (B) of TIL 2369 T cells after co-culture with autologous Mel 2369 cells or 293-A1 cells pulsed with PPP1R3B 172wt or 172mut peptide. (C) Healthy donor T cells were transduced with mut PPP1R3B TCR. These T cells were co-cultured with Mel 2369, Mel 2556 (HLA-A*01+) cells or 293-A1 cells pulsed with PPP1R3B 172wt or 172mut peptide. (D) In a 51Cr-release assay, health donor T cells transduced with mut PPP1R3B TCR or un-transduced T cells were incubated with autologous Mel 2369 cells or Mel 2556 cells.
Immune surveillance plays a significant role in host defense against pathogens, and may also play a role in limiting or preventing tumor recurrence. To examine the T cell reactivity against mutated PPP1R3B epitope in the long-term clinical response observed in patient 2369, PBMCs obtained from this patient following adoptive transfer were analyzed for their ability to secrete IFN-γ after the stimulation with PPP1R3B 172mut peptide. As shown in Fig. 7, the PBMCs obtained more than five years following treatment generated significant levels of IFN-γ in response to the mutant but not the WT PPP1R3B peptide. This result provides the evidence for long-term persistence of adoptively transferred T cells against this mutated epitope.
FIGURE 7.
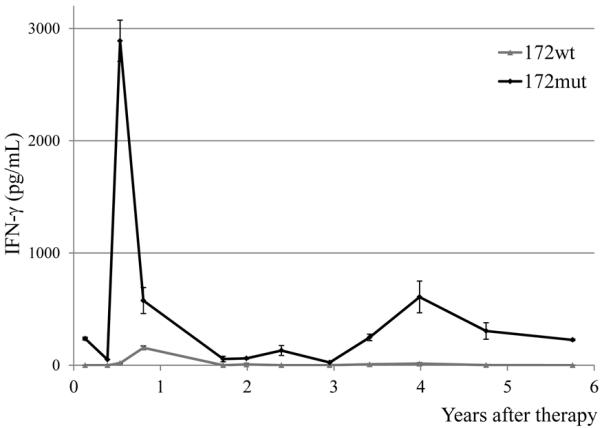
The long-term persistence of mutated PPP1R3B-reactive T cells. Patient 2369 PBMC samples were thawed and incubated overnight in medium with IL-2, followed by stimulation with PPP1R3B 172wt or 172mut peptides. The secretion of IFN-γ was determined by ELISA after overnight culture.
Discussion
In this study, we identified a dominant T cell population, which is responsible for the majority of Mel 2369-reactive T cells within TIL 2369. Importantly, TIL 2369 recognized mutated PPP1R3B antigen, but not the wild-type counterpart. This is consistent with the observation that adoptive cell therapy using this TIL product did not induce severe toxicity in patient 2369. One potential explanation is that only the mutated epitope, but not the wild-type one, can bind to the MHC molecules. However, this is only true when the point mutations occur at the anchor residues of the epitopes, which can dramatically change the binding affinity of the epitopes (30). Instead, the PPP1R3B mutation in Mel 2369 does not take place at the anchor residues, and both the PPP1R3B 172wt and 172mut peptides are good binders. Alternatively, the amino acids altered by mutation may influence antigen processing, including proteasomal cleavage and TAP-mediated transport. Again, that is generally true only for the C-terminus of the epitope, but not likely to affect antigen processing if it is in the middle of the epitope. Future studies to detect naturally presented peptides on MHC molecules from tumor cell surface might be able to address this possibility. More likely, the mutated amino acid in PPP1R3B represents a TCR contact residue, and the specificity of TCR may contribute to the recognition of this neo-antigen. Negative selection in the thymus presumably resulted in deletion of T cells that recognize the wild type PPP1R3B peptide.
It has been proposed that the immune system controls tumor growth and shapes tumor immunogenicity, a process named cancer immunoediting, which has been proposed to comprise three phases: elimination, equilibrium and escape (7). Recent murine studies illustrated that mutated as well as foreign antigens present in tumors isolated from immunodeficient mice are responsible for the rejection of these tumors when transplanted into immunocompetent mice in the “elimination phase” (30, 31). Subsequently, rare variants of tumors that can outgrow in immunocompetent mice generally have lost or down-regulated expression of these dominant tumor rejection antigens in the “escape phase”. While the immunoediting process has primarily been demonstrated in murine models, human studies also suggest that immunotherapy can lead to a loss of HLA or antigen expression on tumors that recur following initial regression, indicating that immunoediting may result from effective in vivo anti-tumor immune responses (32, 33).
In contrast to these observations, the tumor in patient 2369 failed to recur following initial regression. In addition, T cells recognizing mutated PPP1R3B epitope persist beyond five years. To explain the disappearance of the tumor and the long-term persistence of these T cells, one possibility is that low levels of tumor cells persist for several years following treatment but fail to be detected in the patient. These cells continue to express the mutated PPP1R3B antigen, which is subsequently recognized by T cells. Therefore, the tumor and immune system interact and stay in the “equilibrium phase”. The alternative hypothesis is that T cells which have killed the last tumor cell do not encounter this mutated antigen for several years but are maintained through antigen-independent mechanisms. However, it has not been clearly established that whether the maintenance of T cell memory is necessarily dependent on constitutive stimulation by cognate antigen (34, 35). Understanding the detailed mechanisms of how tumor and immune system interact with each other in human will be an interest topic to study in future.
A recent study demonstrated that melanoma cells stimulated with TNF-α were poorly recognized by T cells specific for melanocyte differentiation antigens due to the inflammation-induced reversible de-differentiation (36). This is consistent with our hypothesis in this study that adoptive cell therapy targeting non-essential, melanocyte differentiation antigens is likely less effective than targeting essential, tumor-specific antigens. The results presented here provide the most direct evidence to date that targeting a tumor-specific, mutated antigen in an adoptive cell therapy could mediate a complete, durable regression of a human cancer without recurrence seven years following treatment. Mutated gene products, especially gene products that play essential roles in carcinogenesis, may represent particularly potent immunotherapy targets. The development of techniques that facilitate the identification of mutated tumor antigens may allow more effective tumor targeting and avoid the autoimmune toxicity observed in previous adoptive therapy protocols targeting tumor-associated antigens which are also expressed in normal tissues (37).
Supplementary Material
Acknowledgments
We are grateful to J.S. Crystal, M. Garcia, C. Gross, K. Hanada, Q. J. Wang, J. R. Wunderlich, Z. Zheng and S. Zhu for technical support and suggestions.
Grant Support: This research was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health.
References
- 1.Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S, Eggermont AM, Flaherty KT, Gimotty PA, Kirkwood JM, McMasters KM, Mihm MC, Jr., Morton DL, Ross MI, Sober AJ, Sondak VK. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med. 2012;4:127–128. doi: 10.1126/scitranslmed.3003634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, Morton KE, Laurencot CM, Steinberg SM, White DE, Dudley ME. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, Wieckowski S, Bouzourene H, Deplancke B, Romero P, Rufer N, Speiser DE. Exhaustion of tumor-specific CD8 T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 8.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, Wunderlich J, Restifo NP, Thomasian A, Downey SG, Smith FO, Klapper J, Morton K, Laurencot C, White DE, Rosenberg SA. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26:332–342. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riddell SR, Watanabe KS, Goodrich JM, Li CR, Agha ME, Greenberg PD. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science. 1992;257:238–241. doi: 10.1126/science.1352912. [DOI] [PubMed] [Google Scholar]
- 12.Price DA, West SM, Betts MR, Ruff LE, Brenchley JM, Ambrozak DR, Edghill-Smith Y, Kuroda MJ, Bogdan D, Kunstman K, Letvin NL, Franchini G, Wolinsky SM, Koup RA, Douek DC. T cell receptor recognition motifs govern immune escape patterns in acute SIV infection. Immunity. 2004;21:793–803. doi: 10.1016/j.immuni.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 13.Wonderlich J, Shearer G, Livingstone A, Brooks A. Induction and measurement of cytotoxic T lymphocyte activity. Curr Protoc Immunol. 2006;Chapter 3(Unit 3):11. doi: 10.1002/0471142735.im0311s72. [DOI] [PubMed] [Google Scholar]
- 14.Kessler JH, Benckhuijsen WE, Mutis T, Melief CJ, van der Burg SH, Drijfhout JW. Competition-based cellular peptide binding assay for HLA class I. Curr Protoc Immunol. 2004;Chapter 18(Unit 18):12. doi: 10.1002/0471142735.im1812s61. [DOI] [PubMed] [Google Scholar]
- 15.Wei X, Walia V, Lin JC, Teer JK, Prickett TD, Gartner J, Davis S, Stemke-Hale K, Davies MA, Gershenwald JE, Robinson W, Robinson S, Rosenberg SA, Samuels Y. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet. 2011;43:442–446. doi: 10.1038/ng.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen PT. Protein phosphatase 1--targeted in many directions. J Cell Sci. 2002;115:241–256. doi: 10.1242/jcs.115.2.241. [DOI] [PubMed] [Google Scholar]
- 17.Doherty MJ, Cadefau J, Stalmans W, Bollen M, Cohen PT. Loss of the hepatic glycogen-binding subunit (GL) of protein phosphatase 1 underlies deficient glycogen synthesis in insulin-dependent diabetic rats and in adrenalectomized starved rats. Biochem J. 1998;333(Pt 2):253–257. doi: 10.1042/bj3330253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doherty MJ, Moorhead G, Morrice N, Cohen P, Cohen PT. Amino acid sequence and expression of the hepatic glycogen-binding (GL)-subunit of protein phosphatase-1. FEBS Lett. 1995;375:294–298. doi: 10.1016/0014-5793(95)01184-g. [DOI] [PubMed] [Google Scholar]
- 19.Gasa R, Jensen PB, Berman HK, Brady MJ, DePaoli-Roach AA, Newgard CB. Distinctive regulatory and metabolic properties of glycogen-targeting subunits of protein phosphatase-1 (PTG, GL, GM/RGl) expressed in hepatocytes. J Biol Chem. 2000;275:26396–26403. doi: 10.1074/jbc.M002427200. [DOI] [PubMed] [Google Scholar]
- 20.Munro S, Cuthbertson DJ, Cunningham J, Sales M, Cohen PT. Human skeletal muscle expresses a glycogen-targeting subunit of PP1 that is identical to the insulin-sensitive glycogen-targeting subunit G(L) of liver. Diabetes. 2002;51:591–598. doi: 10.2337/diabetes.51.3.591. [DOI] [PubMed] [Google Scholar]
- 21.Rousset M, Zweibaum A, Fogh J. Presence of glycogen and growth-related variations in 58 cultured human tumor cell lines of various tissue origins. Cancer Res. 1981;41:1165–1170. [PubMed] [Google Scholar]
- 22.Namiot Z, Stasiewicz J, Szalaj W, Skwarski L, Jodczyk J, Gorski J. Gastric cancer with special references to WHO and Lauren's classifications: glycogen and triacylglycerol concentrations in the tumor. Neoplasma. 1989;36:363–368. [PubMed] [Google Scholar]
- 23.Takahashi S, Satomi A, Yano K, Kawase H, Tanimizu T, Tuji Y, Murakami S, Hirayama R. Estimation of glycogen levels in human colorectal cancer tissue: relationship with cell cycle and tumor outgrowth. J Gastroenterol. 1999;34:474–480. doi: 10.1007/s005350050299. [DOI] [PubMed] [Google Scholar]
- 24.Pescador N, Villar D, Cifuentes D, Garcia-Rocha M, Ortiz-Barahona A, Vazquez S, Ordonez A, Cuevas Y, Saez-Morales D, Garcia-Bermejo ML, Landazuri MO, Guinovart J, del Peso L. Hypoxia promotes glycogen accumulation through hypoxia inducible factor (HIF)-mediated induction of glycogen synthase 1. PLoS One. 2010;5:e9644. doi: 10.1371/journal.pone.0009644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen GM, Zhang FL, Liu XL, Zhang JW. Hypoxia-inducible factor 1-mediated regulation of PPP1R3C promotes glycogen accumulation in human MCF-7 cells under hypoxia. FEBS Lett. 2010;584:4366–4372. doi: 10.1016/j.febslet.2010.09.040. [DOI] [PubMed] [Google Scholar]
- 26.Armstrong CG, Doherty MJ, Cohen PT. Identification of the separate domains in the hepatic glycogen-targeting subunit of protein phosphatase 1 that interact with phosphorylase a, glycogen and protein phosphatase 1. Biochem J. 1998;336(Pt 3):699–704. doi: 10.1042/bj3360699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E, Ghosh P, Zhang H, Zeid R, Ren X, Cibulskis K, Sivachenko AY, Wagle N, Sucker A, Sougnez C, Onofrio R, Ambrogio L, Auclair D, Fennell T, Carter SL, Drier Y, Stojanov P, Singer MA, Voet D, Jing R, Saksena G, Barretina J, Ramos AH, Pugh TJ, Stransky N, Parkin M, Winckler W, Mahan S, Ardlie K, Baldwin J, Wargo J, Schadendorf D, Meyerson M, Gabriel SB, Golub TR, Wagner SN, Lander ES, Getz G, Chin L, Garraway LA. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–506. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, Saksena G, Stransky N, Onofrio RC, Winckler W, Ardlie K, Wagle N, Wargo J, Chong K, Morton DL, Stemke-Hale K, Chen G, Noble M, Meyerson M, Ladbury JE, Davies MA, Gershenwald JE, Wagner SN, Hoon DS, Schadendorf D, Lander ES, Gabriel SB, Getz G, Garraway LA, Chin L. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994;152:163–175. [PubMed] [Google Scholar]
- 30.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, Hundal J, Wendl MC, Demeter R, Wylie T, Allison JP, Smyth MJ, Old LJ, Mardis ER, Schreiber RD. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–404. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482:405–409. doi: 10.1038/nature10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenberg SA, Yang JC, Robbins PF, Wunderlich JR, Hwu P, Sherry RM, Schwartzentruber DJ, Topalian SL, Restifo NP, Filie A, Chang R, Dudley ME. Cell transfer therapy for cancer: lessons from sequential treatments of a patient with metastatic melanoma. J Immunother. 2003;26:385–393. doi: 10.1097/00002371-200309000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khong HT, Wang QJ, Rosenberg SA. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. J Immunother. 2004;27:184–190. doi: 10.1097/00002371-200405000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Antia R, Ganusov VV, Ahmed R. The role of models in understanding CD8+ T-cell memory. Nat Rev Immunol. 2005;5:101–111. doi: 10.1038/nri1550. [DOI] [PubMed] [Google Scholar]
- 35.Jameson SC, Masopust D. Diversity in T cell memory: an embarrassment of riches. Immunity. 2009;31:859–871. doi: 10.1016/j.immuni.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, Fatho M, Lennerz V, Wolfel T, Holzel M, Tuting T. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature. 2012;490:412–416. doi: 10.1038/nature11538. [DOI] [PubMed] [Google Scholar]
- 37.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee CC, Restifo NP, Schwarz SL, Cogdill AP, Bishop RJ, Kim H, Brewer CC, Rudy SF, VanWaes C, Davis JL, Mathur A, Ripley RT, Nathan DA, Laurencot CM, Rosenberg SA. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.