

Abstract
First discovered as a structure-specific endonuclease that evolved to cut at the base of single-stranded flaps, flap endonuclease (FEN1) is now recognized as a central component of cellular DNA metabolism. Substrate specificity allows FEN1 to process intermediates of Okazaki fragment maturation, long-patch base excision repair, telomere maintenance, and stalled replication fork rescue. For Okazaki fragments, the RNA primer is displaced into a 5′ flap and then cleaved off. FEN1 binds to the flap base and then threads the 5′ end of the flap through its helical arch and active site to create a configuration for cleavage. The threading requirement prevents this active nuclease from cutting the single-stranded template between Okazaki fragments. FEN1 efficiency and specificity are critical to the maintenance of genome fidelity. Overall, recent advances in our knowledge of FEN1 suggest that it was an ancient protein that has been fine-tuned over eons to coordinate many essential DNA transactions.
Keywords: endonucleases, genome fidelity, genome stability, long-patch base excision repair, Okazaki fragment maturation
INTRODUCTION
Flap endonucleases are key components in the DNA transactions of cells in organisms from bacteria to humans. A fitting tribute to these nucleases is that the first Nobel Prize in DNA replication, awarded to Kornberg and Ochoa for characterization of Escherichia coli DNA polymerase I, also honored the discoverers of flap endonuclease 1 (FEN1). In this review, we attempt to summarize the new developments in the vast FEN1 literature since our last review (1) and to bring into focus why these enzymes are so vital during various cellular DNA transactions.
All cells have double-stranded DNA (dsDNA) genomes that need accurate replication and repair. DNA replication begins at sites on the genome acting as origins and proceeds bidirectionally by the creation of replication forks. As the antiparallel strands of the DNA separate for duplication, one strand is copied in the direction of the fork opening to synthesize the leading strand. The other strand is copied in the opposite direction and so is made as RNA-primed discontinuous segments, or Okazaki fragments, that later join to form the lagging strand. In E. coli and other prokaryotes, the fork moves more than 1,000 nucleotides (nt) per second. The RNA primers must be removed from each 1,200-nt Okazaki fragment, and the fragments must join to keep up this pace. In eukaryotes, the rate of synthesis is slower, but the Okazaki fragments are only 150–200 nts long, with sizes determined by nucleosome spacing (2). This means that each time the human genome is replicated, 10–20 million fragments are processed.
FEN1 NUCLEASE IS CENTRAL TO ACCURATE EUKARYOTIC OKAZAKI FRAGMENT MATURATION
Initial reconstitution of eukaryotic DNA replication using Simian virus 40 as a model system revealed the many steps involved (3). Replication on both the leading and lagging strands is initiated by DNA polymerase α/primase (Pol α), which synthesizes an RNA primer approximately 12 nts long that is further extended with approximately 20 nts of DNA. This initiator primer is made at low fidelity because Pol α lacks a proofreading 3′ nuclease. DNA polymerases ε (Pol ε)and δ (Pol δ) then take over the synthesis of the leading and the lagging strand, respectively. This polymerase switch is coordinated by ATP-dependent replication factor C, the clamp loader, which attaches proliferating cell nuclear antigen (PCNA), the processivity clamp, and either Pol ε or Pol δ. On the lagging strand, Pol δ elongates the initiator primer and closes the gap between two Okazaki fragments. On encountering the downstream Okazaki fragment, Pol δ commences strand-displacement synthesis, whereby it unanneals the 5′ end of the preceding Okazaki fragment into a single-stranded 5′ flap structure. FEN1 recognizes this structure, binds to the base of the flap, and precisely cleaves it, removing the RNA and some portion of the initiator DNA to make a nick. To complete the maturation process, DNA ligase I (Lig I) seals the nick. Okazaki fragments processed in this manner undergo the short flap pathway for maturation.
Sometimes FEN1 can disengage from the core replication complex, allowing for the displacement and creation of longer flaps (4). Additionally, helicases such as the 5′ helicase, Pif1, can bind the displaced flap and lengthen it further (4). If the displaced flap contains complementary sequences, it can fold back on itself, forming a hairpin, which is refractory to proper loading of FEN1 for cleavage. Also, the single-strand binding protein, replication protein A (RPA), can bind and coat a long flap, forming a complex, which also inhibits cleavage by FEN1 (5–7). Dna2 nuclease/helicase is a binding and functional partner of FEN1 in cells. Work from the Campbell laboratory (8) has shown that overexpression of Dna2 compensates for catalytic defects in mutant forms of FEN1 in the cell and vice versa. Additionally, double mutants of nuclease-defective Dna2 and 3′ nuclease–deficient Pol δ,which cause greater strand-displacement activity, are lethal in Saccharomyces cerevisiae. These results imply that Dna2 works with FEN1 to specifically process flaps.
Dna2 can displace RPA from the flap, cleaving periodically and leaving a terminal product flap 5–6 nts in length. This flap is too short to rebind RPA and is readily available for cleavage by FEN1. This is the long flap pathway. Based in part on the genetic evidence in S. cerevisiae that whereas Dna2 is absolutely essential for cell survival, Rad27 (scFEN1) is not, Seo and colleagues (5) proposed that Okazaki fragments generally achieve lengths that require Dna2 for processing. However, recent reports from the Campbell laboratory (9) suggest that whereas enzymes with similar functions (e.g., Exo1) can inefficiently back up FEN1 for fragment processing, no other protein backs up Dna2's function in the double-strand break (DSB) repair pathway. Dna2's absence, therefore, causes checkpoint activation and ultimately cell death (9–11). Reconstitution experiments from our laboratory have shown that although most of the flaps are processed through the short flap pathway, a few mature via the long flap pathway (4). Additionally, posttranslational modification of the proteins appears to regulate the relative flow through these pathways, as discussed below (12).
Flaps that contain a hairpin structure cannot be processed via either the short or the long flap pathway and must use a different pathway. Biochemical reconstitutions from our laboratory showed that when stable hairpins are created on the 5′ displaced flaps, they can be removed with the help of Pif1, a 5′ helicase (13). Although we originally proposed that Pif1 would unwind the hairpin, allowing for binding by RPA and processing of the flap through the long flap pathway, our actual results suggest otherwise and agree with an original proposal by Seo and colleagues (14). Pif1 unwinds the entire Okazaki segment in vitro when a stable hairpin is created, allowing for resynthesis by Pol δ (14), although we do not know whether this process occurs in cells.
BACTERIAL, ARCHAEAL, AND MAMMALIAN FEN1
The initial discovery of a replication-dependent 5′ exonuclease came from studies using purified Pol I from E. coli and Thermus aquaticus, wherein the nuclease was found to be an integral part of the polymerase complex (15, 16). Proteolysis of purified Pol I separated the polymerase and 3′ –5′ exonuclease (large C-terminal fragment) and the 5′ –3′ exonuclease (small N-terminal fragment), allowing for better characterization of the individual components of the complex. (17). The polymerase and 5′ nuclease act together to carry out a process called nick translation, in which the 5′ side of a nick in DNA is degraded while the 3′ side is extended. Investigators found that the 5′ –3′ exonuclease produces mono- and oligonucleotides as well as excises mismatched bases, revealing that the mechanism involves short flap creation and cleavage (18, 19). In the late 1980s and early 1990s, various research groups discovered many mammalian FEN1 homologs following the initial isolation of a 5′ –3′ exonuclease from HeLa cell extracts (20, 21). Cloning and characterization of the mouse nuclease by Lieber and colleagues (22) led to the naming of the 5′ –3′ nuclease as a flap endonuclease because it specifically cleaved 5′ flaps. Following the isolation and characterization of mammalian FEN1, researchers also identified archaeal, plant (23), and Xenous laevis FEN1 (24).
RNase H
Ribonuclease H (RNase H) encompasses a class of enzymes that excise the RNA from RNA/DNA hybrids. Because removal of both the initiator RNA during DNA replication and misincorporated ribonucleotides during repair is vital for maintaining genome fidelity, these enzymes are extremely important to normal cellular function. There are two distinct classes of RNase H enzymes: type 1 (RNase HI in prokaryotes, RNase H1 in eukaryotes) and type 2 (RNase HII in prokaryotes, RNase H2 in eukaryotes). Whereas RNase H1 may primarily function in mitochondrial DNA replication, RNase H2 finds its role in nuclear DNA replication and repair. RNase H2 functions differently from RNase H1 by cleaving 5′ of single ribonucleotides embedded in the DNA duplex (25). Our group termed RNase H2 (mistakenly called RNase H1 at the time) as a junction RNase H for its ability to hydrolyze RNA primers during Okazaki fragment maturation (26) and the specific ability to cut efficiently between the last two ribonucleotides before the DNA but not between the last ribonucleotide and the first deoxynucleotide. Similar to Eder et al. (27), we showed that RNase H2 leaves behind a terminal ribonucleotide that requires the activity of FEN1. Although biochemical studies showed an important role for RNase H2 along with FEN1 in the maturation of Okazaki fragments (26, 27), genetic studies in S. cerevisiae showed that deletion of either RNase H1 or RNase H2 did not result in a significantly different phenotype, suggesting that the RNase H pathway is redundant for the removal of RNA during replication (28, 29). Recently, Bubeck et al. found that PCNA specifically directs RNase H2 and not RNase H1 to nuclear replication forks (30). PCNA also stimulates the removal of RNA from Okazaki substrates and misincorporated ribonucleotides (30). Although Pol δ can strand-displace the initiator primer and FEN1/Dna2 can efficiently process these flaps, catalysis by RNase H2 before displacement and FEN1 after minimal displacement appears to be the most efficient means of removing the RNA primer.
BIOCHEMICAL PROPERTIES AND SUBSTRATE PREFERENCE
Initial biochemical experiments showed that FEN1 is a sequence-independent, structure-specific endonuclease that can cleave single-stranded flaps up to 200 nts in length (16). The nuclease activity of FEN1 is functional only in the presence of magnesium (Mg2+) and manganese (Mn2+) ions and is not supported by other cations such as zinc (Zn2+)and calcium (Ca2+). Optimum cleavage by FEN1 occurs in a concentration range of 1 mM to 10 mM of Mg2+ (22). The presence of salts diminishes the nuclease activity of FEN1, with cleavage greatly reduced at 50 mM of NaCl (22). FEN1 cleavage activity also functions over a broad range of temperatures (from 25°C to 85°C), with mammalian FEN1 cleaving best at 37°C.
The most favored substrate configuration for FEN1 [bacterial (31), yeast (32), archae-bacteria (33), and human (34, 35)] is a double flap with a characteristic single-nucleotide 3′ flap. Crystal structures revealed a closed chamber in FEN1 that fits the 3′ flap and helps orient the nuclease on its substrate (36, 37). The need for the double-flap configuration suggests the complexity of motions needed for eukaryotic nick translation. Pol δ carries out strand-displacement synthesis by physically pushing away the encountered strand while extending the primer. Because the protein and not the extending strand forces the displacement, the steady-state structure of the substrate has a short gap between the primer terminus and the first annealed nucleotide of the displaced strand. This structure must reconfigure to a double flap before FEN1 can act. To allow for the required redistribution of base pairs, the polymerase probably backs up and releases the substrate so the FEN1-substrate complex can form. The term nick translation, which implies sequential addition and removal of single nucleotides, is clearly an oversimplification of the actual process.
TRACKING VERSUS THREADING
Cleavage by FEN1 was inhibited in the presence of a secondary structure in the 5′ flap or when a complementary oligonucleotide was annealed to the 5′ end of the flap. Later results also revealed that a flap containing a biotin-conjugated streptavidin at its 5′ end, a branch or bubble structure, or certain chemical adducts also inhibited cleavage activity in FEN1 (38). These observations suggested that the nuclease activity of FEN1 depends on the presence of a free 5′ flap end. This led to the proposal of a tracking mechanism in which FEN1 must enter a free 5′ end and then move along the flap to its base for cleavage. The tracking requirement is an attractive model because it ensures the protection of the genome. Because FEN1 needs to track from the 5′ end and then cleave, the requirement would shield the single-stranded region between the Okazaki fragments from indiscriminate nuclease activity. Protection would occur even though the gap between Okazaki fragments presents a structure that has elements of a FEN1-preferred substrate with junctions of single- and double-strand regions. Dna2, the interacting partner of FEN1, displays the same tracking requirement as FEN1.
In contrast to the popular tracking model, Joyce and colleagues (39) suggested that eubacterial FEN1 first recognized the dsDNA junction and then threaded the flap through its active site for cleavage. Giving credence to this model, recent biochemical and crystal structure data have refined the sequence of events in the recognition, binding, and cleavage steps of FEN1 (40, 41). Results show that FEN1 can still bind flap substrates blocked at the 5′ end, indicating that the nuclease initially recognizes the flap base (40). FEN1 binding is then further stabilized by a threading process in which the 5′ end and then the entire length of the flap pass through the nuclease. Revisiting of the Dna2 mechanism showed that it has the same elements (42). This threading requirement also applies to the helicase function, making it one the few helicases that must enter its substrate from an end (43). The threading mechanism is consistent with the interaction of FEN1 with PCNA, which strongly stimulates nuclease activity, and with the myriad of FEN1 protein-binding partners. These interactions portray FEN1 as part of larger functional protein complexes, consistent with the idea that it stays at the flap base with a complex and does not wander to find the flap 5′ end.
Why is there a threading requirement? Substrate-binding analyses of FEN1 suggest that the threading requirement slows FEN1 association and dissociation with its substrate. Moreover, additional studies indicate that flaps shorter than 3 nts have no effect on binding stability or dissociation kinetics (40). This suggests that FEN1 does not usually employ threading because in most cases short flaps are created and removed. These properties are still consistent with the interpretation that the threading requirement is a protective mechanism that has evolved to prevent FEN1 and Dna2 from cleaving the single-stranded region between Okazaki fragments. In that way, the slower tracking process is a necessary side effect of the protective mechanism, employed only with the relatively few flaps that become long.
FEN1 also has a gap cleavage or gap endonuclease (GEN) activity (44). This activity would appear to be at odds with the threading requirement. Researchers have suggested that GEN activity is regulated so that it is employed selectively to spare gapped regions that must remain intact, but it can also participate in genome cleavage in apoptosis (44–46).
FEN1 CRYSTAL STRUCTURE
Human FEN1, a metallonuclease, is composed of a nuclease domain [made up of N-terminal (N), intermediate (I), and C-terminal (C) sections] and an extended C-terminal region, which is responsible for important interactions with proteins including PCNA and the Werner protein (WRN) (44, 47–49). Early ideas about the structure of FEN1 emerged from analyses of its bacteriophage homolog, T5 5′ exonuclease, a protein with a helical arch that allows for passage of single-stranded DNA (ssDNA) (36). Consistent with the biochemical data, subsequent crystal structure data from Archaeoglobus fulgidus showed that FEN1 preferentially binds to a 1-nt 3′ overhang and 5′ flap by forming a hydrophobic wedge around the 3′ flap (37). This formation orients the enzyme to the base of the flap and guides it in a manner that allows FEN1 to cleave precisely 1 nt into the downstream double-stranded region, allowing the generation of a nick (37).
Further work by Sakurai and colleagues (50) on human FEN1 confirmed the primary recognition of the 3′ flap by FEN1; however, because of the disordered structure of the active site, they could not distinguish between the threading and tracking mechanisms. Recent crystal structures of human FEN1 in complex with model substrates have allowed us to visualize the structural features that correspond to the unique substrate interactions of the FEN1 superfamily of proteins (41). The protein was shown to have a left-handed boxing glove–like structure. On binding the preferred substrate with a 1-nt 3′ flap and longer 5′ flap, the enzyme interacts tightly through the palm and fingers with the double-stranded regions on either side of the flaps, with a total interaction distance of more than 16 nt. The active site bearing the dual Mg2+ atoms resides at the flap base and is sharply bent at 100°. The structural results show two major protein-DNA interfaces on either side of the bend. Binding occurs to both flaps, ensuring recognition of a true double-strand flap substrate. A substantial number of DNA-protein contacts in the 3′ flap–binding pocket attest to why the 3′ flap is so important for keeping the DNA in proper register with the protein for catalysis. There are protein contacts to the 5′ flap only near its base; to the gateway structure, which is present in superfamily members FEN1, EXO1, XPG, and GEN1; and to the cap, which is present only in FEN1 and EXO1. The cap allows a strand end to have access to the gateway but denies access to the middle of a strand or a double strand. The cap is part of a larger structure called the helical arch, which changes from disordered to ordered upon substrate binding. These features create the threading requirement for FEN1. The absence of the cap in some nucleases possibly allows them to access DNA bubbles and recombination intermediates. Figure 1 outlines the mechanism of FEN1 cleavage based on the crystal structure data.
Figure 1.
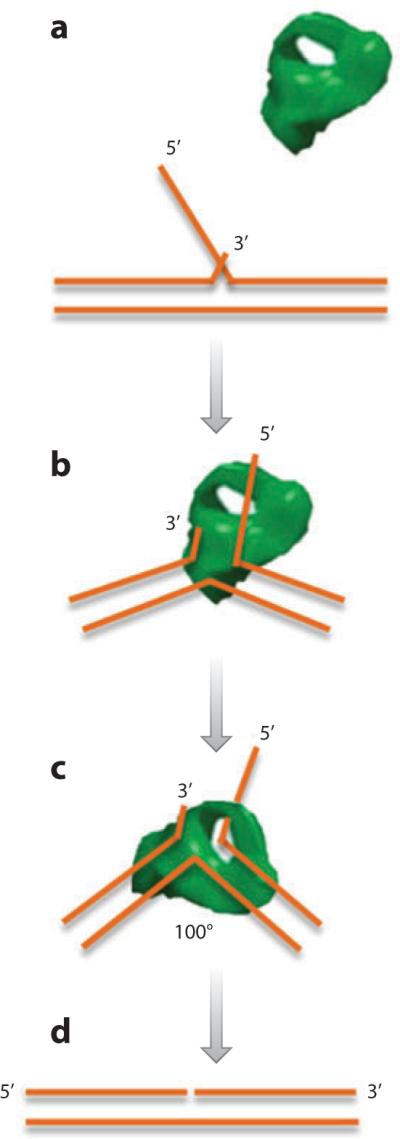
Flap endonuclease 1 (FEN1) substrate recognition and cleavage. FEN1 (a) recognizes the displaced flap, (b) binds to the base of the flap, (c) bends the substrate into a 100° angle to arrange the one-nucleotide 3′ overhang and 5′ displaced flap in the active site, and (d) precisely cleaves the flap, generating a nick. Based on structure information from Reference 41 and adapted with permission from Susan E. Tsutakawa, Lawrence Berkeley National Laboratory, Berkeley, CA 94720.
Amino acid residues in the putative active site are positioned to use an electrostatic mechanism to promote endonucleolytic phosphodiester hydrolysis. Moreover, the structure reveals how the downstream double strand is distorted out of B-form conformation so that the active site can cleave 1 nt into the annealed downstream helix. The structural model implicates tyrosine 40, lysine 93, and arginine 100 in catalysis, a conclusion confirmed by mutational analysis. The mechanism is consistent with biochemical evidence that FEN1 binds first to the flap base and that specific amino acid-DNA interactions do not extend into the 5′ flap (40). Because the flap is not bound directly but must pass through an opening, the structural features also explain why some flap adducts are tolerated. The lack of specific flap binding also explains why both DNA and RNA flaps are cleaved. Additionally, the affinity for the downstream double strands provides a model for how FEN1 can be available at the substrate site even before the polymerase relinquishes the DNA 3′ end and how the protein can allow access for DNA ligase after flap cleavage.
FEN1 IN LONG-PATCH BASE EXCISION REPAIR
Cellular DNA is constantly exposed to physical and chemical damaging agents and naturally occurring breakdowns, both of which structurally alter the DNA, resulting in genome instability. By-products of metabolism in the form of reactive oxygen species modify mammalian DNA structure at a very high rate, producing 1,000 to 100,000 lesions per cell per day (51–53). The primary means for removing base lesions in mammals is base excision repair (BER) (54, 55). This process involves two subpathways: single-nucleotide BER (SN-BER) and long-patch BER (LP-BER). These differ in the size of the repair patch and in the enzymes utilized to restore the lesion site (56, 57). Both subpathways are initiated by a DNA glycosylase, which recognizes the damaged base and excises it by cleaving the glycosidic bond between the altered base and the deoxyribose phosphate backbone, creating an abasic [apurinic/apyrimidinic (AP)] site (58). In addition, the spontaneous hydrolytic loss of purines creates~10,000 AP sites daily (59, 60). All AP sites are further processed by AP endonuclease 1, which cleaves the phosphodiester backbone, leaving a 3′ hydroxyl (3′-OH) and a 5′ deoxyribose phosphate (5′-dRP) moiety (61, 62). The cell then employs SN-BER. SN-BER uses DNA polymerase β (Pol β), a multifunctional enzyme containing a synthesis function in a core 30-kDa domain and a lyase activity in an 8-kDa domain (63, 64). The lyase activity excises the dRP moiety, and the polymerase then replaces it with the correct nucleotide complementary to the template, producing a nick that is sealed by DNA ligase III (Lig III). However, if the dRP moiety is either oxidized or reduced, the lyase cannot function (65). Alternatively, acetylation can posttranslationally modify Pol β, resulting in inhibition of the lyase (66). When the lyase is unable to function, Pol β performs strand-displacement synthesis, similar to Pol δ during Okazaki fragment maturation, and lifts the 5′ dRP moiety into a flap structure, initiating LP-BER. The flap is a substrate for FEN1. Operating with the same specificity as with Okazaki fragment processing, FEN1 binds to the base of the flap and cleaves off the displaced strand, creating a nick that is then sealed in by Lig I or the XRCC1/Lig III complex (67).
In S. cerevisiae and malaria parasites, SN-BER is absent, and LP-BER is the predominant method of lesion correction (68, 69). However, in mammalian cells, SN-BER may be the major repair subpathway, with a small percentage of damage corrected by LP-BER (70). Although the LP-BER repair patch size is technically difficult to measure in vivo because of the constant removal and repair of lesions, many indirect approaches using plasmid DNA containing defined damage sequences suggest a repair patch length of 2–12 nt (71).
Experiments show that in both cells and reconstituted systems, FEN1 is a critical enzyme for LP-BER. Although LP-BER functions in a PCNA-independent subpathway utilizing Pol β, it can also operate in a PCNA-dependent subpathway that utilizes Pol δ/ε. For repair purposes, FEN1 and Pol β employ a hit-and-run mechanism. After replacing the 1-nt base lesion, if the lyase function of Pol β is inhibited, the system alternates polymerase and nuclease binding with sequential single-nucleotide addition and cleavage for several additional residues, a hallmark of LP-BER. Reconstitution experiments have verified that Pol β and FEN1 influence each other by stimulating inherent enzymatic activities on their cognate substrates. The heterotrimeric protein complex, Rad9, Rad1, and Hus1 (the 9-1-1 complex), also interacts with many of the BER proteins and, similar to its functional partner, PCNA, stimulates the enzymatic activities of the BER proteins including FEN1.
FEN1 IN TRINUCLEOTIDE REPEATS
The genome contains many sections of repeated sequences that researchers have categorized into minisatellite and microsatellite repeats according to their length. Regions containing these repeats, termed fragile sites, are highly unstablebecause their replication could lead to the formation of unprocessed intermediates that degenerate into DSBs and single-strand breaks (SSBs) (72, 73). Mutations in both RAD27 and DNA2 in S. cerevisiae destabilize minisatellite regions, suggesting that proper processing at Okazaki fragments is crucial for genome maintenance (74). Trinucleotide repeats (TNRs), a category of minisatellite repeats that includes GC trinucleotide repeats, have a tendency to expand rapidly, achieving substantial lengths. The expansion of TNRs is the underlying cause of at least 20 severe neuromuscular and neurodegenerative diseases, including Huntington's disease (HD) and myotonic dystrophy (75).
Many models propose a mechanism for TNR expansion, all suggesting the formation of looped DNA intermediates during both DNA repair and replication. Interestingly, large expansions occur mostly during nondividing cellular conditions (75). Even though none of the DSB repair pathways influence sequence expansion (76, 77), SSB repair has been implicated in expansion. During BER, SSBs are generated by the excision of bases damaged chemically, particularly through oxidation (78). Reports have indicated that in mouse models for HD, TNR expansion occurs simultaneously with the increase of oxidized bases (79). Additionally, the loss of 7,8-dihydro-8-oxoguanine DNA glycosylase, which removes oxidized bases, causes a decrease in TNR lengths in the same mouse model (79). Once the damaged base is removed, either a short or a long patch is resynthesized. When the long-patch pathway is followed, the polymerase performs strand displacement, creating an ssDNA 5′ flap. Although the flap is normally processed by FEN1 5′–3′ endonuclease activity, in vitro studies have shown that when flaps form loops or hairpins, the 5' end may become inaccessible for cleavage (80–82). These structures are particularly prone to form when the flap sequence consists of TNR repeats (83). If these looped structured flaps were ligated, an expanded product would result.
Smaller expansion events are observed in dividing cells (84). Polymerases pause upon encountering TNR sequences, especially when the sequences are located on the lagging-strand template (84–86). Polymerase slippage can result in misalignment of the daughter strand and the subsequent generation of an expanded sequence. Evidence in dividing yeast suggests a replication restart model for TNR expansion in which the polymerase stalls at long stretches of TNR sequences and causes the replication fork to back up (87). In this case, misaligned TNR loops can cause the polymerase to reverse and utilize the daughter strand as a template for synthesis, forming a four-way chicken-foot intermediate (88).
Although patients with HD do not have mutations in FEN1 that impair its function, other evidence suggests that FEN1 has evolved to oppose the expansion of repeat sequences (89). Analyses of substrates with overlapping TNR flaps exposed to DNA ligase I and FEN1 show that increasing the concentration of FEN1 prevents the formation of hairpin and bubble intermediates that can be ligated into expanded products (90). Studies in S. cerevisiae demonstrate that absence of FEN1 greatly increases TNR expansions, and genetic analysis implicates the exonucleolytic and gap cleavage activities in the protective effects of FEN1 (91). Liu & Wilson (92) proposed that one mechanism of repeat expansion is the formation of hairpins in the single-strand intermediates of LP-BER. These structures can produce strand slippage, moving the flap base on the template so that FEN1 cuts in the wrong location (Figure 2). This produces an extra length of repeat sequence than can be ligated into the strand. A similar process can occur with Okazaki fragment intermediates. Mutations in the mismatch-repair system have been known to suppress TNR expansions (93, 94). Recent in vitro tests suggest that the mismatch-repair complex Msh2-Msh3 can stabilize bubble and hairpin structures, promoting improper flap structure and incorrect FEN1 cuts (95). This appears to be an undesirable side effect of the normal mismatch-repair system.
Figure 2.
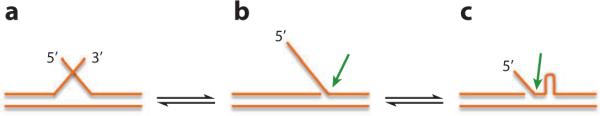
Alternate FEN1 cleavage. Substrates with trinucleotide repeats or other self-complementary sequences can equilibrate to contain (a) both 5′ and 3′flaps, (b)only a 5′ flap, or (c) a configuration producing an expansion intermediate by creating a hairpin and a flap with an alternate FEN1 cleavage site. The green arrows indicate the sites of FEN1 cleavage.
FEN1-INTERACTING PARTNERS
Eukaryotic FEN1 nucleases interact with many protein partners, as indicated by immuno-precipitation and catalytic activity changes in FEN1 and sometimes in the partner. As many as 20 protein partners have been described including PCNA, Dna2, replication protein A (RPA), Bloom syndrome protein (BLM), and WRN. The number is so large, and the molecular weight of FEN1 so small at an average of 42 kDa, that FEN1 is unlikely to bind more than 2–3 partners at a time. This suggests that FEN1 participates in a sequential handoff process in which its role in protein complexes continuously changes to match the necessary sequence of reactions of the pathways in which it is participating.
A general feature of partner interaction is that it augments the nuclease activity of FEN1 by as much as 50-fold. Most partners increase activity to near the maximum possible level when multiple partners are present. This suggests that FEN1 can assume either high or very low activity conformational states. A possible reason for this is that FEN1 has evolved to work only in proper complexes for specific tasks and would display activity detrimental to genome integrity if it were to act alone.
Interaction with PCNA (96–98) is consistent with the role of PCNA as not only a sliding clamp for efficient processive actions of DNA replication and repair proteins but also an exchange platform that coordinates the order of action of these proteins. Binding of FEN1 and Dna2 (99, 100) is consistent with the proposed sequential action of these nucleases in processing long flaps. The interaction with BLM (101) implicates this hexameric helicase in the DNA replication and repair pathways of FEN1. Taken together with the known role of BLM in disrupting recombination intermediates, this interaction suggests that BLM frees long flaps from forming troublesome structures by binding ectopic sites or having the flaps fold back on themselves.
POSTTRANSLATIONAL MODIFICATIONS OF FEN1
Considering FEN1's central role in DNA replication and repair, it is not surprising that its activity is finely regulated by many posttranslational modifications (102). Human FEN1 can be modified by phosphorylation, acetylation, methylation, and more recently by ubiquitination and small ubiquitin-related modifier (SUMO)ylation (103–105).
Researchers first showed that human FEN1 is acetylated in vitro on the C-terminal domain by the acetyltransferase p300 (105). Mass spectrometry analysis of FEN1 acetylated in vitro identified four modified lysine (K) sites on the C-terminal domain (K354, K375, K377, and K380). Recent characterization of the human acetylome analyzing three different cell types by mass spectrometric techniques confirmed that FEN1 is also acetylated in vivo on sites K80, K267, and K375 (106). Biochemical analysis of FEN1 acetylated to saturation reduced catalytic activity by approximately 90% (105). This decreased activity coincided with reduced binding to FEN1's substrate. Interestingly, more acetylation of FEN1 was observed after UV damage to cells. For many years, it was unclear why the cell would intentionally downregulate FEN1 catalytic activity because reduction of FEN1 expression by only 50% in haploinsufficient cells led to genome instability (107). An initial speculative justification for acetylation of FEN1 was that the decreased activity of the nuclease would prevent premature maturation of Okazaki fragments (105). More recently, the rationale for this downregulation has become more evident in the context of the complete pathways of Okazaki fragment processing and LP-BER. Nearly every protein in these pathways is naturally modified by acetylation. Acetylation strongly stimulates Dna2 nuclease, helicase, and ATPase activities (108), in addition to stimulating Pif1 helicase activity (L. Balakrishnan & R. Bambara, unpublished observation). Acetylation also augments RPA-binding activity to ssDNA (L. Balakrishnan, M. Wold & R. Bambara, unpublished observation) and greatly increases the efficiency of strand-displacement synthesis by DNA Pol δ (L. Balakrishnan, B. vanLoon, U. Hubscher & R. Bambara, unpublished observation). The acetylated proteins should displace flaps more efficiently during Okazaki fragment maturation and LP-BER. Acetylated Dna2 should act more efficiently on the flaps, keeping them short. However, acetyl-FEN1 should create nicks for ligation less frequently than the unmodified nuclease does. The overall effect is that a longer patch of the 5′ end region of Okazaki fragments and a longer patch of damaged DNA in LP-BER are removed and replaced. Each Okazaki fragment is initiated by DNA Pol α, which lays down a 9–14-nt RNA followed by approximately 20 nt of DNA. Pol α does not have a 3′ proofreading nuclease and so is two orders of magnitude less accurate than the Pol δ that completes synthesis of the fragment. Consequently, there is an increasing gradient of probability from the 3′ to the 5′ end that an Okazaki fragment will contain a mismatch error. Therefore, replacing a longer patch of Pol α product with Pol δ product should increase the fidelity of DNA replication. Moreover, replacing a longer patch of damaged DNA in LP-BER increases the probability that the damage will be fully removed. In this manner, the cell employs an acetylation-based regulatory process that allows it to trade efficiency of DNA repair for fidelity. The choice of which direction of trade is more desirable may depend on nutritional or developmental states of the cell or on whether the enzymes are acting on transcriptionally active DNA.
FEN1 was also phosphorylated at serine 187 in the late S phase by the cyclin A or Cdk2/cyclin E complex (103). FEN1 phosphorylated in vitro bound to its substrate with a similar affinity as the unmodified form; however, its endonuclease activity was inhibited (103). Additionally, PCNA was unable to interact with the phosphorylated form of FEN1 (103). Guo et al. (109) showed that phosphorylation of FEN1 regulates its localization to the nucleolus and influences its role in ribosomal DNA replication and repair. Creation of phosphorylation mutants (either constitutively phosphorylated or phosphorylation inhibitory) increased cellular UV damage sensitivity, UV repair capacity, and ultimately cell survival (109).
Recently investigators showed the methylation of FEN1 at the arginine 192 residue (110). Interestingly, methylation of FEN1 prevented its phosphorylation. Methylated FEN1 retained its interaction with PCNA. The Shen group (111) postulates that FEN1 is dynamically methylated and phosphorylated to regulate its activity on the replication fork. During Okazaki fragment processing, methylated FEN1 initially interacts with PCNA and replaces Pol δ to gain access to the flap. Following flap removal, FEN1 is phosphorylated, which causes it to lose its interaction with PCNA and leave the substrate to provide access for DNA ligase I.
To determine the fate of FEN1 after completion of replication, Shen and colleagues (104) analyzed the levels of FEN1 during different cell cycle phases and showed that it peaked during the S phase and dramatically decreased during the G2/M phase. Because there was no decrease in the FEN1 transcript level, they questioned whether the reduction in FEN1 expression was posttranslational-modification dependent. They determined that ubiquitination and SUMOylation modify FEN1. The UBE1/UBE2M/PRP19 complex ubiquitinated FEN1 at K354 (104). FEN1 was modified by SUMO3 at K168 (104). Phosphorylation of FEN1 stimulated its ubiquitination, which in turn stimulated SUMOlyation and degradation by the proteasome pathway (104). This cascade of modifications alters the levels of FEN1 in the cell, potentially to decrease the risk of having an active nuclease in the cell during phases when this activity is not required. Thus, the activity and expression are tightly controlled in the cell by programmed regulation of multiple posttranslational modifications (104).
FEN1 IN MITOCHONDRIA AND TELOMERES
Although FEN1 may have a predominantly nuclear localization, researchers have long suspected its presence in mitochondria. In view of the highly oxidative environment of mitochondria, BER in mitochondria should require a FEN1-like endonuclease. In 2008, FEN1's level of involvement in the repair process was debated, with the Demple and Shen groups (112) suggesting that FEN1 plays a central role in the LP-BER process in mitochondria and the Mitra group (113) maintaining that other mitochondrial nucleases, along with FEN1, effected repair. Irrespective of its level of involvement, both groups established the presence of mammalian FEN1 in the mitochondria (112, 113). Construction of a Rad27-green fluorescent protein fusion integrated into the yeast genome allowed tracking of the cellular distribution of FEN1 by fluorescence microscopy and showed that although most of the proteins concentrated in the nucleus, FEN1 also associates with the mitochondria (114). The role of FEN1 in mitochondrial DNA replication has not been confirmed (113).
FEN1 has also been implicated in maintaining telomere stability. Early studies in rad27 yeast strains showed that the lengths of the telomere repeats destabilized, specifically when the cells were grown at 37°C (115, 116). These mutants also accumulated single-stranded G overhangs at the lagging telomere ends, possibly contributing to telomere shortening and an accelerated senescence phenotype (115, 116). During the S and G2 phases of the cell cycle, human FEN1 localized to the telomere and associated with telomeric-repeat binding factor 2 (TRF2), a component of the shelterin complex (117, 118). FEN1 also forms a complex with telomerase via telomeric DNA (119). Deficiency of FEN1 in mouse embryonic fibroblasts caused an increase in telomere end-to-end fusions (119). Depletion of FEN1 in BJ fibroblasts led to an increase in γ-H2AX foci and loss of sister telomeres replicated by lagging-strand synthesis (120). Genetic rescue experiments revealed that the nuclease activity of FEN1 and its ability to interact with WRN and TRF2 are essential for it to function at telomeres (120). Mutations affecting the GEN activity of FEN1 and the WRN interaction sites promoted telomere instability, suggesting that both of these functions of FEN1 are essential for its role at chromosome ends (121). FEN1 also processed flaps created on substrates that form G4 quartets (122). Although the exact role of FEN1 at telomeres needs clarification, taking into account its proposed interactions and functions, we infer that FEN1 collaborates with other proteins at the telomeres to reinitiate stalled replication forks, thereby contributing to higher genome stability.
NATURALLY OCCURRING FEN1 MUTANTS
Although FEN1 is not essential for the viability of S. cerevisiae and Schizosaccharomyces pombe, it is required for normal cell growth and proliferation (123). Several mutations have been made to the active site, localization site, and interaction sites of FEN1 to elucidate its role in replication and repair. Naturally occurring mutations in FEN1 have also been helpful in defining its role in DNA transactions. Two functional variants of FEN1 in germ line cells (69G → A and 4150G → T) have been associated with an increased frequency of lung and gastrointestinal cancers (124, 125). The 69G → A polymorphism resides in the promoter region of the FEN1 transcript, and 4150G → T occurs in the 3′ untranslated region of the FEN1 transcript. Both of these polymorphisms are linked to decreased mRNA expression of FEN1 (125). These single-nucleotide polymorphisms may contribute to cancer risk, given the role of FEN1 as a tumor suppressor (described in the section below), and could be used as markers for detection of lung and gastrointestinal cancers.
Direct DNA sequencing of the FEN1 gene derived from tumors or tumor-derived cell lines revealed mutations that alter the nuclease activity of FEN1 (126). Detection of in-frame mutations in lung cancer tissues, a missense mutation in melanoma, and a silent mutation in esophageal cancer, all of which are not present in the corresponding normal tissues, suggests these mutations are somatic in nature (126). To further establish the etiological significance of the somatic mutations, the Shen group (126) established the first mouse model that contains an E160D mutation in the FEN1 gene that alters Mg2+ binding to FEN1 and also specifically decreases the EXO and GEN activities of FEN1. Elimination of those nuclease activities of FEN1 in this mouse model led to an increase in frequent spontaneous mutations (126). Additionally, this model showed an increase in the accumulation of incompletely digested DNA fragments in apoptotic cells (126). Furthermore, these mutant mice showed signs of chronic inflammation and were predisposed to autoimmunity (126). Another independent study confirmed that the E160D mutation in FEN1 indeed increased susceptibility to cancer (127).
ROLE OF FEN1 IN CANCER
A reduction of FEN1 activity in haploinsufficient cells caused deleterious effects including rapid tumor progression (107). Although FEN1 is generally thought of as a tumor suppressor gene, many reports emphasize its role in supporting growth and proliferation in cancer cells. Normally, FEN1 expression is proliferation dependent and silenced in terminally differentiated cells (128, 129). Because cancer progression involves deregulation in the expression of many proteins and FEN1 is central to DNA replication, FEN1 is unsurprisingly overexpressed in multiple cancer types.
Using a cancer-profiling assay that compares tumor tissues with normal tissues, Singh et al. (130) showed that FEN1 is overexpressed in breast cancer tissues. This overexpression was linked to increased mRNA levels, most likely as a consequence of FEN1-promoter hypomethylation (130). Additionally, Singh et al. (130) showed that the mRNA levels of FEN1 were overexpressed in other types of cancer, including uterine, kidney, ovarian, and colon. Using global gene-profiling assays, FEN1 was also identified as a novel gene that was upregulated in pancreatic adenocarcinoma (131). Comparison of normal lung cells with small and nonsmall lung cancer cells showed an increased level of FEN1 gene expression in the cancer cells (132). The level of FEN1 mRNA transcripts was also increased in gastric cancer cells compared with normal cells (133). The FEN1 protein level was increased in most but not all glioblastoma and astrocystoma tumors, suggesting that it could be potentially used as a tumor marker (134).
The DNA of cancerous cells is constantly exposed to endogenous stress as a result of rapid DNA synthesis and defects in DNA repair systems. Researchers previously reported that the expression of FEN1 is upregulated in mouse fibroblasts undergoing genotoxic stress (135). We currently do not know whether the levels of FEN1 are altered during chemotherapy or radiation. Glioblastoma cell lines depleted of FEN1 showed increased damage sensitivity to methylating agents such as methyl methanesulfonate and temozolomide. Considering how integral FEN1 is to repair pathways, the high expression levels of FEN1 may contribute to altered drug resistance. As proof of this concept, one study showed that breast cancer cells having a lower FEN1 expression level were more susceptible to apoptosis compared with cells with a high expression level (136). However, because FEN1 plays a role in DNA replication, LP-BER, and the final steps of nucleotide excision repair, we must fully understand how altered levels of FEN1 can confer resistance to anticancer drugs and determine whether FEN1 can be used as a tumor marker.
Predictably, lack of FEN1 expression causes genome instability and cancer predisposition. Homozygous knockout of FEN1 genes in mice causes embryonic lethality (137). Heterozygous knockout of FEN1 along with heterozygous knockout of the Adenomatous polyposis coli gene, a classic tumor suppressor, results in adenocarcinoma and decreased survival rates (137). In cancer cells expressing a normal level of FEN1, the activity of the nuclease may be downregulated by a variety of factors such as genetic mutations of the active site, impairment in protein-protein interactions, improper cell localization, and effects of posttranslational modifications. The mutation E160D, reported naturally in human cancers, alters cation (Mg2+) binding in the active site, thereby resulting in altered substrate binding and cleavage properties. The Shen group (126) created a mouse line harboring E160D and showed that this mutation affected the EXO and GEN activities of the nuclease, consistent with previously observed defects in apoptosis (126, 138). Additionally, they observed that the mutant mice were susceptible to cancer and also to autoimmunity and chronic inflammation (126). These findings strongly suggest the role of FEN1 as a tumor suppressor as it is absolutely essential in the maintenance of genome fidelity.
CONCLUSIONS
FEN1 is one of the most ancient and centrally important proteins in the cell. Usually just more than 40 kDa, it has evolved for efficient form and function. It has adapted to interact with a succession of approximately 20 protein-binding partners and is regulated by numerous posttranslational modifications. It is a guardian of genome integrity, but if mutated or improperly regulated, it is key to cancer progression. Its structure-specific substrate specificity and threading mechanism are marvels of protein machinery. FEN1 is a testimony to the level of elegance that a protein can achieve after eons of strong evolutionary pressure.
FUTURE ISSUES
The role of posttranslational modification of FEN1 in important cellular regulatory processes that preserve genome integrity deserves considerably more investigation.
Functional interactions of FEN1 with telomere-binding proteins that act in telomer-especific DNA replication and repair should be characterized.
The role of FEN1 in cancer progression is an important new topic.
Recent evidence suggests that properly functioning mismatch-repair proteins interfere with FEN1 in a way that promotes triplet repeat expansion. The potentially undesirable interactions of replication and repair proteins deserve consideration.
ACKNOWLEDGMENTS
The work from our laboratory was funded by National Institutes of Health grants GM098328 (to L.B.) and GM24441 (to R.A.B.). We are indebted to our past and present collaborators, who have helped us through the many years of research on understanding and defining the properties of the FEN1 protein. We apologize to our many colleagues whose work we were not able to cite in this review owing to space limitations.
Footnotes
DISCLOSURE STATEMENT The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: a central component of DNA metabolism. Annu. Rev. Biochem. 2004;73:589–615. doi: 10.1146/annurev.biochem.73.012803.092453. [DOI] [PubMed] [Google Scholar]
- 2.Smith DJ, Whitehouse I. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature. 483:434–38. doi: 10.1038/nature10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li JJ, Kelly TJ. Simian virus 40 DNA replication in vitro. Proc. Natl. Acad. Sci. USA. 1984;81:6973–77. doi: 10.1073/pnas.81.22.6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossi ML, Bambara RA. Reconstituted Okazaki fragment processing indicates two pathways of primer removal. J. Biol. Chem. 2006;281:26051–61. doi: 10.1074/jbc.M604805200. [DOI] [PubMed] [Google Scholar]
- 5.Bae SH, Bae KH, Kim JA, Seo YS. RPA governs endonuclease switching during processing of Okazaki fragments in eukaryotes. Nature. 2001;412:456–61. doi: 10.1038/35086609. [DOI] [PubMed] [Google Scholar]
- 6.Maga G, Frouin I, Spadari S, Hubscher U. Replication protein A as a “fidelity clamp” for DNA polymerase α. J. Biol. Chem. 2001;276:18235–42. doi: 10.1074/jbc.M009599200. [DOI] [PubMed] [Google Scholar]
- 7.Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- 8.Budd ME, Campbell JL. The pattern of sensitivity of yeast dna2 mutants to DNA damaging agents suggests a role in DSB and postreplication repair pathways. Mutat. Res. 2000;459:173–86. doi: 10.1016/s0921-8777(99)00072-5. [DOI] [PubMed] [Google Scholar]
- 9.Budd ME, Antoshechkin IA, Reis C, Wold BJ, Campbell JL. Inviability of a DNA2 deletion mutant is due to the DNA damage checkpoint. Cell Cycle. 2011;10:1690–98. doi: 10.4161/cc.10.10.15643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balakrishnan L, Bambara RA. The changing view of Dna2. Cell Cycle. 2011;10:2620–21. doi: 10.4161/cc.10.16.16545. [DOI] [PubMed] [Google Scholar]
- 11.Burgers PM. It's all about flaps: Dna2 and checkpoint activation. Cell Cycle. 2011;10:2417–18. doi: 10.4161/cc.10.15.15851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balakrishnan L, Bambara RA. Eukaryotic lagging strand DNA replication employs a multi-pathway mechanism that protects genome integrity. J. Biol. Chem. 286:6865–70. doi: 10.1074/jbc.R110.209502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pike JE, Henry RA, Burgers PM, Campbell JL, Bambara RA. An alternative pathway for Okazaki fragment processing: resolution of fold-back flaps by Pif1 helicase. J. Biol. Chem. 285:41712–23. doi: 10.1074/jbc.M110.146894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryu GH, Tanaka H, Kim DH, Kim JH, Bae SH, et al. Genetic and biochemical analyses of Pfh1 DNA helicase function in fission yeast. Nucleic Acids Res. 2004;32:4205–16. doi: 10.1093/nar/gkh720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klett RP, Cerami A, Reich E. Exonuclease VI, a new nuclease activity associated with E. coli DNA polymerase. Proc. Natl. Acad. Sci. USA. 1968;60:943–50. doi: 10.1073/pnas.60.3.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lyamichev V, Brow MA, Dahlberg JE. Structure-specific endonucleolytic cleavage of nucleic acids by eubacterial DNA polymerases. Science. 1993;260:778–83. doi: 10.1126/science.7683443. [DOI] [PubMed] [Google Scholar]
- 17.Brutlag D, Atkinson MR, Setlow P, Kornberg A. An active fragment of DNA polymerase produced by proteolytic cleavage. Biochem. Biophys. Res. Commun. 1969;37:982–89. doi: 10.1016/0006-291x(69)90228-9. [DOI] [PubMed] [Google Scholar]
- 18.Setlow P, Kornberg A. Deoxyribonucleic acid polymerase: two distinct enzymes in one polypeptide. II. A proteolytic fragment containing the 5′ leads to 3′ exonuclease function. Restoration of intact enzyme functions from the two proteolytic fragments. J. Biol. Chem. 1972;247:232–40. [PubMed] [Google Scholar]
- 19.Setlow P, Brutlag D, Kornberg A. Deoxyribonucleic acid polymerase: two distinct enzymes in one polypeptide. I. A proteolytic fragment containing the polymerase and 3′ leads to 5′ exonuclease functions. J. Biol. Chem. 1972;247:224–31. [PubMed] [Google Scholar]
- 20.Guggenheimer RA, Nagata K, Kenny M, Hurwitz J. Protein-primed replication of plasmids containing the terminus of the adenovirus genome. II. Purification and characterization of a host protein required for the replication of DNA templates devoid of the terminal protein. J. Biol. Chem. 1984;259:7815–25. [PubMed] [Google Scholar]
- 21.Guggenheimer RA, Nagata K, Lindenbaum J, Hurwitz J. Protein-primed replication of plasmids containing the terminus of the adenovirus genome. I. Characterization of an in vitro DNA replication system dependent on adenoviral DNA sequences. J. Biol. Chem. 1984;259:7807–14. [PubMed] [Google Scholar]
- 22.Harrington JJ, Lieber MR. The characterization of a mammalian DNA structure-specific endonuclease. EMBO J. 1994;13:1235–46. doi: 10.1002/j.1460-2075.1994.tb06373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimura S, Kai M, Kobayashi H, Suzuki A, Morioka H, et al. A structure-specific endonuclease from cauliflower (Brassica oleracea var. botrytis) inflorescence. Nucleic Acids Res. 1997;25:4970–76. doi: 10.1093/nar/25.24.4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bibikova M, Wu B, Chi E, Kim KH, Trautman JK, Carroll D. Characterization of FEN-1 from Xenopus laevis. cDNA cloning and role in DNA metabolism. J. Biol. Chem. 1998;273:34222–29. doi: 10.1074/jbc.273.51.34222. [DOI] [PubMed] [Google Scholar]
- 25.Cerritelli SM, Crouch RJ. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 2009;276:1494–505. doi: 10.1111/j.1742-4658.2009.06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murante RS, Henricksen LA, Bambara RA. Junction ribonuclease: an activity in Okazaki fragment processing. Proc. Natl. Acad. Sci. USA. 1998;95:2244–49. doi: 10.1073/pnas.95.5.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eder PS, Walder RY, Walder JA. Substrate specificity of human RNase H1 and its role in excision repair of ribose residues misincorporated in DNA. Biochimie. 1993;75:123–26. doi: 10.1016/0300-9084(93)90033-o. [DOI] [PubMed] [Google Scholar]
- 28.Frank G, Qiu J, Somsouk M, Weng Y, Somsouk L, et al. Partial functional deficiency of E160D flap endonuclease-1 mutant in vitro and in vivo is due to defective cleavage of DNA substrates. J. Biol. Chem. 1998;273:33064–72. doi: 10.1074/jbc.273.49.33064. [DOI] [PubMed] [Google Scholar]
- 29.Qiu J, Qian Y, Frank P, Wintersberger U, Shen B. Saccharomyces cerevisiae RNase H(35) functions in RNA primer removal during lagging-strand DNA synthesis, most efficiently in cooperation with Rad27 nuclease. Mol. Cell. Biol. 1999;19:8361–71. doi: 10.1128/mcb.19.12.8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bubeck D, Reijns MA, Graham SC, Astell KR, Jones EY, Jackson AP. PCNA directs type 2 RNase H activity on DNA replication and repair substrates. Nucleic Acids Res. 39:3652–66. doi: 10.1093/nar/gkq980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu Y, Grindley ND, Joyce CM. Coordination between the polymerase and 5′-nuclease components of DNA polymerase I of Escherichia coli. J. Biol. Chem. 2000;275:20949–55. doi: 10.1074/jbc.M909135199. [DOI] [PubMed] [Google Scholar]
- 32.Kao HI, Henricksen LA, Liu Y, Bambara RA. Cleavage specificity of Saccharomyces cerevisiae flap endonuclease 1 suggests a double-flap structure as the cellular substrate. J. Biol. Chem. 2002;277:14379–89. doi: 10.1074/jbc.M110662200. [DOI] [PubMed] [Google Scholar]
- 33.Kaiser MW, Lyamicheva N, Ma W, Miller C, Neri B, et al. A comparison of eubacterial and archaeal structure-specific 5′-exonucleases. J. Biol. Chem. 1999;274:21387–94. doi: 10.1074/jbc.274.30.21387. [DOI] [PubMed] [Google Scholar]
- 34.Friedrich-Heineken E, Henneke G, Ferrari E, Hübscher U. The acetylatable lysines of human Fen1 are important for endo- and exonuclease activities. J. Mol. Biol. 2003;328:73–84. doi: 10.1016/s0022-2836(03)00270-5. [DOI] [PubMed] [Google Scholar]
- 35.Storici F, Henneke G, Ferrari E, Gordenin DA, Hübscher U, Resnick MA. The flexible loop of human FEN1 endonuclease is required for flap cleavage during DNA replication and repair. EMBO J. 2002;21:5930–42. doi: 10.1093/emboj/cdf587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ceska TA, Sayers JR, Stier G, Suck D. A helical arch allowing single-stranded DNA to thread through T5 5′-exonuclease. Nature. 1996;382:90–93. doi: 10.1038/382090a0. [DOI] [PubMed] [Google Scholar]
- 37.Chapados BR, Hosfield DJ, Han S, Qiu J, Yelent B, et al. Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell. 2004;116:39–50. doi: 10.1016/s0092-8674(03)01036-5. [DOI] [PubMed] [Google Scholar]
- 38.Bornarth CJ, Ranalli TA, Henricksen LA, Wahl AF, Bambara RA. Effect of flap modifications on human FEN1 cleavage. Biochemistry. 1999;38:13347–54. doi: 10.1021/bi991321u. [DOI] [PubMed] [Google Scholar]
- 39.Xu Y, Potapova O, Leschziner AE, Grindley ND, Joyce CM. Contacts between the 5′ nuclease of DNA polymerase I and its DNA substrate. J. Biol. Chem. 2001;276:30167–77. doi: 10.1074/jbc.M100985200. [DOI] [PubMed] [Google Scholar]
- 40.Gloor JW, Balakrishnan L, Bambara RA. Flap endonuclease 1 mechanism analysis indicates flap base binding prior to threading. J. Biol. Chem. 2010;285:34922–31. doi: 10.1074/jbc.M110.165902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsutakawa SE, Classen S, Chapados BR, Arvai AS, Finger LD, et al. Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell. 2011;145:198–211. doi: 10.1016/j.cell.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stewart JA, Campbell JL, Bambara RA. Dna2 is a structure-specific nuclease, with affinity for 5′-flap intermediates. Nucleic Acids Res. 2010;38:920–30. doi: 10.1093/nar/gkp1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balakrishnan L, Polaczek P, Pokharel S, Campbell JL, Bambara RA. Dna2 exhibits a unique strand end-dependent helicase function. J. Biol. Chem. 2010;285:38861–68. doi: 10.1074/jbc.M110.165191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng L, Zhou M, Chai Q, Parrish J, Xue D, et al. Novel function of the flap endonuclease 1 complex in processing stalled DNA replication forks. EMBO Rep. 2005;6:83–89. doi: 10.1038/sj.embor.7400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng L, Shen B. Okazaki fragment maturation: Nucleases take centre stage. J. Mol. Cell Biol. 2011;3:23–30. doi: 10.1093/jmcb/mjq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng L, Jia J, Finger LD, Guo Z, Zer C, Shen B. Functional regulation of FEN1 nuclease and its link to cancer. Nucleic Acids Res. 2011;39:781–94. doi: 10.1093/nar/gkq884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brosh RM, Jr, von Kobbe C, Sommers JA, Karmakar P, Opresko PL, et al. Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. EMBO J. 2001;20:5791–801. doi: 10.1093/emboj/20.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brosh RM, Jr, Driscoll HC, Dianov GL, Sommers JA. Biochemical characterization of the WRN-FEN-1 functional interaction. Biochemistry. 2002;41:12204–16. doi: 10.1021/bi026031j. [DOI] [PubMed] [Google Scholar]
- 49.Karanja KK, Livingston DM. C-terminal flap endonuclease (rad27) mutations: lethal interactions with a DNA ligase I mutation (cdc9-p) and suppression by proliferating cell nuclear antigen (POL30)in Saccharomyces cerevisiae. Genetics. 2009;183:63–78. doi: 10.1534/genetics.109.103937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sakurai S, Kitano K, Morioka H, Hakoshima T. Crystallization and preliminary crystallographic analysis of the catalytic domain of human flap endonuclease 1 in complex with a nicked DNA product: use of a DPCS kit for efficient protein-DNA complex crystallization. Acta Crystallogr. Sect. F. 2008;64:39–43. doi: 10.1107/S1744309107065372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drinkwater NR, Miller EC, Miller JA. Estimation of apurinic/apyrimidinic sites and phosphotriesters in deoxyribonucleic acid treated with electrophilic carcinogens and mutagens. Biochemistry. 1980;19:5087–92. doi: 10.1021/bi00563a023. [DOI] [PubMed] [Google Scholar]
- 52.Nakamura J, Swenberg JA. Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res. 1999;59:2522–26. [PubMed] [Google Scholar]
- 53.Roberts KP, Sobrino JA, Payton J, Mason LB, Turesky RJ. Determination of apurinic/apyrimidinic lesions in DNA with high-performance liquid chromatography and tandem mass spectrometry. Chem. Res. Toxicol. 2006;19:300–9. doi: 10.1021/tx0502589. [DOI] [PubMed] [Google Scholar]
- 54.Friedberg EC, Aguilera A, Gellert M, Hanawalt PC, Hays JB, et al. DNA repair: from molecular mechanism to human disease. DNA Repair. 2006;5:986–96. doi: 10.1016/j.dnarep.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 55.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 56.Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, et al. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 1996;271:9573–78. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- 57.Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) EMBO J. 1997;16:3341–48. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hazra TK, Hill JW, Izumi T, Mitra S. Multiple DNA glycosylases for repair of 8-oxoguanine and their potential in vivo functions. Prog. Nucleic Acid Res. Mol. Biol. 2001;68:193–205. doi: 10.1016/s0079-6603(01)68100-5. [DOI] [PubMed] [Google Scholar]
- 59.Lindahl T, Andersson A. Rate of chain breakage at apurinic sites in double-stranded deoxyribonucleic acid. Biochemistry. 1972;11:3618–23. doi: 10.1021/bi00769a019. [DOI] [PubMed] [Google Scholar]
- 60.Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–18. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 61.Doetsch PW, Cunningham RP. The enzymology of apurinic/apyrimidinic endonucleases. Mutat. Res. 1990;236:173–201. doi: 10.1016/0921-8777(90)90004-o. [DOI] [PubMed] [Google Scholar]
- 62.Haukanes BI, Doetsch PW, Olsen LC, Huq I, Krokan HE, Helland DE. Damage specific mammalian endonucleases. Basic Life Sci. 1990;53:191–202. doi: 10.1007/978-1-4613-0637-5_15. [DOI] [PubMed] [Google Scholar]
- 63.Matsumoto Y, Kim K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science. 1995;269:699–702. doi: 10.1126/science.7624801. [DOI] [PubMed] [Google Scholar]
- 64.Piersen CE, Prasad R, Wilson SH, Lloyd RS. Evidence for an imino intermediate in the DNA polymerase β deoxyribose phosphate excision reaction. J. Biol. Chem. 1996;271:17811–15. doi: 10.1074/jbc.271.30.17811. [DOI] [PubMed] [Google Scholar]
- 65.Wilson DM, 3rd, Barsky D. The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat. Res. 2001;485:283–307. doi: 10.1016/s0921-8777(01)00063-5. [DOI] [PubMed] [Google Scholar]
- 66.Conway DH, Hasan SK, Simpson ME. Target-controlled propofol requirements at induction of anaesthesia: effect of remifentanil and midazolam. Eur. J. Anaesthesiol. 2002;19:580–84. doi: 10.1017/s0265021502000935. [DOI] [PubMed] [Google Scholar]
- 67.Prasad R, Singhal RK, Srivastava DK, Molina JT, Tomkinson AE, Wilson SH. Specific interaction of DNA polymerase β and DNA ligase I in a multiprotein base excision repair complex from bovine testis. J. Biol. Chem. 1996;271:16000–7. doi: 10.1074/jbc.271.27.16000. [DOI] [PubMed] [Google Scholar]
- 68.Budd ME, Campbell JL. The roles of the eukaryotic DNA polymerases in DNA repair synthesis. Mutat. Res. 1997;384:157–67. doi: 10.1016/s0921-8777(97)00024-4. [DOI] [PubMed] [Google Scholar]
- 69.Prakash S, Sung P, Prakash L. DNA repair genes and proteins of Saccharomyces cerevisiae. Annu. Rev. Genet. 1993;27:33–70. doi: 10.1146/annurev.ge.27.120193.000341. [DOI] [PubMed] [Google Scholar]
- 70.Dianov G, Sedgwick B, Daly G, Olsson M, Lovett S, Lindahl T. Release of 5′-terminal deoxyribose-phosphate residues from incised abasic sites in DNA by the Escherichia coli RecJ protein. Nucleic Acids Res. 1994;22:993–98. doi: 10.1093/nar/22.6.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sattler U, Frit P, Salles B, Calsou P. Long-patch DNA repair synthesis during base excision repair in mammalian cells. EMBO Rep. 2003;4:363–67. doi: 10.1038/sj.embor.embor796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Handt O, Sutherland GR, Richards RI. Fragile sites and minisatellite repeat instability. Mol. Genet. Metab. 2000;70:99–105. doi: 10.1006/mgme.2000.2996. [DOI] [PubMed] [Google Scholar]
- 73.Vergnaud G, Denoeud F. Minisatellites: mutability and genome architecture. Genome Res. 2000;10:899–907. doi: 10.1101/gr.10.7.899. [DOI] [PubMed] [Google Scholar]
- 74.Lopes J, Debrauwère H, Buard J, Nicolas A. Instability of the human minisatellite CEB1 in rad27 and dna2-1 replication-deficient yeast cells. EMBO J. 2002;21:3201–11. doi: 10.1093/emboj/cdf310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat. Rev. Genet. 2010;11:786–99. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Richards RI, Holman K, Kozman H, Kremer E, Lynch M, et al. Fragile X syndrome: genetic localisation by linkage mapping of two microsatellite repeats FRAXAC1 and FRAXAC2 which immediately flank the fragile site. J. Med. Genet. 1991;28:818–23. doi: 10.1136/jmg.28.12.818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Savouret C, Brisson E, Essers J, Kanaar R, Pastink A, et al. CTG repeat instability and size variation timing in DNA repair-deficient mice. EMBO J. 2003;22:2264–73. doi: 10.1093/emboj/cdg202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bjelland S, Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat. Res. 2003;531:37–80. doi: 10.1016/j.mrfmmm.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 79.Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–52. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Spiro C, Pelletier R, Rolfsmeier ML, Dixon MJ, Lahue RS, et al. Inhibition of FEN-1 processing by DNA secondary structure at trinucleotide repeats. Mol. Cell. 1999;4:1079–85. doi: 10.1016/s1097-2765(00)80236-1. [DOI] [PubMed] [Google Scholar]
- 81.Liu Y, Prasad R, Beard WA, Hou EW, Horton JK, et al. Coordination between polymerase β and FEN1 can modulate CAG repeat expansion. J. Biol. Chem. 2009;284:28352–66. doi: 10.1074/jbc.M109.050286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Henricksen LA, Veeraraghavan J, Chafin DR, Bambara RA. DNA ligase I competes with FEN1 to expand repetitive DNA sequences in vitro. J. Biol. Chem. 2002;277:22361–69. doi: 10.1074/jbc.M201765200. [DOI] [PubMed] [Google Scholar]
- 83.Vallur AC, Maizels N. Complementary roles for exonuclease 1 and Flap endonuclease 1 in maintenance of triplet repeats. J. Biol. Chem. 2010;285:28514–19. doi: 10.1074/jbc.M110.132738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yoon SR, Dubeau L, de Young M, Wexler NS, Arnheim N. Huntington disease expansion mutations in humans can occur before meiosis is completed. Proc. Natl. Acad. Sci. USA. 2003;100:8834–38. doi: 10.1073/pnas.1331390100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Viguera E, Canceill D, Ehrlich SD. Replication slippage involves DNA polymerase pausing and dissociation. EMBO J. 2001;20:2587–95. doi: 10.1093/emboj/20.10.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Delagoutte E, Goellner GM, Guo J, Baldacci G, McMurray CT. Single-stranded DNA-binding protein in vitro eliminates the orientation-dependent impediment to polymerase passage on CAG/CTG repeats. J. Biol. Chem. 2008;283:13341–56. doi: 10.1074/jbc.M800153200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pomerantz RT, O'Donnell M. Direct restart of a replication fork stalled by a head-on RNA polymerase. Science. 2010;327:590–92. doi: 10.1126/science.1179595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fouche N, Ozgur S, Roy D, Griffith JD. Replication fork regression in repetitive DNAs. Nucleic Acids Res. 2006;34:6044–50. doi: 10.1093/nar/gkl757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Otto CJ, Almqvist E, Hayden MR, Andrew SE. The “flap” endonuclease gene FEN1 is excluded as a candidate gene implicated in the CAG repeat expansion underlying Huntington disease. Clin. Genet. 2001;59:122–27. doi: 10.1034/j.1399-0004.2001.590210.x. [DOI] [PubMed] [Google Scholar]
- 90.Liu Y, Zhang H, Veeraraghavan J, Bambara RA, Freudenreich CH. Saccharomyces cerevisiae flap endonuclease 1 uses flap equilibration to maintain triplet repeat stability. Mol. Cell. Biol. 2004;24:4049–64. doi: 10.1128/MCB.24.9.4049-4064.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Singh P, Zheng L, Chavez V, Qiu J, Shen B. Concerted action of exonuclease and Gap-dependent endonuclease activities of FEN-1 contributes to the resolution of triplet repeat sequences (CTG)n-and (GAA)n-derived secondary structures formed during maturation of Okazaki fragments. J. Biol. Chem. 2007;282:3465–77. doi: 10.1074/jbc.M606582200. [DOI] [PubMed] [Google Scholar]
- 92.Liu Y, Wilson SH. DNA base excision repair: a mechanism of trinucleotide repeat expansion. Trends Biochem. Sci. 2012;37:162–72. doi: 10.1016/j.tibs.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Manley K, Shirley TL, Flaherty L, Messer A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat. Genet. 1999;23:471–73. doi: 10.1038/70598. [DOI] [PubMed] [Google Scholar]
- 94.Owen BA, Yang Z, Lai M, Gajec M, Badger JD, 2nd, et al. (CAG)n-hairpin DNA binds to Msh2-Msh3 and changes properties of mismatch recognition. Nat. Struct. Mol. Biol. 2005;12:663–70. doi: 10.1038/nsmb965. [DOI] [PubMed] [Google Scholar]
- 95.Kantartzis A, Williams GM, Balakrishnan L, Roberts RL, Surtees JA, Bambara RA. Msh2-Msh3 interferes with Okazaki fragment processing to promote trinucleotide repeat expansions. Cell Rep. 2012;2:216–22. doi: 10.1016/j.celrep.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tom S, Henricksen LA, Bambara RA. Mechanism whereby proliferating cell nuclear antigen stimulates flap endonuclease 1. J. Biol. Chem. 2000;275:10498–505. doi: 10.1074/jbc.275.14.10498. [DOI] [PubMed] [Google Scholar]
- 97.Li X, Li J, Harrington J, Lieber MR, Burgers PM. Lagging strand DNA synthesis at the eukaryotic replication fork involves binding and stimulation of FEN-1 by proliferating cell nuclear antigen. J. Biol. Chem. 1995;270:22109–12. doi: 10.1074/jbc.270.38.22109. [DOI] [PubMed] [Google Scholar]
- 98.Wu X, Li J, Li X, Hsieh CL, Burgers PM, Lieber MR. Processing of branched DNA intermediates by a complex of human FEN-1 and PCNA. Nucleic Acids Res. 1996;24:2036–43. doi: 10.1093/nar/24.11.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Budd ME, Campbell JL. A yeast gene required for DNA replication encodes a protein with homology to DNA helicases. Proc. Natl. Acad. Sci. USA. 1995;92:7642–46. doi: 10.1073/pnas.92.17.7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Budd ME, Campbell JL. A yeast replicative helicase, Dna2 helicase, interacts with yeast FEN-1 nuclease in carrying out its essential function. Mol. Cell. Biol. 1997;17:2136–42. doi: 10.1128/mcb.17.4.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Imamura O, Campbell JL. The human Bloom syndrome gene suppresses the DNA replication and repair defects of yeast dna2 mutants. Proc. Natl. Acad. Sci. USA. 2003;100:8193–98. doi: 10.1073/pnas.1431624100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nazarkina Zh K, Lavrik OI, Khodyreva SN. Flap endonuclease-1 and its role in the processes of DNA metabolism in eucaryotic cells. Mol. Biol. (Mosk) 2008;42:405–21. [PubMed] [Google Scholar]
- 103.Henneke G, Koundrioukoff S, Hübscher U. Phosphorylation of human Fen1 by cyclin-dependent kinase modulates its role in replication fork regulation. Oncogene. 2003;22:4301–13. doi: 10.1038/sj.onc.1206606. [DOI] [PubMed] [Google Scholar]
- 104.Guo Z, Kanjanapangka J, Liu N, Liu S, Liu C, et al. Sequential posttranslational modifications program FEN1 degradation during cell-cycle progression. Mol. Cell. 2012;47:444–56. doi: 10.1016/j.molcel.2012.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hasan S, Stucki M, Hassa PO, Imhof R, Gehrig P, et al. Regulation of human flap endonuclease-1 activity by acetylation through the transcriptional coactivator p300. Mol. Cell. 2001;7:1221–31. doi: 10.1016/s1097-2765(01)00272-6. [DOI] [PubMed] [Google Scholar]
- 106.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 107.Kucherlapati M, Yang K, Kuraguchi M, Zhao J, Lia M, et al. Haploinsufficiency of Flap endonuclease (Fen1) leads to rapid tumor progression. Proc. Natl. Acad. Sci. USA. 2002;99:9924–29. doi: 10.1073/pnas.152321699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Balakrishnan L, Stewart J, Polaczek P, Campbell JL, Bambara RA. Acetylation of Dna2 endonuclease/helicase and flap endonuclease 1 by p300 promotes DNA stability by creating long flap intermediates. J. Biol. Chem. 2010;285:4398–404. doi: 10.1074/jbc.M109.086397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Guo Z, Qian L, Liu R, Dai H, Zhou M, et al. Nucleolar localization and dynamic roles of flap endonuclease 1 in ribosomal DNA replication and damage repair. Mol. Cell. Biol. 2008;28:4310–19. doi: 10.1128/MCB.00200-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Guo Z, Zheng L, Xu H, Dai H, Zhou M, et al. Methylation of FEN1 suppresses nearby phosphorylation and facilitates PCNA binding. Nat. Chem. Biol. 2010;6:766–73. doi: 10.1038/nchembio.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zheng L, Jia J, Finger LD, Guo Z, Zer C, Shen B. Functional regulation of FEN1 nuclease and its link to cancer. Nucleic Acids Res. 2011;39:781–94. doi: 10.1093/nar/gkq884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu P, Qian L, Sung JS, de Souza-Pinto NC, Zheng L, et al. Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria. Mol. Cell. Biol. 2008;28:4975–87. doi: 10.1128/MCB.00457-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Szczesny B, Tann AW, Longley MJ, Copeland WC, Mitra S. Long patch base excision repair in mammalian mitochondrial genomes. J. Biol. Chem. 2008;283:26349–56. doi: 10.1074/jbc.M803491200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kalifa L, Beutner G, Phadnis N, Sheu SS, Sia EA. Evidence for a role of FEN1 in maintaining mitochondrial DNA integrity. DNA Repair. 2009;8:1242–49. doi: 10.1016/j.dnarep.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Parenteau J, Wellinger RJ. Accumulation of single-stranded DNA and destabilization of telomeric repeats in yeast mutant strains carrying a deletion of RAD27. Mol. Cell. Biol. 1999;19:4143–52. doi: 10.1128/mcb.19.6.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Parenteau J, Wellinger RJ. Differential processing of leading- and lagging-strand ends at Saccharomyces cerevisiae telomeres revealed by the absence of Rad27p nuclease. Genetics. 2002;162:1583–94. doi: 10.1093/genetics/162.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Verdun RE, Karlseder J. The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell. 2006;127:709–20. doi: 10.1016/j.cell.2006.09.034. [DOI] [PubMed] [Google Scholar]
- 118.Muftuoglu M, Wong HK, Imam SZ, Wilson DM, 3rd, Bohr VA, Opresko PL. Telomere repeat binding factor 2 interacts with base excision repair proteins and stimulates DNA synthesis by DNA polymerase β. Cancer Res. 2006;66:113–24. doi: 10.1158/0008-5472.CAN-05-2742. [DOI] [PubMed] [Google Scholar]
- 119.Sampathi S, Bhusari A, Shen B, Chai W. Human flap endonuclease I is in complex with telomerase and is required for telomerase-mediated telomere maintenance. J. Biol. Chem. 2009;284:3682–90. doi: 10.1074/jbc.M805362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Saharia A, Guittat L, Crocker S, Lim A, Steffen M, et al. Flap endonuclease 1 contributes to telomere stability. Curr. Biol. 2008;18:496–500. doi: 10.1016/j.cub.2008.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sharma S, Sommers JA, Gary RK, Friedrich-Heineken E, Hübscher U, Brosh RM., Jr. The interaction site of Flap Endonuclease-1 with WRN helicase suggests a coordination of WRN and PCNA. Nucleic Acids Res. 2005;33:6769–81. doi: 10.1093/nar/gki1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vallur AC, Maizels N. Distinct activities of exonuclease 1 and flap endonuclease 1 at telomeric g4 DNA. PLoS ONE. 2010;5:e8908. doi: 10.1371/journal.pone.0008908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kang HY, Choi E, Bae SH, Lee KH, Gim BS, et al. Genetic analyses of Schizosaccharomyces pombe dna2+ reveal that Dna2 plays an essential role in Okazaki fragment metabolism. Genetics. 2000;155:1055–67. doi: 10.1093/genetics/155.3.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu L, Zhou C, Zhou L, Peng L, Li D, et al. Functional FEN1 genetic variants contribute to risk of hepatocellular carcinoma, esophageal cancer, gastric cancer and colorectal cancer. Carcinogenesis. 2011;33:119–23. doi: 10.1093/carcin/bgr250. [DOI] [PubMed] [Google Scholar]
- 125.Yang M, Guo H, Wu C, He Y, Yu D, et al. Functional FEN1 polymorphisms are associated with DNA damage levels and lung cancer risk. Hum. Mutat. 2009;30:1320–28. doi: 10.1002/humu.21060. [DOI] [PubMed] [Google Scholar]
- 126.Zheng L, Dai H, Zhou M, Li M, Singh P, et al. Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nat. Med. 2007;13:812–19. doi: 10.1038/nm1599. [DOI] [PubMed] [Google Scholar]
- 127.Larsen E, Kleppa L, Meza TJ, Meza-Zepeda LA, Rada C, et al. Early-onset lymphoma and extensive embryonic apoptosis in two domain-specific Fen1 mice mutants. Cancer Res. 2008;68:4571–79. doi: 10.1158/0008-5472.CAN-08-0168. [DOI] [PubMed] [Google Scholar]
- 128.Warbrick E, Coates PJ, Hall PA. Fen1 expression: a novel marker for cell proliferation. J. Pathol. 1998;186:319–24. doi: 10.1002/(SICI)1096-9896(1998110)186:3<319::AID-PATH184>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 129.Kim IS, Lee MY, Lee IH, Shin SL, Lee SY. Gene expression of flap endonuclease-1 during cell proliferation and differentiation. Biochim. Biophys. Acta. 2000;1496:333–40. doi: 10.1016/s0167-4889(00)00029-x. [DOI] [PubMed] [Google Scholar]
- 130.Singh P, Yang M, Dai H, Yu D, Huang Q, et al. Overexpression and hypomethylation of flap endonuclease 1 gene in breast and other cancers. Mol. Cancer Res. 2008;6:1710–17. doi: 10.1158/1541-7786.MCR-08-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Iacobuzio-Donahue CAAR, Maitra A, Adsay NV, Shen-Ong GL, Berg K, et al. Highly expressed genes in pancreatic ductal adenocarcinomas: a comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Res. 2003;63:8614–22. [PubMed] [Google Scholar]
- 132.Sato M, Girard L, Sekine I, Sunaga N, Ramirez RD, et al. Increased expression and no mutation of the Flap endonuclease (FEN1) gene in human lung cancer. Oncogene. 2003;22:7243–46. doi: 10.1038/sj.onc.1206977. [DOI] [PubMed] [Google Scholar]
- 133.Kim JM, Sohn HY, Yoon SY, Oh JH, Yang JO, et al. Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells. Clin. Cancer Res. 2005;11:473–82. [PubMed] [Google Scholar]
- 134.Nikolova T, Christmann M, Kaina B. FEN1 is overexpressed in testis, lung and brain tumors. Anticancer Res. 2009;29:2453–59. [PubMed] [Google Scholar]
- 135.Christmann M, Tomicic MT, Origer J, Kaina B. Fen1 is induced p53 dependently and involved in the recovery from UV-light-induced replication inhibition. Oncogene. 2005;24:8304–13. doi: 10.1038/sj.onc.1208994. [DOI] [PubMed] [Google Scholar]
- 136.Johansson VM, Miniotis MF, Hegardt C, Jonsson G, Staaf J, et al. Effect of polyamine deficiency on proteins involved in Okazaki fragment maturation. Cell Biol. Int. 2008;32:1467–77. doi: 10.1016/j.cellbi.2008.08.018. [DOI] [PubMed] [Google Scholar]
- 137.Larsen E, Gran C, Saether BE, Seeberg E, Klungland A. Proliferation failure and gamma radiation sensitivity of Fen1 null mutant mice at the blastocyst stage. Mol. Cell. Biol. 2003;23:5346–53. doi: 10.1128/MCB.23.15.5346-5353.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shen B, Nolan JP, Sklar LA, Park MS. Functional analysis of point mutations in human flap endonuclease-1 active site. Nucleic Acids Res. 1997;25:3332–38. doi: 10.1093/nar/25.16.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]