

Abstract
We tested independent and interactive contributions of a recently noted and promising Insulin Degrading Enzyme polymorphism (IDE, rs6583817) and Type 2 diabetes (T2D) to executive function performance, both concurrently and longitudinally. Regarding normal neurocognitive decline and Alzheimer’s disease (AD), T2D is a known risk factor and this IDE variant may contribute risk or risk reduction through the minor (A) or major (G) allele.
We compared normal aging and T2D groups (baseline n=574, ages 53–95) over two longitudinal waves (M interval=4.4 years). We used confirmatory factor analysis, latent growth curve modeling, and path analysis.
A confirmed single-factor model of four executive function tasks established the cognitive phenotype. This IDE variant predicted both concurrent group differences and differential change in cognitive performance. Furthermore, the IDE major allele reduced risk of cognitive decline. T2D predicted performance only concurrently.
Both IDE and T2D are associated with executive function levels in older adults, but only IDE moderated two-wave change. Previously linked to AD, this IDE variant should be further explored for its potential influence on cognitive phenotypes of normal aging.
Keywords: Aging, Executive Functions, Insulin Degrading Enzyme, Type 2 Diabetes, Victoria Longitudinal Study
1. Introduction
Increasingly, mechanisms associated with neurocognitive phenotypes of normal aging, preclinical (impaired) aging, and neurodegenerative diseases are understood as exerting influence either independently or interactively (e.g., Lindenberger et al., 2008). These mechanisms include both risk-elevating and risk-reducing influences that range across potentially modifying domains such as neurobiological (e.g., genetic), bio-health (e.g., metabolic conditions), and environmental (e.g., lifestyle activities) (e.g., Nagel et al., 2008; Raz et al., 2008; Raz et al., 2011). We examine independent and interactive associations of two factors receiving growing attention for their influence on normal cognitive aging and Alzheimer’s disease (AD). Specifically, we test associations of a recently noted Insulin Degrading Enzyme polymorphism (IDE rs6583817) and Type 2 diabetes (T2D) on both level and actual two-wave change for a latent variable of executive function in a large sample of older adults spanning 40 years of aging. Among other interesting IDE polymorphisms, this IDE variant has been identified as carrying the strongest association with AD (Carrasquillo et al., 2010) and may be particularly promising as a marker of normal or preclinical neurocognitive changes. Correspondingly, T2D has been linked with AD risk and accelerated neurocognitive deficits (e.g., EF) in normal aging, and genetic susceptibility, but further research on genetic influences on level and (especially) longitudinal change in associated cognitive decline is required (Seaquist et al., 2012). We assembled a 2-wave longitudinal data set from the Victoria Longitudinal Study (VLS) and performed structural and latent growth curve analyses to distinguish potential patterns, influences, and interactions among the neurobiological (IDE variant) and metabolic-health (T2D) factors on EF level and 4-year change.
Executive functions (EF) are involved in monitoring, organizing, and regulating complex cognitive operations, especially those requiring planning, problem solving, and goal directed components (West, 1996). As linked to aging-related changes in prefrontal cortex, EF performance declines with aging (Turner and Spreng, 2012). Such decrements may (a) be observed in terms of both level and structure (dimensionality) of EFs, (b) create difficulties for cognitive performance, (c) be affected by risk (or protection) factors from biological (e.g., genetic), neurobiological (e.g., dopaminergic), health (e.g., diabetes), and environmental (e.g., lifestyle) domains, (d) predict mild cognitive impairment and sporadic AD, and (e) be exacerbated or moderated by combinations exerting increasing influence with aging (e.g., de Frias et al., 2006; Grober et al., 2008; Lindenberger et al., 2008; Luszcz, 2011; Nathan et al., 2001; Rapp and Reischies, 2005; Wishart et al., 2011; Yeung et al., 2009). In addition to gradual changes in level of performance, the structure of EF varies systematically across the lifespan. The observed pattern includes unitary (single-factor) models for children (Wiebe et al., 2008; Wiebe et al., 2010), differentiated three-factor models for prefrontal mature young adults (Miyake et al., 2000), and de-differentiated single-factor models for typical aging adults (e.g., Adrover-Roig et al., 2012; de Frias et al., 2006; but see exception reported by de Frias et al., 2009, for older adults with sustained cognitive and brain health). Current emphases in EF and aging research call for studying longitudinal trajectories as potentially modified by neurobiological, metabolic, and health covariates (Luszcz, 2011).
Accordingly, we identified two biological and health factors theoretically related to the EF cognitive phenotype. First, genetic involvement in individual differences in executive functioning is apparent and complex (Friedman et al., 2008; Kremen et al., 2009). We selected and tested the IDE gene because several variants of this gene have been linked to risk of developing both T2D and AD (Bartl et al., 2011). IDE has the function of degrading hormones and bioactive peptides. It was first recognized as the most important proteolytic enzyme for insulin and has since been identified in the processing of other glycemia regulating peptides (amylin and glucagon; Bennett et al., 2000; Shen et al., 2006) and of a 4 kDa neuropeptide product of the human precursor protein, Amyloid Beta (Aβ; Kurochkin and Goto, 1994). In the genetics realm, the IDE locus has demonstrated a linkage peak for both T2D and late onset Alzheimer’s disease (Bartl et al., 2011; Carrasquillo et al., 2010; Farris et al., 2003; Grarup et al., 2007). Although IDE has also been associated with an increased risk of T2D, specific polymorphisms have not as yet been clearly identified and may in fact involve several SNPs located in the IDE area (Bartl et al., 2011; Duggirala et al., 1999; Karamohamed et al., 2003; Rudovich et al., 2009; Vionnet et al., 2000; Zeggini et al., 2008). IDE risk alleles are associated with lower capacity to degrade insulin possibly resulting in insulin resistance (Bartl et al., 2011), which could in turn lead to the compensating hyperinsulinema associated with T2D and implicated cognitive deficits (Awad et al., 2004; Umegaki, 2012). A different IDE haplotype has been associated with decreased risk of T2D, suggesting an increase in insulin degradation (Kwak et al., 2008). In fact, the diversion of the limited supply of IDE to the degradation of insulin may be linked to increased Aβ levels (Qiu and Folstein, 2006), which is a hallmark of AD (Blomqvist et al., 2005; Kurochkin and Goto, 1994).
Arguably compatible with the observed associations of IDE with Aβ degradation and its location on chromosome 10q (a chromosomal region consistently linked to late-onset AD; Lendon and Craddock, 2001), some IDE polymorphisms are risk factors for neurodegeneration in the form of AD, associations with MCI, or even normal cognitive decline (Bertram et al., 2000; Björk et al., 2007; Ertekin-Taner et al., 2004; Wang et al., 2012; see also Abraham et al., 2001; Boussaha et al., 2002; Vardy et al., 2012). However, some variants of IDE (specifically, rs6583817, rs5786996, and rs4646953) are associated with increased levels of IDE and decreased levels of Aβ, suggesting the possibility of lowered risk for AD (Bartl et al., 2011; Belbin et al., 2011). Conceivably—but not previously studied—differences in risk of AD outcome status may be preceded by differences in risk of level and change in cognitive decline. Carrasquillo and colleagues (2010) reported four IDE variants with significant relationships to IDE transcription. The present variant, IDE rs6583817, had the highest association. Notably, the minor allele for this variant correlated with elevated IDE expression, reduced Aβ levels, and reduced risk of AD status. Whereas the relationship of IDE to sporadic AD status has been explored, little research has examined associations of IDE polymorphisms on neurocognitive performance (Okereke et al., 2009; see also Vardy et al., 2012), and none for this particular and promising IDE variant. We selected this IDE polymorphism to examine whether the minor allele (A) or the major allele (G) would be associated with normal aging-related preservation of (a) performance level of the EF phenotype and (b) trajectory of 4-year change in a large sample of older adults.
For the second factor we selected T2D, which has been linked to increased risk of AD (Arvanitakis et al., 2004; Profenno et al., 2010; Qiu and Folstein, 2006) and changes in the non-AD brain (e.g., exacerbated insulin dysregulation, disrupted Aβ clearance) that are associated with decrements in neurocognitive performance (Awad et al., 2004; Cholerton et al., 2011; Nilsson, 2006; Okereke et al., 2009; Seaquist et al., 2012; Strachan et al., 2011). For example, older adults with T2D have exhibited lower performance on speed-related EF shifting and inhibition tasks in both cross sectional and short-term longitudinal studies (Biessels et al., 2008; Fischer et al., 2009; Yeung et al., 2009). These decrements have been linked to mediators from functional, vascular, genetic, and other biological domains (McFall et al., 2010; Strachan et al., 2011). Regarding AD, insulin resistance (hyperinsulinemia) reduces the effectiveness with which IDE degrades Aβ oligomers in the brain, a link to the neuropathogenesis of AD. When regulated in normal aging, cerebral insulin performs multiple cognitive-supportive functions, such as increasing neurotransmitter levels, enhancing glucose utilization, and promoting lipid metabolism. However, when the aging brain experiences chronic dysregulation (or reduced levels) of insulin uptake, cognitive deficits may be exacerbated to levels that are consistent with those of clinical impaired patients (Cholerton et al., 2011). Insulin receptors are distributed in brain regions such as the hippocampus and frontal lobe, with the latter opening the possibility that EF may be a target cognitive phenotype for research integrating T2D, IDE polymorphisms, and human aging.
The aim of this study is to examine potential independent and interactive contributions of selected genetic (IDE rs6583817) and metabolic-health (T2D) markers to concurrent performance and longitudinal change in executive functioning among a large sample of older adults. We test the relationship with sensitive statistical techniques as implemented concurrently and across two longitudinal waves. Notably, the approach includes a unique combination of a risk factor (T2D) and a potential risk-reduction factor (IDE) theoretically related to executive functioning, an important cognitive phenotype of aging. We pursue four specific research goals. First, using structural equation modeling in the context of a large sample of older adults, we estimate a latent variable model for EF using four manifest measures related to two potential EF dimensions. Second, using confirmatory factor analysis we test for longitudinal measurement invariance. Third, we used two-wave longitudinal data over a 40-year age band of aging (ages 53–95) to estimate latent growth mixture models examining the interindividual differences in intraindividual change in EF associated (independently) with T2D and IDE. Fourth, we used path analyses to explore the potentially interactive effects of T2D and IDE on EF change. Based on previous reports, we hypothesized an interaction between the two factors in which the IDE rs6583817 would moderate the harmful effects of T2D on EF performance and change.
2. Method
2.1 Participants
Data were assembled from the Victoria Longitudinal Study (VLS), a large-scale study of biomedical, genetic, health, cognitive, and neuropsychological aspects of aging (see Dixon and de Frias, 2004, for methodological details). The VLS and all present data collection procedures are in full and certified compliance with prevailing human research ethics guidelines and boards. Written informed consent was obtained from all participants. Using standard procedures (e.g., Dixon et al., 2012; Small et al., 2011), we assembled a two-wave longitudinal data set. We began with a Wave 1 (W1) data set of n = 694 adults (ages 53–100 years), all with genetic data. We then applied T2D inclusion and participant exclusionary criteria, targeting conditions that could modify EF performance independent of the factors modeled in this study. We excluded n=119 participants with unconfirmed T2D status, MMSE scores<26, reported severe health conditions with cognitive implications (i.e., high blood pressure), moderate or severe stroke, anti-psychotic medication, and incomplete EF data. Not excluded were participants on anti-hypertension medications. An additional participant (n=1) provided data only for W2. From the remainder we applied the standard VLS multi-level diagnostic regimen for classifying T2D (see McFall et al., 2010; Yeung et al., 2009). Specifically, inclusion into the T2D group required all of the following conditions: (a) W1 self-report of T2D diagnosis, (b) W1 specified method of treatment (i.e., oral medication, insulin, diet and exercise, no control), (c) W1 objective evidence of reported medication, and (d) W2 validation of T2D status (repeating the three previous steps).
The final W1 sample included n =574 adults (M age = 70.1, SD = 8.54, range = 53.2 – 95.2 years; n = 384 [66.9%] women). The diagnostic procedure identified n = 46 [8.0%] adults with T2D (M age = 71.4, SD = 7.97, range = 55.4 – 90.5, n = 24 [52.2%] women). The remainder constituted the normal aging group of n = 528 (M age = 70.0, SD = 8.59, range = 53.2 – 95.2 years; n = 360 [68.2%] women). At W2 n = 474 of W1 adults were available (M age = 74.3, SD = 8.46, range = 57.3 – 94.5 years, n = 316 [66.7%] women). This included n=101 non-returning adults (M age at W1 = 71.3, SD = 8.74, range = 55.05 – 95.2 years, n = 69 [68.3%] women, and n=1 adult without W1 data. Returning participants were similar to non-returning participants in age, education, and gender distribution. In addition, genotype percentages within the three allelic combinations were quite similar (returnees/non-returnees): AA (13.5/14.9%), AG (64.6/67.3%), GG (21.9/17.8%) and T2D proportions were almost identical (7.8/8.9%). The standard T2D diagnostic procedure identified n = 37 (7.8%) adults with T2D (M age = 76.0, SD = 7.76, range = 60.5 – 91.1 years; n = 21 [56.8%] women). The remainder constituted the W2 normal aging group of n = 437 (M age = 74.2, SD = 8.51, range = 57.3 – 94.5 years; n = 295 [67.5%] women). Table 1 presents additional demographic data organized by genetic allelic combination and wave. The 4-year retention rate was about 80% with equivalent ratios of the two groups at both waves. The longitudinal statistical models do not implement listwise deletion, so all participants contributed data at either one or both waves.
Table 1.
Descriptive Statistics for Sample by IDE genotype and Longitudinal Wave
| IDE genotype | ||||
|---|---|---|---|---|
|
| ||||
| G+ (G/G & G/A) | G− (A/A) | |||
|
| ||||
| W1 | W2 | W1 | W2 | |
|
|
||||
| na | 495 (122 & 373) | 410 (104 & 306) | 79 | 64 |
| Age M(SD) | 69.8 (8.48) | 73.9 (8.36) | 72.3 (8.65) | 76.9 (8.75) |
| Range | 53.2 – 95.2 | 57.3 – 94.1 | 54.6 – 89.3 | 58.9 – 94.5 |
| Gender (% women) | 67.7 | 67.8 | 62.0 | 59.4 |
| T2D (% with T2D) | 7.9 | 7.8 | 8.9 | 7.8 |
| Hayling M(SD) | 5.62 (1.42) | 5.49 (1.49) | 5.47 (1.42) | 5.23 (1.40) |
| Stroopb M(SD) | 1.25 (.706) | 1.31 (.910) | 1.41 (.828) | 1.44 (.892) |
| Brixton M(SD) | 4.96 (2.13) | 5.42 (2.00) | 4.56 (2.17) | 4.98 (2.12) |
| Color Trailsb M(SD) | 91.9 (28.9) | 99.1 (38.6) | 99.3 (35.0) | 107.1 (41.7) |
| EF factor scores M(SD) | .008 (.805) | .051 (1.20) | −.244 (.873) | −.331 (1.38) |
Note. Hardy-Weinberg equilibrium χ2 = 54.09 at W1, therefore the genotypic distribution for IDE is not in Hardy-Weinberg equilibrium. W1 = Wave 1. W2 = Wave 2.
For G+ n is for total G (G/G & G/A).
Lower scores indicate better performance.
2.2 Executive Function Measures
2.2.1 Hayling sentence completion test
This task, which indexed inhibition (Burgess and Shallice, 1997), consisted of two sets of 15 sentences, each having the last word missing. Section A required completing the sentence quickly, and measured initiation speed. Section B required completing the sentence with an unconnected word quickly, and measured response suppression. Response speed on both sections and errors on Section 2 were used to derive an overall scaled score for each participant on a scale ranging from 1 (impaired) to 10 (very superior).
2.2.2 Stroop test
This task taps inhibitory processes by requiring the respondent to ignore the automatic response of reading a printed word and to instead name the color of ink in which the word is printed (Taylor et al., 1997). The performance score was the interference index and reflected slowing in response to interference in Part C ([Part Ctime − Part Atime]/Part Atime). Lower scores indicated better performance.
2.2.3 Brixton spatial anticipation test
This task (Burgess and Shallice, 1997) was a rule-attainment (or shifting) task based on the Wisconsin Card Sorting Task (Berg, 1948). Participants are required to deduce simple and changing patterns, measuring their ability to abstract logical rules (Andrés and Van der Linden, 2000). The total errors were recorded and these errors (maximum 54) were converted to scaled scores. An overall standardized scaled score based on a scale ranging from 1 (impaired) to 10 (very superior) was used for analysis.
2.2.4 Color trails test (Part 2 CTT-2)
Indexing shifting, the CTT (D’Elia et al., 1996) was similar to the Trail Making Test (Reitan and Wolfson, 1992) but minimized the influence of language. Part 2 required participants to connect numbers from 1 to 25 alternating between pink and yellow circles and disregarding the numbers in circles of the alternate color. The latency score for Part 2 was used for analysis. Lower scores indicate better performance.
2.3 DNA Extraction and IDE Genotyping
Saliva was collected according to standard procedures from Oragene DNA Genotek and stored at room temperature in the Oragene® disks until DNA extraction. DNA was manually extracted using the manufacturer’s protocol and quantified using a NanoDrop® ND-1000 Spectrophotometer (Wilmington, DE). Genotyping was carried out by using a PCR-RFLP strategy to analyze the allele status for IDE (rs6583817). Briefly, SNP-containing PCR fragments were amplified from 25 ng of genomic DNA using specific primers (Fwd: 5′-AATATATGGGCAAATATTAAGTGCAC-3′; Rev: CAGTTGTGGGAATATATT CCTGAG-3′). Reactions were setup in 96-well plates using the QIAgility robotic system (QIAgen). RFLP analysis was performed on a high resolution DNA screening cartridge on a QIAxcel capillary electrophoresis system (QIAgen) using the protocol OL700 after digestion of the PCR amplicons with the restriction enzymes DdeI (NE Biolabs) for 4 hours at 37°C. The analysis was confirmed upon migration of the restriction fragments on 10 or 15% acrylamide gels for the SNP.
For genetic analyses the IDE genotypes were categorized by the presence of an A allele (A+ = A/A, homozygous minor allele, and G/A, heterozygous allele) or the absence of an A allele (A− = G/G, homozygous major allele). No effect on EF performance was observed; therefore, the alternative configuration (presence or absence of an G allele) was used for analyses. IDE genotypes were categorized by the presence of a G allele (G+ = G/G, homozygous major allele, and G/A, heterozygous allele) or the absence of a G allele (G− = A/A, homozygous minor allele).
2.4 Statistical Analyses
Statistical model fit for all analyses was determined using standard indexes: (a) χ2 for which a good fit would produce a non-significant test (p > .05) indicating that the data are not significantly different from the estimates associated with the model, (b) the comparative fit index (CFI) for which fit is judged by a value of ≥ .95 as good and ≥ .90 as adequate, (c) root mean square error of approximation (RMSEA) for which fit is judged by a value of ≤ .05 as good and ≤ .08 as adequate, and (d) standardized root mean square residual (SRMR) for which fit is judged by a value of ≤ .08 as good (Kline, 2011).
2.4.1 Analyses for Research Goal 1 (EF Latent Model) and Research Goal 2 (Invariance Testing Across Two Waves)
First, we used Mplus 6 (Muthén and Muthén, 2010) to conduct confirmatory factor analysis. We tested two models (a) a single factor model and (b) a 2-factor model consisting of inhibition (Hayling, Stroop) and shifting (Brixton, CTT). Second, we tested longitudinal (two-wave) measurement invariance including (a) configural invariance, the same indicator variables load onto the latent variable used to test the model across time, (b) metric invariance, factor loadings are constrained to be equal for each latent variable indicating that the latent variable is measuring the same construct, (c) scalar invariance, indicator intercepts are constrained to be equal allowing mean differences to be evident at the latent mean level, and (d) residual invariance, indicator residuals are constrained to be equal accounting for error variability and thus group differences are based on their common variability. We estimated factor scores for EF in Mplus and used these in subsequent latent growth models.
2.4.2 Analyses for Research Goal 3 (EF Latent Growth Models) and Research Goal 4 (Path Analyses with IDE and T2D)
We coded age as a continuous factor and computed latent growth models with individually-varying ages. We centered the age variable at 75, an inflection point for many cognitive domains (Small et al., 2011). To identify the functional form of change, we determined the best-fitting unconditional growth model by testing in sequence (a) a fixed intercept model, which assumes no inter- or intraindividual variation, (b) a random intercept model, which models interindividual variation but no intraindividual change, (c) a random intercept fixed slope model, which allows interindividual variation in initial performance but assumes all individuals change at the same rate, and (d) a random intercept random slope model, which models interindividual variation in both initial performance and change over time (Singer and Willett, 2003). After the best unconditional growth model was determined, predictors of change were examined by regressing intercept and slope separately on IDE genotype (Model 1) and T2D status (Model 2). Next, we added intercept and slope regression pathways for IDE genotype, T2D status, and IDE genotype xT2D status to test our moderation hypothesis (Model 3).
3. Results
3.1 Research Goal 1 (EF Latent Model) and Research Goal 2 (Invariance Testing Across Two Waves)
We performed confirmatory factor analyses for EF. Regarding research goal 1, the one-factor EF model fit the data well for both W1 and W2. In contrast, the two-factor model could not be estimated at either wave, resulting in the absence of a positive definite variance-covariance matrix (see Table 2 for model goodness of fit indexes). Therefore, as observed in earlier research, we accepted the single-factor model for normal older adults. Regarding research goal 2, we conducted invariance testing on the single-factor model. The model holding indicator factor loadings equal across W1 and W2 fit the data well, thus indicating metric invariance. Fixing intercepts to be equal across time resulted in significantly poorer fit to the data according to the χ2 difference test, although the other fit indexes were adequate. We conducted tests of partial scalar invariance by freeing intercepts for each indicator in turn. These analyses supported a model with intercepts fixed to be equal across time for inhibition (Stroop and Hayling) but not shifting (Color Trails and Brixton) tasks. Overall, we observed metric invariance for the single-factor EF model, but only partial scalar invariance. This result showed that the model measured the same EF construct across time, but that the manifest variables marking EF shifting exhibited mean differences across time outside of the latent differences. Latent variable reliability was indicated in three ways: (a) significant factor loadings for all four manifest variables at each wave of data (range = .31 to .75), (b) metric invariance across the two waves of data (χ2 = 23.97, df = 20, p = .244; RMSEA = .019 (.000–.042); CFI = .995; SRMR = .029, and (c) adequate bivariate correlations across indicator variables for the two waves (range r = 0.4 to 0.7).
Table 2.
Goodness of Fit Indexes for Executive Function Confirmatory Factor Analysis Models and Measurement Invariance Testing
| AIC | BIC | χ2 | df | p | RMSEA | CFI | SRMR | |
|---|---|---|---|---|---|---|---|---|
| Model | ||||||||
| One factor EF (W1) | 5866.575 | 5914.454 | 3.529 | 3 | .309 | .019 (.000–.075) | .995 | .023 |
| Two factor EFa (W1) | Residual covariance matrix not positive definite. | |||||||
| One factor EF (W2) | 5214.349 | 5260.123 | 1.756 | 3 | .6249 | .000 (.000–.063) | 1.00 | .028 |
| Two factor EFa (W2) | Residual covariance matrix not positive definite. | |||||||
| One factor EF (W1 and W2) | 10505.045 | 10626.967 | 11.821 | 16 | .7562 | .000 (.000–.028) | 1.00 | .019 |
| Equal indicator loadings | 10504.623 | 10613.482 | 17.398 | 19 | .5629 | .000 (.000–.033) | 1.00 | .027 |
| Equal intercepts | 10563.543 | 10659.339 | 82.318 | 22 | <.001 | .069 (.054–.085) | .931 | .054 |
| Equal intercepts STRP & HAY | 10509.190 | 10613.695 | 23.966 | 20 | .2439 | .019 (.000–.042) | .995 | .029 |
Note. AIC = Akaike information criteria. BIC = Bayesian information criteria. RMSEA = Root Mean Square Error of Approximation. CFI = Comparative Fit Index. SRMR = Standardized Root Mean Square Residual. EF = Executive Function. W1 = Wave 1. W2 = Wave 2. STRP = Stroop. HAY = Hayling.
3.2 Research Goal 3 (EF Latent Growth Model) and Research Goal 4 (Path Analyses with IDE and T2D)
We performed latent growth modeling using estimated EF factor scores. The best fitting unconditional growth model for EF was established as a random intercept, random slope latent growth model (see Table 3 for model goodness of fit indexes). Next, building on this model of change over time, three additional models were tested to determine if IDE genotype or T2D status predicted EF initial performance or change. Finally, we tested a moderation model to examine if IDE genotype mitigates the negative effects of T2D status.
Table 3.
Goodness of Fit Indexes for Executive Function Latent Growth Models
| Model | H0 value | −2LL | Parameters Free | AIC | BIC | Intercept | Slope | ||
|---|---|---|---|---|---|---|---|---|---|
| M | S | M | S | ||||||
| Fixed intercept | −1470.253 | 2940.506 | 3 | 2946.506 | 2959.57 | .022 | - | - | - |
| Random intercept | −971.301 | 1942.602 | 4 | 1950.601 | 1968.019 | −.028 | .668** | - | - |
| Random intercept Fixed slope | −905.567 | 1811.134 | 5 | 1821.134 | 1842.906 | −.199** | .473** | −.036** | - |
| Random intercept Random slope | −722.631 | 1445.262 | 7 | 1459.262 | 1489.742 | .014 | .785** | −.022** | .002** |
| b* | b* | ||||||||
| Model 1 (A+/A−)a | −722.244 | 1444.488 | 9 | 1462.489 | 1501.678 | −.062 | −.001 | ||
| Model 1 (G+/G−)b | −719.114 | 1438.228 | 9 | 1456.228 | 1495.417 | .306** | .018* | ||
| Model 2c | −716.752 | 1433.504 | 9 | 1451.503 | 1490.693 | −.325* | −.003 | ||
| Model 3d | |||||||||
| IDE, T2D | −713.260 | 1426.520 | 11 | 1448.521 | 1496.419 | .302*/−.326* | .018*/−.003 | ||
| IDE, T2D, & IDE × T2D | −712.257 | 1424.514 | 13 | 1450.514 | 1507.120 | .305*/−.366/.040 | .016/−.025/.026 | ||
Note. H0 = Loglikelihood value; −2LL = −2 (H0) = −2 log likelihood; AIC = Akaike information criterion; BIC = Bayesian information criterion; M = Mean; S = Sample variance; b* reported as IDE/T2D and IDE/T2D/IDE × T2D.
Model 1 = EF intercept and slope regressed on IDE testing the A allele was non-significant and was dropped from analyses.
Model 1 testing the G allele was retained for all further analyses.
Model 2 = EF intercept and slope regressed on T2D.
Model 3 = EF intercept and slope regressed on IDE and T2D and then EF intercept and slope regressed on IDE, T2D, and IDE × T2D. Model 3 had an intercept-slope correlation of .037**.
p < .05.
p < .01.
As shown in Table 3, Model 1 was used to test if IDE predicted EF initial performance (at the age 75 centering point) or change. The intercept and slope of EF was regressed on IDE genotype. We first tested the presence of the IDE minor allele (A+ or A−), which produced no difference in initial performance of EF (Ms = .063 and .001 respectively, p > .05) or rate of EF decline (Ms = −.021 and −.022 respectively, p > .05). However, the IDE major allele (G+ or G−) predicted performance of EF (see Figure 1). Specifically, at the stipulated intercept (age 75) adults with a G allele (the G+ group) performed significantly better (M = .058) than those without a G allele (G− group; M = −.248). In addition, as shown in Figure 1, IDE genotype predicted linear change in EF performances. Specifically, adults with a G allele exhibited significantly less decline (M = −.019) in EF performance than those without a G allele (M = −.037). Furthermore, a significant dose-response effect for EF performance level at age 75 years was observed. Participants with A/A performed the poorest (M = −.133), those with A/G performed better (M = .005), and those with G/G performed best (M = .143, p = .043). There was no dosage affect for EF change (p > .05).
Figure 1.
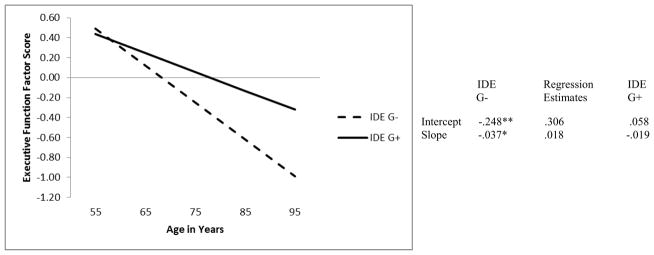
Predicted growth curve for executive function factor scores using IDE genotype (i.e., G− = no G allele, G+ = at least one G allele) as a predictor with age as a continuous variable centered at 75 years. *p < .05. **p <.01.
Model 2 tested if T2D predicted EF initial performance or change. The intercept and slope of EF was regressed on T2D status. T2D status predicted initial performance of EF (Figure 2). Specifically, adults with T2D performed significantly worse (M = −.285) than adults without T2D (M =.040) (see age 75 model intercept in the figure). However, adults with and without T2D showed no differences in the rate of EF decline (Ms = −.024 and −.021, respectively; p > .05). Model 3 tested whether the effect of T2D on EF is lessened in individuals with at least one G IDE allele. First, EF intercept and slope were each regressed on both IDE and T2D status. Second, EF intercept and slope were regressed on IDE, T2D status, and IDE × T2D status. This model produced non-significant results for the IDE × T2D status variable for both initial EF performance, b* = .040, p = .924 and change over time, b* = .026, p = .631. In addition, the previously significant effect of IDE on EF change was no longer significant, b* = .016, p = .052, and the effect of T2D status on initial EF performance was no longer significant, b* = −.366, p = .343. Therefore, the presence of an IDE G allele did not moderate the negative effects of T2D on EF.
Figure 2.
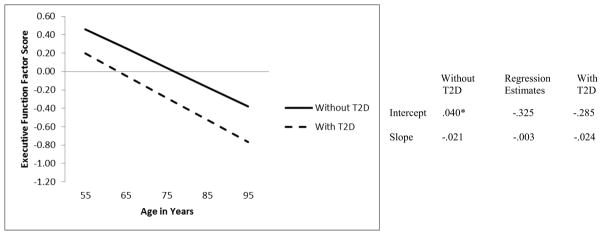
Predicted growth curve for executive function factor scores using T2D status (i.e., Without T2D, With T2D) as a predictor with age as a continuous variable centered at 75 years. *p < .05. **p <.01
4. Discussion
The aim of this research was to explore (a) the concurrent and longitudinal associations of one novel genetic (IDE rs6583817) and one metabolic-health (T2D) factor and (b) whether these factors acted independently or interactively in influencing concurrent level of performance and two-wave change in a key cognitive phenotype (EF) of aging. Of special interest was whether the possession of a G allele of this IDE variant would (a) exert a risk reduction or positive effect on EF performance and change or (b) moderate the countervailing expectation for a risk elevation or negative effect of T2D. Our expectations reflected these independent and interactive possibilities. We examined these issues using a large sample of older adults followed longitudinally over four years, with data analyzed using leading edge latent growth mixture models.
Regarding research goals 1 (EF latent model) and 2 (two-wave invariance testing), the confirmatory factor analyses revealed that, as expected, a single-factor model fit the data best. This model exhibited metric invariance overall and partial scalar invariance for the inhibition dimension. Our findings provide further evidence for a single-factor EF latent variable for normal aging, and are consistent with the differentiation/de-differentiation theory of typical aging in the EF phenotype. However, we note that this theory has not been tested on the same people over an extended longitudinal period (de Frias et al., 2006; Luszcz, 2011) and we could not test the EF factor structure at the genetic level (Kremen et al., 2009; Vasilopoulos et al., 2012). Moreover, recent neurocognitive evidence indicates that it may apply differentially across the spectrum of cognitively impaired, normal, and sustained healthy aging, as associated with individualized lifetime levels of biological vulnerability and environmental risk or protective factors (de Frias et al., 2009; Dixon, 2010; Lindenberger et al., 2008).
For research goals 3 (EF latent growth model) and 4 (path analyses with IDE and T2D), we observed that there is indeed a potential protective effect the IDE G allele on the EF phenotype—both concurrently and longitudinally. Concurrently, at the age 75 centering point (Figure 1), the presence of an IDE G allele (our IDE G+ group) was associated with better EF performance. In fact, the dosage effect showed that each additional G allele resulted in better concurrent EF performance. Longitudinally, adults with a G allele exhibited reduced EF decline rates as compared with adults who did not possess the G allele. This IDE (rs6583817) variant was previously associated with increased level of IDE transcription and reduced risk of AD in a synthetic in vitro system (Belbin et al., 2011; Carrasquillo et al., 2010). The present research is the first to link this IDE polymorphism to actual cognitive performance and change in non-demented older adults. The results indicated a potential protection function for normal cognitive aging, as associated with the IDE major allele (G). We did not observe such an association for the minor allele (A), which had been reported as a potential protection factor for classification with neurodegenerative disease (i.e., AD). The increase of IDE mRNA related to the minor allele (A) reported by Carrasquillo and colleagues would translate to a decrease in both insulin and Aβ. Conceivably, a decrease in Aβ would result in a decreased risk of AD. At the same time, a decrease in insulin would have a deleterious effect on EF, as increases in insulin have been linked to better EF performance (Awad et al., 2004). The minor and major allele of this IDE variant may be correspondingly specialized, with (a) the former affecting AD-related neurobiology and (b) the latter associated with prefrontal neurobiological changes, as phenotypically reflected in EF performance and change. Further research can examine whether (a) IDE targets two main peptides (insulin and Aβ) with the cognitive effects potentially contingent on brain region and physiological condition (e.g., aging), (b) IDE (rs6583817) major and minor alleles differentially affect other basic cognitive resources (e.g., speed) or risk of AD, (c) other IDE polymorphisms affect EF performance in normal aging, (d) other IDE variants interact with (or counteract) the present IDE variant, and (e) this IDE variant interacts with other biological (e.g., vascular) markers.
As expected from complementary univariate (manifest variable) studies (Biessels et al., 2008; Yeung et al., 2009), our findings show a clear link between T2D and decreased EF performance. This study contributes the novel information that T2D predicts level of performance but not accelerated decline in EF. The inference from this result is that T2D may have a (a) relatively early (perhaps pre-diagnosis) impact on EF or (b) moderated impact, by multiple individual-level factors (biological, environmental, severity, therapeutic) or (c) later acceleration in EF decline possibly occurring as a function of pre-clinical neurodegenerative decline. The present results should be interpreted in the context of a sample of a population that would include older adults with mild to moderate cases of T2D, and for whom access to national health care may indicate that the disease is relatively controlled. Our finding that the IDE variant did not moderate the effects of T2D may be in part due to this lack of severe T2D. Further research using other IDE variants and with participants with more severe T2D symptomatology would elucidate another facet of the long term effects of T2D on cognitive phenotypes in aging.
There are several strengths and limitations associated with this study. First, the VLS data set includes only one of several possible IDE genotypes with linkages to common neurodegenerative (AD) and bio-health (T2D) conditions. However, the genotype tested (rs6583817) has only recently been investigated in relation to AD and has unique promise both in terms of strength of association and the potential valence of influence. Future research should examine moderating and interacting influences (both risk and risk-reduction) of other IDE variants, genomic (e.g., APOE) factors, environmental-lifestyle (e.g., physical exercise) influences, and biological (e.g., pulse pressure) modifiers (e.g., Lindenberger et al., 2008). Second, our T2D participants were identified with a strict and standard multi-step process but the assessment did not include continuously distributed and relevant biomarkers (e.g., glycated haemoglobin [HbA1c]), which were unavailable but could have provided additional theoretical and diagnostic information (e.g., Raz et al., 2008). However, the present diagnostic procedures are well-developed, documented, validated, and appropriate to samples of older adults with relatively managed (mild to moderate) T2D conditions. Third, our goal was to examine a subgroup of aging adults who have not yet begun detectable transitions in neurodegenerative disease. Therefore, as noted, the present VLS sample is community-dwelling, fairly well-educated, and with access to national health care services. Not only is this likely to represent a growing proportion of the aging population, it provides a conservative test of the hypotheses concerning health-biological influences on prefrontal-related executive functions in older adults. That the sample may reflect late-life survivorship is reflected in the fact that about 8% had T2D and the allelic distribution of IDE showed some selectivity, in that the AG heterozygotes were present in a greater than expected frequency, relative to the GG homozygotes. Fourth, we focused on a prominent cognitive phenotype (EF) but both other basic (e.g., speed) and complex (e.g., episodic memory) are related to AD and EF—and possibly to this IDE variant in aging—and should be studied in future research. However, we note that among the strengths of this study are (a) that the EF factor was comprised of four standard and strong neuropsychological manifest variables empirically contributing to a latent variable and (b) that the EF phenotype was examined longitudinally with age as a continuous variable, resulting in an investigation of concurrent and longitudinal EF performance and change across a band of approximately 40 years.
In sum, the present study examined the effects of IDE (rs6583817) and T2D on EF level and change independently and together. We found that IDE and T2D are independently and differentially associated with EF performance in older adults. The associations are (at cross-section) in a common direction in that both factors produce expected risk-related group differences at the age intercept. The associations are differential in that the longitudinal EF decline patterns show (a) similar functions for both healthy and T2D participants but (b) steeper decline (for the IDE G− group) and unique substantial preservation (for the IDE G+ group). Our specific analyses of IDE interactions with T2D showed that this variant neither protected (the G+) nor exacerbated (the G−) the observed decrements and declines of EF by T2D versus normal aging groups. Possession of an IDE G allele exhibited a previously unobserved positive effect on both EF level at age 75 years and change across time. Our research is the first to link IDE to cognitive performance and change in non-demented older adults.
Verification for Neurobiology of Aging.
There are no actual or potential conflicts of interest, as queried specifically and completely in sections (a), (b), and (c) of this item.
This research has been supported by grants from the Alberta Health Services (to authors David Westaway, Jack Jhamandas, and Roger Dixon) and the National Institute on Aging (National Institutes of Health; R37 AG008235) to author Roger Dixon.
The data presented in this study have not been previously published or submitted elsewhere, and these data will not be submitted elsewhere while under consideration by this journal.
All research has been approved continuously by relevant institutional review boards. Certificates are available and on file in the University of Alberta Research Services Office and the U.S. National Institutes of Health. All participants have completed and signed informed consent forms.
All authors have participated in multiple phases of this research and all have participated in the process of preparing and submitting this manuscript. All authors approve of their inclusion on the authorship list.
Acknowledgments
The present research is supported by grants from (a) the Alberta Health Services (University Hospital Foundation) to authors DW, JJ, and RAD and (b) the National Institutes of Health (National Institute on Aging; R37 AG008235) to RAD. Both RAD and DW are also supported by the Canada Research Chairs program. We thank the volunteer participants and the VLS staff for their many contributions. We acknowledge the University of Alberta Centre for Prions and Protein Folding Diseases for laboratory and technical support. We acknowledge the specific contributions of Correne DeCarlo, Stuart MacDonald, and Bonnie Whitehead to the VLS genetics initiative. More information about the VLS may be found at: http://www.ualberta.ca/~vlslab/.
Footnotes
Disclosure Statement
The authors state that there is no actual or potential conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham R, Myers A, Wavrant-DeVrieze F, Hamshere ML, Thomas HV, Marshall H, Compton D, Spurlock G, Turic D, Hoogendoorn B, Kwon JM, Petersen RC, Tangalow E, Norton J, Morris JC, Bullock R, Liolitsa D, Lovestone S, Hardy J, Goate A, O’Donovan M, Williams J, Owen MJ, Jones L. Substantial linkage disequilibrium across the insulin-degrading enzyme locus but no association with late-onset Alsheimer’s disease. Hum Genet. 2001;109:646–652. doi: 10.1007/s00439-001-0614-1. [DOI] [PubMed] [Google Scholar]
- Adrover-Roig D, Sesé A, Barceló F, Palmer A. A latent variable approach to executive control in healthy ageing. Brain Cogn. 2012;78:284–299. doi: 10.1016/j.bandc.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Andrés P, Van der Linden M. Age-related differences in supervisory attentional system functions. J Gerontol B Psychol Sci Soc Sci. 2000;55:373–380. doi: 10.1093/geronb/55.6.p373. [DOI] [PubMed] [Google Scholar]
- Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61:661–666. doi: 10.1001/archneur.61.5.661. [DOI] [PubMed] [Google Scholar]
- Awad N, Gagnon M, Messier C. The relationship between impaired glucose tolerance, type 2 diabetes, and cognitive function. J Clin Exp Neuropsyc. 2004;26:1044–1080. doi: 10.1080/13803390490514875. [DOI] [PubMed] [Google Scholar]
- Bartl J, Scholz C-J, Hinterberger M, Jungwirth S, Wichart I, Rainer MK, Kneitz S, Danielczyk W, Tragl KH, Fischer P, Riederer P, Grunblatt E. Disorder-specific effects of polymorphisms at opposing ends of the insulin degrading enzyme gene. BMC Med Genet. 2011;12:151. doi: 10.1186/1471-2350-12-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belbin O, Crump M, Bisceglio GD, Carrasquillo MM, Morgan K, Younkin SG. Multiple insulin degrading enzyme variants alter in vitro reporter gene expression. PLoS ONE. 2011;6:e21429. doi: 10.1371/journal.pone.0021429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RG, Duckworth WC, Hamel FG. Degradation of amylin by insulin-degrading enzyme. J Biol Chem. 2000;275:36621–36625. doi: 10.1074/jbc.M006170200. [DOI] [PubMed] [Google Scholar]
- Berg EA. A simple objective technique for measuring flexibility in thinking. J Gen Psychol. 1948;39:15–22. doi: 10.1080/00221309.1948.9918159. [DOI] [PubMed] [Google Scholar]
- Bertram L, Blacker D, Mullin K, Keeney D, Jones J, Basu S, Yhu S, McInnis MG, Go RCP, Vekrellis K, Selkoe DJ, Saunders AJ, Tanzi RE. Evidence for genetic linkage of Alzheimer’s disease to chromosome 10q. Science. 2000;290:2302. doi: 10.1126/science.290.5500.2302. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Deary IJ, Ryan CM. Cognition and diabetes: A lifespan perspective. Lancet Neurol. 2008;7:184–190. doi: 10.1016/S1474-4422(08)70021-8. [DOI] [PubMed] [Google Scholar]
- Björk BF, Katzov H, Kehoe P, Fratiglioni L, Winblad B, Prince JA, Graff C. Positive association between risk for late-onset Alzheimer disease and genetic variation in IDE. Neurobiol Aging. 2007;28:1374–1380. doi: 10.1016/j.neurobiolaging.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Blomqvist MEL, Chalmers K, Andreasen N, Bogdanovic N, Wilcock GK, Cairns NJ, Feuk L, Brookes AJ, Love S, Blennow K, Kehoe PG, Prince JA. Sequence variants of IDE are associated with the extent of β-amyloid deposition in the Alzheimer’s disease brain. Neurobiol Aging. 2005;26:795–802. doi: 10.1016/j.neurobiolaging.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Boussaha M, Hannequin D, Verpillat P, Brice A, Frebourg T, Campion D. Polymorphisms of insulin degrading enzyme gene are not associated with Alzheimer’s disease. Neurosci Lett. 2002;329:121–123. doi: 10.1016/s0304-3940(02)00586-4. [DOI] [PubMed] [Google Scholar]
- Burgess PW, Shallice T. The Hayling and Brixton tests. Thames Valley Test Company; England: 1997. [Google Scholar]
- Carrasquillo MM, Belbin O, Zou F, Allen M, Ertekin-Taner N, Ansari M, Wilcox SL, Kashino MR, Ma L, Younkin LH, Younkin SG, Younkin CS, Dincman TA, Howard ME, Howell CC, Stanton CM, Watson CM, Crump M, Vitart V, Hayward C, Hastie ND, Rudan I, Campbell H, Polasek O, Brown K, Passmore P, Craig D, McGuinness B, Todd S, Kehoe PG, Mann DM, Smith AD, Beaumont H, Warden D, Holmes C, Heun R, Kölsch H, Kalsheker N, Pankratz VS, Dickson DW, Graff-Radford NR, Petersen RC, Wright AF, Younkin SG, Morgan K. Concordant association of insulin degrading enzyme gene (IDE) variants with IDE mRNA, Aβ, and Alzheimer’s disease. PLoS ONE. 2010;5:e8764. doi: 10.1371/journal.pone.0008764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholerton B, Baker CD, Craft S. Insulin resistance and pathological brain ageing. Diabetic Med. 2011;28:1463–1475. doi: 10.1111/j.1464-5491.2011.03464.x. [DOI] [PubMed] [Google Scholar]
- D’Elia LA, Satz P, Uchiyama CL, White T. Color Trails Test: Professional Manual. Psychological Assessment Resources; FL: 1996. [Google Scholar]
- de Frias CM, Dixon RA, Strauss E. Structure of four executive functioning tests in healthy older adults. Neuropsychology. 2006;20:206–214. doi: 10.1037/0894-4105.20.2.206. [DOI] [PubMed] [Google Scholar]
- de Frias CM, Dixon RA, Strauss E. Characterizing executive functioning in older special populations: From cognitively elite to cognitively impaired. Neuropsychology. 2009;23:778–791. doi: 10.1037/a0016743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon RA. An epidemiological approach to cognitive health in aging. In: Bäckman L, Nyberg L, editors. Memory, Aging, and the Brain. Psychology Press; England: 2010. pp. 144–166. [Google Scholar]
- Dixon RA, de Frias CM. The Victoria Longitudinal Study: From characterizing cognitive aging to illustrating changes in memory compensation. Aging Neuropsychol C. 2004;11:346–376. [Google Scholar]
- Dixon RA, Small BJ, MacDonald SWS, McArdle JJ. Yes, memory declines with aging – but when, how, and why? In: Naveh-Benjamin M, Ohta N, editors. Memory and Aging. Psychology Press; New York: 2012. pp. 325–347. [Google Scholar]
- Duggirala R, Blangero J, Almasy L, Dyer TD, Williams KL, Leach RJ, O’Connell P, Stern MP. Linkage of type 2 diabetes mellitus and of age on onset to a genetic location on chromosome 10q in Mexican Americans. Am J Hum Genet. 1999;64:1127–1140. doi: 10.1086/302316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertekin-Taner N, Aölen M, Fadale D, Scanlin L, Younkin L, Petersen RC, Graff-Radford N, Younkin SG. Genetic variants in a haplotype block spanning IDE are significantly associated with plasma Aβ42 levels and risk for Alzheimer Disease. Hum Mut. 2004;23:334–342. doi: 10.1002/humu.20016. [DOI] [PubMed] [Google Scholar]
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guénette S. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. P Natl Acad Sci USA. 2003;100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AL, de Frias CM, Yeung SE, Dixon RA. Short-term longitudinal trends in cognitive performance in older adults with Type 2 diabetes. J Clin Exp Neuropsych. 2009;31:809–822. doi: 10.1080/13803390802537636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman N, Miyake A, Young S, DeFries J, Corley R, Hewitt J. Individual differences in executive functions are almost entirely genetic in origin. J Exp Psychol Gen. 2008;137:201–225. doi: 10.1037/0096-3445.137.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grarup N, Rose CS, Andersson EA, Andersen G, Nielsen AL, Albrechtsen A, Clausen JO, Rasmussen SS, Jørgenen T, Sandbæk A, Lauritzen T, Schmitz O, Hansen T, Pedersen O. Studies of association of variants near the HHEX, CDKN2A/B, and IGF2BP2 genes with type 2 diabetes and impaired insulin release in 10,705 Danish subjects: Validation and extension of genome-wide association studies. Diabetes. 2007;56:3105–3111. doi: 10.2337/db07-0856. [DOI] [PubMed] [Google Scholar]
- Grober E, Hall CB, Lipton RB, Zonderman AB, Resnick SM, Kawas C. Memory impairment, executive dysfunction, and intellectual decline in preclinical Alzheimer’s disease. J Int Neuropsych Soc. 2008;14:266–278. doi: 10.1017/S1355617708080302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamohamed S, Demissie S, Volcjak J, Liu C, Heard-Costa N, Liu J, Shoemaker CM, Panhuysen CI, Meigs JB, Wilson P, Atwood LD, Cupples LA, Herbert A. Polymorphisms in the insulin-degrading enzyme gene are associated with type 2 diabetes in men from the NHLBI Framingham Heart Study. Diabetes. 2003;52:1562–1567. doi: 10.2337/diabetes.52.6.1562. [DOI] [PubMed] [Google Scholar]
- Kline RB. Principles and Practice of Structural Equation Modeling. 3. Guilford; New York: 2011. [Google Scholar]
- Kremen WS, Jacobson KC, Panizzon MS, Xian H, Eaves LJ, Eisen SA, Tsuang MT, Lyons MJ. Factor structure of planning and problem-solving: A behavioral genetic analysis of the Tower of London task in middle-aged twins. Behav Genet. 2009;39:133–144. doi: 10.1007/s10519-008-9242-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurochkin IV, Goto S. Alzheimer’s β-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994;345:33–37. doi: 10.1016/0014-5793(94)00387-4. [DOI] [PubMed] [Google Scholar]
- Kwak SH, Cho YM, Moon MK, Kim JH, Park BL, Cheon HS, Shin HD, Jang HC, Kim SY, Lee HK, Park KS. Association of polymorphisms in the insulin-degrading enzyme gene with type 2 diabetes in the Korean population. Diabetes Res Clin Pr. 2008;79:284–290. doi: 10.1016/j.diabres.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Lendon C, Craddock N. Susceptibility gene(s) for Alzheimer’s disease on chromosome 10. TRENDS Neurosci. 2001;24:557–559. doi: 10.1016/s0166-2236(00)01912-3. [DOI] [PubMed] [Google Scholar]
- Lindenberger U, Nagel IE, Chicherio C, Li SC, Keekeren HR, Bäckman L. Age-related decline in brain resources modulates genetic effects on cognitive functioning. Front Neurosci. 2008;2:234–244. doi: 10.3389/neuro.01.039.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luszcz M. Executive function and cognitive aging. In: Schaie KW, Willis SL, editors. The Handbook of the Psychology of Aging. 7. Academic Press; California: 2011. pp. 59–72. [Google Scholar]
- McFall GP, Geall BP, Fischer AL, Dolcos S, Dixon RA. Testing covariates of type 2 diabetes-cognition associations in older adults: Moderating or mediating effects? Neuropsychology. 2010;24:547–562. doi: 10.1037/a0019246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake A, Friedman NP, Emerson MJ, Witzkin AH, Howerter A, Wager TD. The unity and diversity of executive functions and their contributions to complex “frontal lobe” tasks: A latent variable analysis. Cognitive Psychol. 2000;41:49–100. doi: 10.1006/cogp.1999.0734. [DOI] [PubMed] [Google Scholar]
- Muthén LK, Muthén BO. Mplus User’s Guide. 6. Muthén and Muthén; California: 2010. [Google Scholar]
- Nagel IE, Chicherio C, Li SC, von Oertzen T, Sander T, Villringer A, Heekeren HR, Bäckman L, Lindenberger U. Human aging magnifies genetic effects on executive functioning and working memory. Front Hum Neurosci. 2008;2:1–8. doi: 10.3389/neuro.09.001.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan J, Wilkinson D, Stammers S, Low L. The role of tests of frontal executive function in the detection of mild dementia. Int J Geriatr Psych. 2001;16:18–26. doi: 10.1002/1099-1166(200101)16:1<18::aid-gps265>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Nilsson E. Unpublished doctoral dissertation. Karolinska Institutet; Stockholm: 2006. Diabetes and cognitive functioning: The role of age and comorbidity. [Google Scholar]
- Okereke OI, Selkoe DJ, Pollak MN, Stampfer MJ, Hu FB, Hankinson SE, Grodstein F. A profile of impaired insulin degradation in relation to late-life cognitive decline: A preliminary investigation. Int J Geriatr Psych. 2009;24:177–182. doi: 10.1002/gps.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu WQ, Folstein MF. Insulin, insulin-degrading enzyme and amyloid-β peptide in Alzehimer’s disease: Review and hypothesis. Neurobiol Aging. 2006;27:190–198. doi: 10.1016/j.neurobiolaging.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Profenno LA, Porsteinsson AP, Faraone S. Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol Psychiat. 2010;67:505–512. doi: 10.1016/j.biopsych.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Rapp MA, Reischies FM. Attention and executive control predict Alzheimer disease in late life: Results from the Berlin Aging Study (BASE) Am J Geriat Psychiat. 2005;13:134–141. doi: 10.1176/appi.ajgp.13.2.134. [DOI] [PubMed] [Google Scholar]
- Raz N, Dahle CL, Rodrigue KM, Kennedy KM, Land S. Effects of age, genes, and pulse pressure on executive functions in health adults. Neurobiol Aging. 2011;32:1124–1137. doi: 10.1016/j.neurobiolaging.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Dahle CL, Rodrigue KM, Kennedy KM, Land S, Jacobs BS. Brain-derived neurotrophic factor Val66Met and blood glucose: a synergistic effect on memory. Frontiers in Human Neuroscience. 2008;2:1–6. doi: 10.3389/neuro.09.012.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitan RM, Wolfson D. Neuropsychological Evaluation of Older Children. Neuropsychology Press; Arizona: 1992. [DOI] [PubMed] [Google Scholar]
- Rudovich N, Pivovarova O, Fisher E, Fischer-Rosinsky A, Spranger J, Möhlig M, Schulze MB, Boeing H, Pfeiffer AFH. Polymorphisms with insulin-degrading enzyme (IDE) gene determine insulin metabolism and risk of type 2 diabetes. J Mol Med. 2009;87:1145–1151. doi: 10.1007/s00109-009-0540-6. [DOI] [PubMed] [Google Scholar]
- Seaquist ER, Latteman DF, Dixon RA. Diabetes and the brain: American Diabetes Association Research Symposium. Diabetes. 2012;61:3056–3062. doi: 10.2337/db12-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Joachimiak A, Rosner MR, Tang WJ. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature. 2006;443:870–874. doi: 10.1038/nature05143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer JD, Willett JB. Applied Longitudinal Data Analysis: Modeling Change and Event Occurrence. Oxford University Press; New York: 2003. [Google Scholar]
- Small BJ, Dixon RA, McArdle JJ. Tracking cognition-health changes from 55 to 95 years of age. J Gerontol, B Psychol Sci Soc Sci. 2011;66B:i153–i161. doi: 10.1093/geronb/gbq093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan MWJ, Reynolds RM, Marioni RE, Price JF. Cognitive function, dementia, and type 2 diabetes mellitus in the elderly. Nat Rev Endocrinol. 2011;7:108–114. doi: 10.1038/nrendo.2010.228. [DOI] [PubMed] [Google Scholar]
- Taylor SF, Kornblum S, Lauber EJ, Minoshima S, Koeppe RA. Isolation of specific interference processing in the Stroop task: PET activation studies. NeuroImage. 1997;6:81–92. doi: 10.1006/nimg.1997.0285. [DOI] [PubMed] [Google Scholar]
- Turner GR, Spreng RN. Executive functions and neurocognitive aging: Dissociable patterns of brain activity. Neurobiol Aging. 2012;33:826.el–.e13. doi: 10.1016/j.neurobiolaging.2011.06.005. [DOI] [PubMed] [Google Scholar]
- Umegaki H. Neurodegeneration in diabetes mellitus. In: Ahmad HI, editor. Neurodegenerative Diseases. Landes Bioscience and Springer Science+Business Media; New York: 2012. pp. 258–265. [Google Scholar]
- Vardy ERLC, Brown K, Stopford CL, Thompson JC, Richardson AM, Neary D, Kalsheker N, Morgan K, Mann DM, Snowden JS. Cognitive phenotypes in Alzheimer’s disease and genetic variants in ACE and IDE. Neurobiol Aging. 2012;33:1486.e1–e2. doi: 10.1016/j.neurobiolaging.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Vasilopoulos T, Franz CE, Panisson MS, Xian H, Grant MD, Lyons MJ, Toomey R, Jacobson KC, Kremen WS. Genetic architecture of the Delis-Kaplan executive function system trail making test: Evidence for distinct genetic influences on executive function. Neuropsychology. 2012;26:238–250. doi: 10.1037/a0026768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vionnet N, Hani EH, Dupont S, Gallena S, Francke S, Dotte S, De Matos F, Durand E, Leprêtre F, Lecoeur C, Gallina P, Zekiri L, Dina C, Froguel P. Genomewide search for type 2 diabetes-susceptibility genes in French Whites: A novel susceptibility locus for early-onset diabetes on chromosome 3q27-qter and independent replication of a type 2-diabetes locus on chromosome 1q21-q24. Am J Hum Genet. 2000;67:1470–1480. doi: 10.1086/316887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Shu C, Jia L, Zuo X, Zhang Y, Zhou A, Qin W, Song H, Wei C, Zhang F, Hong Z, Tang M, Wang D-M, Jia J. Exploration of 16 candidate genes identifies the association of IDE with Alzheimer’s disease in Han Chinese. Neurobiol Aging. 2012;33:1014.e1–.e9. doi: 10.1016/j.neurobiolaging.2010.08.004. [DOI] [PubMed] [Google Scholar]
- West RL. An application of prefrontal cortex function theory to cognitive aging, Psychol. Bull. 1996;120:272–292. doi: 10.1037/0033-2909.120.2.272. [DOI] [PubMed] [Google Scholar]
- Wiebe SA, Espy KA, Charak D. Using confirmatory factor analysis to understand executive control in preschool children: I. latent structure. Dev Psychol. 2008;44:575–587. doi: 10.1037/0012-1649.44.2.575. [DOI] [PubMed] [Google Scholar]
- Wiebe SA, Sheffield T, Nelson JM, Clark CAC, Chevalier N, Espy KA. The structure of executive function in 3-year-olds. J Exp Child Psychol. 2010;108:436–452. doi: 10.1016/j.jecp.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart HA, Roth RM, Saykin AJ, Rhodes CH, Tsongalis GJ, Pattin KA, Moore JH, McAllister TW. COMT Val158Met genotype and individual differences in executive function in healthy adults. J Int Neuropsych Soc. 2011;17:174–180. doi: 10.1017/S1355617710001402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SE, Fischer AL, Dixon RA. Exploring effects of Type 2 diabetes on cognitive functioning in older adults. Neuropsychology. 2009;23:1–9. doi: 10.1037/a0013849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, de Bakker PIW, Abecasis GR, Almgren P, Andersen G, Ardlie K, Boström KB, Bergman RN, Bonnycastle LL, Borch-Johnsen K, Burtt NP, Chen H, Chines PS, Daly MJ, Deodhar P, Ding C, Doney ASF, Duren WL, Elliott KS, Erdos MR, Frayling TM, Freathy RM, Gianniny L, Grallert H, Grarup N, Groves CJ, Guiducci C, Hansen T, Herder C, Hitman GA, Hughes TE, Isomaa B, Jackson AU, Jørgensen T, Kong A, Kubalanza K, Kuruvilla FG, Kuusisto J, Langenberg C, Lango H, Lauritzen T, Li Y, Lindgren CM, Lyssenko V, Marvelle AF, Meisinger C, Midthjell K, Mohlke KL, Morken MA, Morris AD, Narisu N, Nilsson P, Owen KR, Palmer CNA, Payne F, Perry JRB, Pettersen E, Platou C, Prokopenko I, Qi L, Qin L, Rayner NW, Rees M, Roix JJ, Sandbæk A, Shields B, Sjögren M, Steinthorsdottir V, Stringham HM, Swift AJ, Thorleifsson G, Thorsteinsdottir U, Timpson NJ, Tuomi T, Tuomilehto J, Walker M, Watanabe RM, Weedon MN, Willer CJ, Illig T, Hveem K, Hu FB, Laakso M, Stefansson K, Pedersen O, Wareham NJ, Barroso I, Hattersley AT, Collins FS, Groop L, McCarthy MI, Boehnke M, Altshuler D Wellcome Trust Case Control Consortium . Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]