

Abstract
Dendritic cells (DCs) have been shown to play a major role in oral tolerance and this function has been associated with their ability to produce anti-inflammatory cytokines and to induce suppressive T regulatory cells. Herein, we demonstrate that upon oral administration of Ag, lamina propia (LP) DCs engage specific T cells and acquire a novel mechanism by which they transfer tolerance against diverse T cell specificities. Indeed, when Ig-MOG carrying the myelin oligodendrocyte glycoprotein (MOG)35–55 epitope was orally administered into either T cell sufficient or deficient mice, only the T cell sufficient hosts yielded CD8α+ and CD8α− LP DCs that were able to transfer tolerance to a variety of MHC class II-restricted effector T cells. Surprisingly, these LP DCs up-regulated programmed cell death ligand 1 (PD-L1) during the initial interaction with MOG-specific T cells and utilized this inhibitory molecule to suppress activation of T cells regardless of Ag specificity. Furthermore, oral Ig-MOG was able to overcome experimental allergic encephalomyelitis (EAE) induced with central nervous system (CNS) homogenate, indicating that the DCs are able to modulate disease involving diverse T cell specificities. This previously unrecognized attribute potentiates DCs against autoimmunity.
INTRODUCTION
Oral tolerance represents a practical and effective approach to counter autoimmunity (1). This is most likely due to the proficiency of the gut immune system to mount T regulatory (Treg) cell responses (2, 3) capable of producing anti-inflammatory cytokines such as TGFβ (4) and IL-10 (5). Dendritic cells (DCs)3 in the gut-associated lymphoid tissues have proven effective in capturing Ag from the intestinal lumen (6, 7) and are conditioned to present oral Ag in a tolerogenic fashion (8). By many accounts DCs have proven efficient in inducing tolerance either by producing IL-10 (9), a powerful anti-inflammatory cytokine, or by Ag-driven induction of Tregs, which can display highly suppressive function against inflammatory T cells (8, 10–12). Despite these attributes, oral delivery of Ag, while effective in preventive settings against diseases such as EAE (1, 13, 14), collagen-induced arthritis (15), type I diabetes (16–18), and colitis (19), has met with challenges in the reversal of ongoing autoimmunity in many animal models of these diseases and translation to human has yet to be achieved. Herein, we report yet another mechanism by which lamina propria (LP) DCs acquire and execute tolerogenic functions that overcome overt autoimmune inflammation. Indeed, Ig-MOG given to sick mice with EAE by oral gavage was able to overcome the disease as it did upon delivery by intraperitoneal injection (20). Intriguingly, however, this was accomplished in an IL-10 independent manner, prompting the question as to whether DCs are endowed with an unknown tolerance mechanism yet to be defined. The initial observation in this gut-priming induced tolerance indicated that only lamina propria, but not mesenteric LN, APCs from Ig-MOG recipient mice can transfer tolerance to T cells. Even more surprising, the LP APCs had to have had contact with effector T cells in an antigen specific manner in order to tolerize target T cells of the same or unrelated specificity. Ultimately, we discovered that the initial antigen-mediated interactions with T cells lead LP DCs to up-regulate the inhibitory molecule programmed death ligand 1 (PD-L1) and utilize it to counter subsequent activation of T cells. These findings reveal yet another DC tolerance attribute specifically acquired via the oral route that could drive broad suppressive function against diverse T cell specificities and overcome overt autoimmunity.
MATERIALS AND METHODS
Mice
C57BL/6, C57BL/6 IL-10−/ −, B6.129S2-H2dlAb1-Ea/J (MHC class II-deficient), and C57BL/6 Rag2−/ − were purchased from The Jackson Laboratory (Bar Harbor, Michigan). C57BL/6 2D2 mice, expressing a transgenic TCR recognizing MOG35-55, have previously been described (21). C57BL/6 FoxP3.GFP.DTR mice expressing the green fluorescence protein (GFP) and human diphtheria toxin receptor (DTR) were previously described (22). C57BL/6 OTII mice, expressing a transgenic TCR recognizing OVA323–339, have been described previously (23). Fcerg1 (FcγRI and III deficient) mice were purchased from Taconic and used as previously described (20). All mice were bred and maintained in our animal care facility for the duration of the experiments. All experimental procedures were performed according to the guidelines of the University of Missouri animal care and use committee.
Antigens
Peptides
The peptides used in this study were purchased from EZBiolab (Westfield, IN). MOG peptide (MEVGWYRSPFSRVVHLYRNGK) encompasses aa residues 35–55 of MOG. OVA peptide (SQAVHAAHAEINEAGR) encompasses aa residues 323–339 of ovalbumin. PLP1 peptide (HSLGKWLGHPDKF) encompasses aa 139–151 of proteolipid protein. CNS homogenate was made from frozen unstripped rat brains (Pelfreez Biologicals) which were homogenized in PBS using a Waring blender and adjusted to 300mg/mL as described previously (24).
Ig chimeras
Ig-MOG (20), and Ig-OVA (25, 26) incorporate MOG and OVA peptide, respectively, inserted within the H chain complementarity determining region 3. Ig-W is the parental IgG2b,κ molecule not encompassing any myelin or other peptide. All chimera transfectants are grown up in large-scale cultures and purified from culture supernatant on affinity chromatography columns made of rat anti-mouse κ-chain coupled to CNBr-activated Sepharose 4B (Amersham Biosciences). Aggregation of Ig chimeras was done using 50%-saturated (NH4)2SO4 as described previously (24).
Cell isolation
CD4+ T cells were isolated from the spleen of 2D2 or OTII transgenic mice by positive selection using the MACS cell separation system (Miltenyi). Bulk splenic and lymph node APCs were obtained by tissue disruption in a collagenase solution and isolation of the cells on a dense BSA gradient. For isolation of bulk APCs from the lamina propria, the intestines were washed clean and the IEL were removed using a CMF-EDTA wash buffer as described (27). The cells were then incubated in a collagenase solution (100U/mL RPMI-10) for 1 hour at 37°C and separated on a dense BSA gradient. CD8α+CD11c+, CD8α−CD4+CD11c+, and CD11b+ cells were purified from the dense-BSA enriched bulk APCs by the differential adherence method previously described (28). Briefly, the APCs were allowed to adhere to petri dishes for 90 min at 37°C, washed, and incubated overnight at 37°C at 7% CO2. The non-adherent dendritic cells were collected and the subsets were purified by positive selection using the MACS separation system.
Depletion of Tregs
C57BL/6 FoxP3.GFP.DTR mice were depleted of T regulatory cells by i.p. injection of 300 ng Diphtheria Toxin (DT) from Corynebacterium diphtheria (Sigma) in PBS once daily for three days.
Induction of EAE
Active EAE
Induction of active EAE has been previously described (20, 24). Briefly, female mice (6–8 wk old) were induced for EAE by s.c. injection of a 200μL IFA/PBS (v/v) solution containing 300μg MOG peptide or 6mg CNS homogenate and 200μg of Mycobacterium tuberculosis H37Ra (Difco) in the footpads and at the base of the limbs. Six hours later, mice were given i.v. 500ng purified Bordetella pertussis toxin (List Biological Laboratories). A second injection of B. pertussis toxin was given after 48 h. The mice were scored daily for clinical signs of EAE as follows: 0, no clinical signs; 1, loss of tail tone; 2, hind limb weakness; 3, hind limb paralysis; 4, forelimb paralysis; and 5, moribund or death.
Induction of EAE in MHC II−/ − mice
MHC II−/ − female mice (6–8 wk old) were given i.p. 1 × 106 splenic or LP APCs along with 10 × 106 purified 2D2 CD4 T cells i.v. The following day, mice were induced for active EAE as above.
Oral Ig treatment of EAE
Mice that were induced for EAE received 300μg sol or agg Ig-MOG or Ig-W and 1mg soy trypsin inhibitor in PBS by oral gavage. Treatments were administered 4 times at 2 day intervals beginning on day 7 post disease induction. For analysis of APCs following oral Ig exposure, mice received a one-time dose of 1mg agg Ig-MOG, Ig-OVA, or Ig-W along with 1mg soy trypsin inhibitor in PBS. APCs were harvested 24 h later for experiments.
Measurement of cell proliferation
5 × 105 purified 2D2 or OTII T cells were cultured with 1 × 105 APCs in the presence of antigen at the indicated concentrations for 48 h. 1μCi 3[H] thymidine was added per well during the last 14.5 h of culture. The cells were harvested on a Trilux 1450 Microbeta Wallac Harvester and incorporated 3[H] thymidine was counted using the Microbeta 270.004 software (EG&G Wallac, Gaithersburg, MD).
Measurement of cytokines by ELISA
IFNγ and IL-17 were detected by ELISA according to BD Pharmingen’s (San Jose, CA) standard protocol. For IFNγ, the capture antibody was rat anti-mouse IFNγ (R4-6A2) and the biotinylated detection antibody was rat anti-mouse IFNγ (XMG1.2). For IL-17 detection, the capture antibody was anti-mouse IL-17A (eBio 17CK15A5) and the biotinylated detection antibody was anti-mouse IL-17A (eBio 17B7). The OD450 was read on a SpectraMAX 190 counter (Molecular Devices, Sunnyvale, CA) and analyzed using SOFTmax PRO 3.1.1 software. Graded amounts of recombinant cytokine were included for construction of the standard curve. Cytokine concentrations were extrapolated from the linear portion of the standard curve.
Flow cytometry
Most antibodies were purchased from BD Biosciences or Ebiosciences. Antibodies used were phycoerythrin (PE)-conjugated PD-L1 (10F.9G2), PD-L2 (TY25), CD80 (16-10A1), CD86 (PO3.1), and I-Ab (NIMR-4); fluorescein isothiocyanate (FITC)-conjugated CD8α (53-6.7) and CD11b (M1/70); PE-Cy7-conjugated CD4 (RM4-5) and CD11c (HL3); allophycocyanin (APC)-conjugated CD4 (RM4-5), CD11b (M1/70), and CD11c (HL3); biotinylated CD103 (2E7), CD40 (3/23), and goat anti-mouse Ig. FcγR’s were blocked prior to staining using mouse IgG (Sigma, St. Louis, MO). Dead cells were excluded using 7-amino-actinomycin D (7AAD) (EMD Biosciences). Cells were collected using a Beckman Coulter CyAn (Brea, CA) and data were analyzed using FlowJo version 8.8.6 (Tree Star) or Summit Software version 4.0 (Dako).
PD-L1 inhibition
Anti-mouse PD-L1 (10F.9G2) and isotype control (LTF-2) antibodies were purchased from BioXCell (West Lebanon, NH). To block PD-L1, APCs (1 × 106 cells/mL) were cultured in DMEM supplemented with 10% fetal calf serum in the presence of 20μg/mL anti-PD-L1 or isotype control for 2 hours at 37°C. Following incubation, APCs were washed twice with PBS and used for experiments.
Statistical analysis
Data were analyzed using a one-sample t test, unpaired two-tailed Student’s t test, or one-way ANOVA as indicated using Prism software v4.0c (Graphpad).
RESULTS
Oral treatment with Ig-MOG suppresses inflammation and attenuates EAE
We have previously shown that the aggregated (agg) form of Ig-MOG is effective against EAE when given to mice via the intraperitoneal route (20). Herein, we sought to test Ig-MOG for suppression of EAE via the oral route which is presumably more effective for induction of T cell tolerance (2, 29) and would be more practical and relevant for human circumstances. Accordingly, C57BL/6 mice were induced for EAE with either MOG peptide (MOG EAE) or CNS homogenate (CNS EAE) and then given Ig-MOG in a soluble (sol) or agg form on day 7, 9, 11 and 13 after disease induction by oral gavage. The animals were simultaneously given soy bean trypsin inhibitor (STI) to minimize gastric degradation of Ig-MOG. The mice were monitored for signs of paralysis until day 30 post disease induction. The results show that mice given sol Ig-MOG +STI had reduced disease severity relative to animals recipient of the control sol Ig-W+STI or STI alone (STI) (Fig. 1A, left). Indeed, for MOG EAE the mean maximal disease severity score (MMS) went down from 2.0 ± 0.5 in the control groups (STI and Ig-W +STI) to 0.6 ± 0.3 in mice recipient of sol Ig-MOG +STI (p < 0.005). Likewise, disease severity of CNS homogenate-induced EAE was reduced from 2.0 ± 0.5 in the STI and Ig-W+STI-treated mice to 1.2 ± 0.3 (p < 0.004) in those recipient of sol Ig-MOG +STI (Fig. 1A, right). Furthermore, the aggregated form of Ig-MOG also reduced disease severity in both MOG peptide (Fig.1B, left) and CNS homogenate (Fig. 1B, right) induced EAE. In fact, the mean maximal disease score was reduced from 1.8 ± 0.4 in the agg Ig-W +STI treated mice to 0.5 ± 0.4 (p < 0.001) for MOG peptide EAE and from 1.8 ± 0.4 to 0.83 ± 0.3 (p < 0.002) for CNS homogenate EAE. Thus, oral treatment with Ig-MOG is effective against EAE whether the chimeras are given in a sol or agg form. Suppression of disease is likely due to modulation of both Th1 and Th17 cells. This is drawn from the finding that IFNγ and IL-17 production by peripheral (PLN) and mesenteric (MLN) lymph node as well as LP CD4 T cells was significantly reduced by treatment with agg Ig-MOG (Fig. 1C). Indeed, MOG-specific IFNγ production was significantly reduced in the PLN, MLN, and LP of oral agg Ig-MOG +STI treated mice compared to STI alone treated mice (Fig. 1C, top row). Similarly, production of IL-17 was significantly reduced compared to STI mice (Fig. 1C, bottom row). This effect was antigen specific, as PLP1 peptide did not elicit cytokine responses from either treatment group. Taken together, these results indicate that oral agg Ig-MOG induces modulation of both Th1 and Th17 T cells in an antigen specific manner.
Figure 1. Oral Ig-MOG diminishes Th1 and Th17 responses and reverses EAE.

Groups of 6- to 8-wk-old C57BL/6 mice were induced for EAE with MOG peptide (MOG EAE) or CNS homogenate (CNS EAE). Seven days later, the mice were treated orally with 300 μg sol (A) or agg (B) Ig-MOG or Ig-W and 1 mg soy trypsin inhibitor (STI) for four times at 2-day intervals. Mice recipient of STI alone (STI) were included for control purposes. Mice were monitored daily for clinical signs of disease. Data is representative of three individual experiments with 5 mice per group. (C) Mice were induced for MOG EAE and treated orally with agg Ig-MOG +STI or STI alone (STI) as in B. Mice were sacrificed 15 days after the last treatment (day 21 peak of disease) and their mesenteric lymph node (MLN), lamina propria (LP), and peripheral lymph node (PLN) including axial and popliteal were harvested and the cells were re-stimulated in vitro with free MOG peptide for 48 hours. IFNγ (top row) and IL-17 (bottom row) cytokine production was analyzed by ELISA. PLP1 peptide was included for control purposes. Data is representative of three individual experiments. Each bar represents the mean ± SD of triplicate wells. * p<0.005
IL-10 displays partial contribution to modulation of EAE by oral agg Ig-MOG
Previous studies have suggested that tolerance in the gut requires local production of suppressive cytokines such as IL-10, along with the expansion, activation, and accumulation of regulatory T cells in the gut associated lymphoid tissues (5). This bodes well with prior observations made with agg Ig-MOG given via the intraperitoneal route because the chimera crosslinks Fc gamma receptors (FcγR’s) on APCs (20) and induces the production of IL-10, a cytokine which sustains both bystander suppression (24) and expansion of regulatory T cells (30). However, given that sol Ig-MOG, which does not stimulate IL-10 production by APCs or expand Tregs (24, 30), had similar effects as the agg form of the chimera when given via the oral route (Fig. 1), one has to probe whether IL-10 contributes to modulation of EAE by oral agg Ig-MOG. Since the aggregated, but not soluble, form of the chimera induces IL-10 production by APCs (20, 24), only agg Ig-MOG was used to test the premises in IL-10-deficient C57BL/6 mice. The findings indicate that IL-10-deficient mice treated with Ig-MOG+STI displayed reduced disease severity compared to mice recipient of STI alone whether EAE was induced with MOG peptide (MMS 2.5 ±0.5 versus 1.2 ± 0.5, p < 0.01) or CNS homogenate (MMS 2.4 ± 0.6 versus 1.4 ± 0.5, p < 0.02) (Fig. 2A). In fact, Ig-MOG treatment had a similar suppressive effect in both IL-10-deficient and sufficient mice (Fig. 2B). Overall, these results suggest that the oral route for delivery of agg Ig-MOG is less dependent on IL-10 and other mechanisms are likely involved in oral T cell tolerance and modulation of EAE.
Figure 2. Evaluation of the contribution of IL-10 to Ig-MOG induced suppression of EAE.

(A) IL-10−/ − C57BL/6 mice were induced for EAE with MOG peptide (left) or CNS homogenate (right), and seven days later were treated orally with agg Ig-MOG + STI four times at 2-day intervals. Mice were monitored daily for disease severity for 30 days. The graphs show the mean clinical scores of mice treated with agg Ig-MOG + STI or STI alone. (B) shows comparison of mean clinical scores of IL-10−/ − and IL-10+/+ C57BL/6 mice induced for EAE with MOG peptide (left) or CNS homogenate (right), and treated orally with agg Ig-MOG + STI as in (A). Data is representative of two independent experiments with 5 mice per group.
Oral Ig-MOG interferes with the presenting function of lamina propria APCs
To further define the mechanism by which oral Ig-MOG modulates effector T cells and suppresses EAE, we sought to determine whether the treatment operates through interference with the presenting function of APCs. Accordingly, mice were given oral Ig-MOG and their SP, LP and PLN as well as MLN APCs were harvested, loaded with MOG peptide in vitro, and tested for stimulation of naïve 2D2 T cells. The results show that SP, PLN and MLN APCs were able to stimulate 2D2 T cells in a fashion similar to APCs emanating from mice that were not exposed to oral Ig-MOG (Fig. 3A and B). Surprisingly, however, the LP APCs displayed a reduced ability to stimulate the naïve T cells (Fig. 3A and B). In fact, T cell proliferation was reduced from 17,098 ± 577 cpm when the stimulation used LP APCs from STI mice to 8,856 ± 3,081 cpm for oral Ig-MOG +STI LP APCs (Fig. 3A). In parallel, IFNγ production by the T cells was also significantly reduced when the stimulation used LP APCs from oral Ig-MOG + STI recipient mice versus LP APCs from STI mice (compare 6.8 ± 0.9 versus 2.4 ± 0.5 ng/mL) (Fig. 3B). As with proliferation, there was no significant difference in IFNγ production by T cells stimulated with SP, PLN, or MLN APCs from oral agg Ig-MOG +STI versus APCs from STI alone treated mice. Given that the relative frequency of the major professional APCs was similar in the spleen and lamina propria (Fig. S1), it is unlikely that cell composition accounts for differential T cell inhibitory effects. Overall, these results suggest that oral agg Ig-MOG affects the stimulatory function of LP APCs. Previously, we have shown that Ig-MOG internalizes into APCs mostly via FcγRs for processing and delivery of MOG peptide to MHC class II molecules (20). Thus, we sought to determine whether the effect of oral Ig-MOG on the function of lamina propria APCs is dependent on FcγR-mediated up-take of the chimera. Accordingly, FcγR−/ − mice were given 1mg oral agg Ig-MOG and 24h later the LP APCs were assessed for stimulation of naïve 2D2 T cells in comparison with FcγR+/+ LP APCs. The results show that contrary to FcγR+/+ APCs, the LP APCs from FcγR−/ − mice were able to present free MOG peptide and stimulate 2D2 T cells similar to FcγR+/+ mice treated with STI alone (Fig. 3C). In fact, T cell proliferation was increased from 37,420 ± 1,419 cpm by FcγR+/+ LP APCs to 65,570 ± 4,026 cpm by the FcγR−/ − LP APCs (Fig. 3C, left panel). Similarly, IFNγ production increased from 13.4 ± 1.8 to 28.3 ± 6.7 ng/ml by the FcγR+/+ versus FcγR−/ − LP APCs (Fig. 3C, right panel). This data indicates that the effects of oral Ig-MOG on LP APCs is dependent on FcγR mediated uptake of Ig-MOG. Overall, the findings indicate that oral exposure to Ig-MOG affects the presenting function of lamina propria APCs. We then sought to test whether such a functional shift would be operational in vivo and counter the encephalitogenicity of MOG-specific 2D2 TCR transgenic T cells (21). Accordingly, C57BL/6 mice were fed 1mg agg Ig-MOG and their LP APCs (referred to as LPIg-MOG) were adoptively transferred into MHCII-deficient mice and tested for suppression of EAE mediated by 2D2 T cells. The results indicate that hosts recipient of LPIg-MOG APCs were resistant to EAE while control hosts given LP APCs (LPIg-W) from donors treated with Ig-W were susceptible to EAE (Fig. 4A). Indeed, the MMS of mice transferred with LPIg-W APCs was 1.7 ± 0.3 compared to 0.7 ± 0.3 for mice recipient of LPIg-MOG APCs (Table I). Furthermore, the animals were able to recover from disease and their cumulative clinical score was lower than those recipient of LPIg-W APCs (Table I). This phenomenon was specific to the LP, because splenic APCs (SPIg-MOG) from Ig-MOG fed mice did not confer resistance to EAE (Fig. 4B) and the pattern of disease was similar to hosts recipient of SPIg-w APCs (Table I). Thus, the functional shift of LP APCs induced by oral Ig-MOG is operational in vivo and counters EAE mediated by 2D2 TCR transgenic T cells. The results are interpreted to point toward an oral Ig-MOG mediated acquisition of tolerogenic function by LP APCs as the cells were unable to stimulate T cells both in vitro and in vivo.
Figure 3. Lamina propria APCs from oral Ig-MOG treated mice display a diminished ability to stimulate MOG specific T cells.
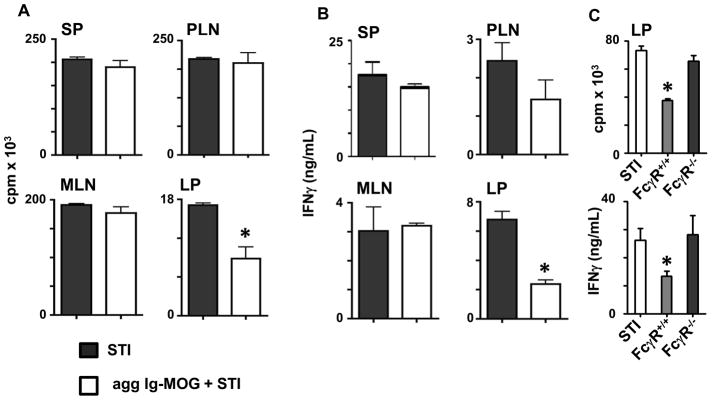
Groups of C57BL/6 mice were given STI alone or 1mg oral agg Ig-MOG + STI and their APCs from different lymphoid tissues were tested for stimulation of MOG-specific 2D2 T cells. (A) shows the proliferative responses of 2D2 CD4 T cells stimulated with MOG peptide-loaded SP, PLN, MLN and LP APC. (B) shows the cytokine responses of the 2D2 T cells described in (A) as measured by ELISA. (C) shows proliferative and IFNγ responses of 2D2 T cells stimulated with MOG peptide-loaded LP APCs that were obtained from FcγR+/+ and FcγR−/ − C57BL/6 mice recipient of 1mg oral agg Ig-MOG+STI. APCs from FcγR+/+ mice treated with STI alone were included for control purposes. Data is representative of two independent experiments and each bar represents the mean ± SD of triplicate wells from 3 to 4 mice. *p < 0.04 (A and B) or < 0.003 for (C).
Figure 4. Effector T cells are required for lamina propria APCs to acquire tolerogenic functions.

MHC II−/ − C57BL/6 mice adoptively transferred with 2D2 T cells and MHCII+/+ APCs were induced for EAE with MOG peptide. Donor APCs were harvested from the LP or SP of C57BL/6 mice (A–B) or Rag2−/ − C57BL/6 mice (C) recipient of agg Ig-MOG + STI or agg Ig-W + STI. Mice were monitored daily for clinical signs of disease. Data is representative of three individual experiments with 5 mice per group. LPIg-MOG, LPIg-W and SPIg-MOG refer to LP or SP APCs from mice treated with oral Ig-MOG or Ig-W.
Table I.
Lamina propria, but not splenic, APCs transfer T cell tolerance against diverse specificitiesa
| Mice | APC origin | Tolerogen | Disease onset | Recovery day | Mean maximal disease score | Cumulative disease score |
|---|---|---|---|---|---|---|
| C57BL/6 | ||||||
| Rag 2+/+ | LP | Ig-MOG | 15.0 ± 1.0 | 34.5 ± 6.5 | 0.7 ± 0.3*1 | 11.5 ± 6.5*2 |
| Ig-W | 14.0 ± 0.6 | 41.3 ± 1.5 | 1.7 ± 0.3 | 34.3 ± 1.1 | ||
| SP | Ig-MOG | 15.0 ± 0.7 | 44.7 ± 0.3 | 1.7 ± 0.2 | 28.3 ± 4.3 | |
| Ig-W | 16.0 ± 0.7 | 43.3 ± 0.3 | 1.7 ± 0.2 | 25.0 ± 0.7 | ||
| Rag 2−/ − | LP | Ig-MOG | 10.2 ± 0.5 | 56.8 ± 1.6 | 2.0 ± 0.0 | 62.6 ± 2.1 |
| Ig-W | 10.3 ± 0.4 | 55.3 ± 1.6 | 2.0 ± 0.0 | 62.9 ± 0.9 | ||
| OTII | ||||||
| Rag 2+/+ | LP | Ig-OVA | 14.3 ± 1.5 | 37.7 ± 0.9 | 1.0 ± 0.0*3 | 12.5 ± 0.8*4 |
| Ig-MOG | 10.6 ± 0.7 | 47.0 ± 3.5 | 2.0 ± 0.0 | 55.0 ± 1.6 | ||
| Ig-W | 11.3 ± 0.7 | 50.0 ± 4.0 | 2.0 ± 0.0 | 59.8 ± 2.9 | ||
| SP | Ig-OVA | 11.0 ± 0.6 | 56.0 ± 0.6 | 2.0 ± 0.0 | 57.8 ± 1.6 | |
Data represent 3 individual experiments with 5 mice per group and are presented as mean ± SE (by unpaired, two-tailed Student’s t test).
indicates statistical analysis between groups by unpaired, two-tailed Student’s t test or one-way ANOVA as indicated.
p=0.047 (t test);
p=0.026 (t test);
p<0.0001 (ANOVA);
p<0.0001 (ANOVA)
The endowment of LP APCs with tolerogenic functions is dependent on T cells
Since Ig-MOG, but not Ig-W, was able to endow LP APCs with tolerogenic function, we hypothesized that antigen contributes to the acquisition of tolerogenic function by LP APCs. In this line of reasoning one would envision that communication with T cells, a phenomenon that requires antigen (presumably MOG peptide of Ig-MOG), influences acquisition of tolerance by LP APCs. To test this premise, oral Ig-MOG was administered to Rag2-deficient (Rag2−/ −) mice, which would be devoid of T cells, and the resulting LP APCs were evaluated for suppression of EAE. The findings show that upon transfer into MHC II−/ − mice along with 2D2 T cells, the hosts were unable to resist EAE induced with MOG peptide (Fig. 4C). Indeed, LP APCs from Rag2−/ − mice fed Ig-MOG (LP-Rag2−/ −IgMOG) did not confer resistance against EAE and the pattern of disease was similar to mice recipient of control LP APCs from Rag2−/ − mice fed Ig-W (LP-Rag2− /−IgW). Also, there was no significant difference among the test and control LP APCs in the onset of disease, the mean maximal or the cumulative scores (Table I). Furthermore, while LP APCs from Rag2+/+ mice (LP-Rag2+/+IgMOG) acquired tolerogenic function and were unable to stimulate proliferation of 2D2 T cells in vitro, those emanating from Rag2−/ − mice (LP-Rag2−/ −IgMOG) did stimulate the 2D2 T cells to a similar extent as LP-Rag2−/ −IgW APCs (Fig. S2)4. Taken together, these results indicate that acquisition of tolerogenic function by LP APCs is dependent on T cells. Also, since Ag was also required (compare Ig-MOG and Ig-W) it is likely that Ag driven interaction of LP APCs with T cells is necessary for the acquisition of tolerogenic function.
Tolerogenic LP APCs display broad suppressive function against diverse T cell specificities
Oral Ig-MOG was able to suppress both MOG peptide and CNS homogenate EAE (Fig. 1). This implies that LP APCs were able to drive tolerance against diverse T cell specificities. To test this premise, we assayed tolerogenic LP APCs for suppression of T cells of unrelated specificity both in vitro and in vivo. Accordingly, we utilized Ig-OVA, carrying ovalbumin (OVA)323–339 peptide (25), to generate LP APCs to target the corresponding OVA-specific OTII T cells (23) or the unrelated MOG-specific 2D2 T cells and vice versa with Ig-MOG. This was accomplished as follows: 2D2 mice were fed 1mg agg Ig-MOG, agg Ig-OVA, or agg Ig-W to generate LP2D2/Ig-MOG, (matching specificity), LP2D2/Ig-OVA (mismatched specificity) and LP2D2/Ig-W, (no Ag control). On the other hand, OTII mice were fed 1 mg agg Ig-MOG, agg Ig-OVA, or agg Ig-W to generate LPOTII/Ig-MOG, (mismatched specificity), LPOTII/Ig-OVA (matched specificity) and LPOTII/Ig-W, (no Ag control). These LP APCs were then tested for suppression of diverse T cell specificities. The results show that LP2D2/Ig-MOG and LPOTII/Ig-OVA APCs were able to suppress both 2D2 and OTII cells (Fig. 5A and B). On the other hand, the mismatched APCs (LP2D2/Ig-OVA and LPOTII/Ig-MOG) could not suppress either transgenic T cell and the proliferation levels were similar to those observed with LP APCs from TCR transgenic mice fed Ig-W, the chimera that does not carry any MOG or OVA (Fig. 5A and B). Again, the SP APCs were not able to display broad suppression under any circumstance (Fig. 5C). These results indicate that while the initial contact (induced by orally fed Ig chimera) between T cells and LP APCs must be Ag specific, the acquired tolerogenic functions are broad and extend to diverse T cell specificities. Similar findings were observed when the LP APCs were tested for tolerogenic function in vivo (Fig. 5D and E). Indeed, LPOTII/Ig-OVA APCs were able to suppress 2D2 T cell-driven EAE induced by immunization of MHC class II-deficient hosts with MOG peptide. Again LPOTII/Ig-MOG, like LPOTII/Ig-W, APCs were unable to suppress MOG/2D2 EAE (Fig. 5D). In fact, LPOTII/Ig-OVA APCs reduced the MMS from 2.0 ± 0 to 1.0 ± 0 relative to LPOTII/Ig-MOG and LPOTII/Ig-W APCs (Table I). The in vivo broad suppressive functions are restricted to LP APCs as SPOTII/Ig-OVA APCs could not suppress MOG/2D2 EAE (Fig. 5E, and Table I). Overall, these results indicate that the acquired tolerogenic functions are broad and extend to diverse T cell specificities as tested by suppression of proliferation in vitro and modulation of EAE in vivo.
Figure 5. Lamina propria APCs display suppressive function against diverse T cell specificities.

OT-II and 2D2 TCR transgenic mice were given oral agg Ig-OVA, agg Ig-MOG, or agg Ig-W along with STI and their LP and SP APCs were tested for transfer of tolerance both in vitro and in vivo. (A) shows proliferative responses of OVA-specific OTII T cells stimulated with OVA peptide presented by the indicated LP APCs (B) shows proliferative responses of 2D2 T cells stimulated with MOG peptide presented by the indicated LP APCs. (C) shows proliferative responses of 2D2 T cells stimulated with MOG peptide presented by the indicated SP APCs. *p<0.04. LP2D2/Ig-MOG refers to LP APCs from 2D2 TCR transgenic mice treated with Ig-MOG; LPOTII/Ig-OVA refers to LP APCs from OTII TCR transgenic mice treated with Ig-OVA; LP2D2/Ig-OVA refers to LP APCs from 2D2 TCR transgenic mice treated with Ig-OVA; LP2D2/Ig-W refers to LP APCs from 2D2 TCR transgenic mice treated with Ig-W; LPOTII/Ig-W refers to LP APCs from OTII TCR transgenic mice treated with Ig-W; Similar nomenclature is used for SP APCs. (D and E) shows the mean clinical score of EAE of MHCII−/ − C57BL/6 mice recipient of 2D2 TCR transgenic T cells and the indicated LP or SP APCs. EAE was induced with MOG peptide after transfer of the T cells and APCs. Mice were graded for disease severity daily using the scale described in Materials and Methods. Data represents 3 individual experiments with 5 mice per group.
Of note, the observation that OT-II LP APCs exposed to oral Ig-MOG could not transfer tolerance to MOG EAE indicates that this phenomenon is not due to carry over of residual Ig-MOG to the host mice, but rather relies upon the APC-T cell interaction during the initial exposure.
Oral Ig-MOG induces a T cell-dependent up-regulation of PD-L1 on LP APCs
The tolerogenic functions of LP APCs could be due to blockade of MHC class II molecules by peptide carryover or to a lack of up-regulation of costimulatory molecules. The fact that LP-OTII/Ig-MOG APCs were able to present the I-Ab-restricted OVA peptide and stimulate OTII T cells indicates that MHC II molecules were not saturated with MOG peptide (also restricted to I-Ab) carryover (Fig. S3). Similarly, LP2D2/Ig-OVA APCs were able to induce proliferation of 2D2 T cells upon stimulation with MOG peptide, again indicating that MHC II blockade was not responsible for the tolerogenic function of LP APCs. In regards to costimulation, the tolerogenic LP APCs generated by feeding Ig-MOG to Rag2-sufficient mice had a pattern of expression of key costimulatory molecules such as CD80, CD86, and CD40, similar to non tolerogenic APCs generated in Rag2-deficient mice (Fig. S4). Thus, it is unlikely that a lack of up-regulation of costimulatory molecules was responsible for acquisition of tolerogenic functions by LP APCs. Moreover, CD103, which marks a subset of LP DCs specialized in tolerance by promoting the generation of T regulatory cells (8) was not affected, indicating that oral Ig-MOG may not affect the function of this subset. In the face of this dilemma, we resorted to explore an alternative mechanism in which LP APCs may up-regulate inhibitory molecules such as PD-L1 and PD-L2 to exercise tolerogenic function. To this end, we assessed the expression of these molecules on LP APCs under tolerizing (Rag2+/+) and non-tolerizing (Rag2−/ −) conditions. The findings indicate that PD-L1, but not PD-L2, expression is up-regulated on the surface of CD4+CD11c+ and CD8α+CD11c+ DC subsets from Rag2+/+, but not Rag2−/ −, mice fed Ig-MOG (Fig. 6A). No significant PD-L1 up-regulation was observed on LP CD11b+ cells. The fold change in mean fluorescence intensity of PD-L1 was 3.3 ± 0.5 and 3.1 ± 0.4 for CD4+ and CD8α+ DCs, respectively, from Rag2+/+ relative to those from Rag2−/ − (which were set to 1) (Fig. 6B). PD-L2 had similar MFIs for both DC subsets from both strains (Fig. 6B). Taken together, these results indicate that the acquired tolerogenic function by LP APCs may lie upon the up-regulation of PD-L1 on the CD8α−CD4+ and CD8α+ DC subsets.
Figure 6. Exposure to oral Ig-MOG induces PD-L1 up-regulation on lamina propria DCs.

LP APCs were harvested from Rag2+/+ and Rag2−/ − C57BL/6 mice 24 h after oral administration of agg Ig-MOG + STI and expression of PD-L1 and PD-L2 was analyzed on specific subsets. (A) shows representative histograms of PD-L1 (top row) and PD-L2 (bottom row) expression on the indicated cell populations. Shaded histograms represent staining with matching isotype control. (B) shows the fold change in MFI for the indicated markers for LP DC subsets of Rag2+/+ relative to Rag2−/ − mice. This was determined by setting the MFI of LP DCs of Rag2−/ −mice to 1. Data is representative of at least three independent experiments. *p < 0.05 by one-sample t test.
PD-L1 sustains the tolerogenic functions of LP APCs
PD-L1 on APCs usually binds its ligand PD-1 and the interactions serve to alleviate T cell hyperactivation (31). Since the LP APCs up-regulated PD-L1 and were unable to stimulate T cells, it is possible that PD-L1 plays a prominent role in the tolerogenic function of the APCs. To test this premise, we performed blockade of PD-L1 with anti-PD-L1 antibody and tested the LP APCs for loss of tolerogenic functions. Indeed, this postulate proved correct and LPIg-MOG APCs that were coated with anti-PD-L1 antibody regained the ability to induce proliferation and the production of IFNγ or IL-17 by 2D2 T cells upon stimulation with MOG peptide in vitro (Fig. 7A–C). This is due to blockade of PD-L1 because LPIg-MOG APCs that were incubated with the isotype control retained their tolerogenic function and could not stimulate the 2D2 T cells (Fig. 7A–C). In fact, proliferation and cytokine production during PD-L1 blockade was significantly higher relative to isotype control but comparable to non-tolerogenic LPIg-W APCs that originated from mice given oral Ig-W. In vivo, when the anti-PD-L1-coated LPIg-MOG APCs were transferred to MHC class II−/ − mice recipient of 2D2 T cells, the hosts developed MOG-peptide-induced EAE similar to mice recipient of non-tolergenic LP-Ig-W APCs while those recipient of isotype-coated LPIg-MOG APCs remained resistant against the disease (Fig. 7D). Indeed the isotype coated LPIg-MOG APCs had a reduced MMS compared to LPIg-W APCs (2.0 ± 0.5 versus 1.16 ± 0.3, p<0.03), while disease induced by anti-PD-L1-coated LPIg-MOG APCs remained severe (MMS 2.3 ± 0.2). Taken together, this data set demonstrates that the tolerogenic function of LP APCs lies on the up-regulation of PD-L1 and operates through its inhibitory functions.
Figure 7. Blockade of PD-L1 on lamina propria APCs nullifies T cell tolerance.

LP APCs were harvested from C57BL/6 mice recipient of agg Ig-W + STI or agg Ig-MOG + STI, incubated with blocking anti-PD-L1 antibody and tested for tolerogenic function. (A–C) shows proliferation (A) and IFNγ (B) and IL-17 (C) production by 2D2 T cells incubated with MOG peptide-loaded LP APCs that were previously blocked with anti-PD-L1 antibody or isotype matched control. *p < 0.05. (D) shows the mean clinical score of EAE induced by MOG peptide in MHCII−/ − mice recipient of 2D2 T cells and LP APCs that were previously blocked with anti-PD-L1 antibody or isotype matched control. Data is representative of three independent experiments with 5 mice per group.
The acquisition of tolerogenic function by LP APCs is independent of T regulatory cells
Since interaction with T cells is required for induction of tolerogenic LP APCs, and T regulatory cells have been shown to play a profound role in oral tolerance (2, 3, 5), we sought to determine whether interaction with Tregs is necessary for the generation of these PD-L1 expressing APCs. This was accomplished by depleting Tregs with diphtheria toxin (DT) and examining LP APCs for PD-L1 expression and suppressive function. The results show that C57BL/6 FoxP3.GFP.DTR mice given DT for 3 consecutive days display Treg depletion in peripheral blood, SP, and LP (Fig. 8A). Mice that were depleted of Tregs and given oral Ig-MOG yielded PD-L1-expressing LP APCs that were able to suppress 2D2 effector T cells (Fig. 8B-D). Indeed, relative to the mice depleted of Tregs but treated with STI alone (STI+DT + No Ig-MOG), the fold change in PD-L1 MFI for LPIg-MOG DCs was similar (2.1 ± 0.1 versus 2.0 ± 0.1) in Treg depleted (STI+ DT+IgMOG) versus non-depleted (STI+NO DT+ Ig-MOG) mice (Fig. 8C). Furthermore, the LPIg-MOG DCs from Treg depleted and non-depleted mice induced equivalent suppression of 2D2 T cells when loaded with MOG peptide (Fig8 D). In fact, T cell proliferation was reduced from 18,013 ± 1077 cpm when the stimulation used non-tolerogenic LP DCs (STI+DT + No Ig-MOG) to 9,826 ± 637 with LPIg-MOG DCs from Treg depleted (STI+ DT+IgMOG) mice, a reduction that is equivalent (9470 ± 223 cpm) to tolerogenic LP DCs from mice that were not depleted of Tregs (STI+NO DT+ Ig-MOG). Thus, the acquisition of tolerogenic function by LP DCs does not require interaction with Tregs.
Figure 8. LP APCs acquire tolerogenic function independent of T regulatory cells.

FoxP3.GFP.DTR C57BL/6 mice were depleted of Tregs by injection of 300 ng diphtheria toxin or saline i.p. for 3 consecutive days. At the time of the third DT treatment, the mice were given 1mg oral agg Ig-MOG + STI or STI alone and their LP APCs were analyzed for PD-L1 expression and tolerogenic function. (A) shows the frequency of Tregs in the peripheral blood (PBL), SP, and LP of mice recipient of 3 injections of DT or control saline. This was determined by staining with anti-CD4 and analyzing GFP (FoxP3) expression on CD4-positive T cells. (B) shows representative histograms of PD-L1 expression on LP APCs 24hrs after oral treatment. (C) shows the fold change in MFI of PD-L1 on LP APCs of the indicated groups. The bars represent the ratio relative to the MFI obtained with the STI + DT treatment which was set to 1. *p < 0.04 by one-sample t test. (D) shows the proliferative responses of 2D2 CD4 T cells stimulated with MOG peptide-loaded LP APCs from the indicated treatment groups. *p < 0.02. Data is representative of three independent experiments.
Both CD8α+ and CD8α−CD4+ LP dendritic cell subsets acquire tolerogenic function upon treatment with oral Ig-MOG
We have previously shown that intraperitoneal (i.p.) agg Ig-MOG drives modulation of myelin-reactive T cells through lack of costimulation and IL-10 production exclusively by the CD8α−CD4+ DC subset (20). However, given that T cell tolerance by oral Ig-MOG is less dependent on IL-10, and that both the CD8α+ and CD8α−CD4+ DC subsets in the LP have up-regulated PD-L1, it is likely that both subsets play a major role in oral tolerance. To test this premise, the LP DCs from mice recipient of oral Ig-MOG were separated into CD8α+ and CD8α−CD4+ subsets and tested for suppression of EAE. Accordingly, LP CD8α+Ig-MOG and LP CD8α−CD4+Ig-MOG were co-transferred with 2D2 TCR transgenic T cells into MHCII−/ − mice and the hosts were induced for EAE with MOG peptide. The results show that LP CD8α+Ig-MOG and LP CD8α−CD4+Ig-MOG, but not splenic CD8α+ counterparts (SP CD8α+Ig-MOG), were able to transfer tolerance against EAE (Fig. 9). In fact, the MMS of mice recipient of CD8α+ or CD8α−CD4+ LP DCs was 1.0 ± 0 whilethose transferred with control SP CD8α+ DCs had a MMS of 2.33 ± 0.1 (p<0.001). Overall, these results indicate that both CD8α+ and CD8α−CD4+ DC subsets acquire tolerogenic function within the lamina propria upon treatment with oral Ig-MOG.
Figure 9. Lamina propria CD8α+ and CD8α− DC subsets transfer tolerance.

EAE was induced by MOG peptide in MHCII−/ − mice recipient of 2D2 T cells and LP or SP DC subsets from agg Ig-MOG fed mice. Mice were monitored for disease severity for 30 days. Data is representative of 2 independent experiments with 5 mice per group.
DISCUSSION
In recent years the Ig delivery system has proven effective against autoimmune diabetes and EAE when applied via the intraperitoneal route (20, 24, 32–36). In fact, a wealth of information has been gained on the mechanisms underlying Ig chimera-mediated tolerance including control of costimulation (37), migration (36), expansion of Tregs (30, 35) and induction of suppressive cytokines (20, 24).
Oral tolerance, despite being the most vital means for preventing immune responses to nutritional essentials and gut floral commensals, remains poorly defined (29, 38). Moreover, given the practicality and the versatility of the gut immune system, a tremendous consideration has lately been given to the oral route for vaccination and stimulation of immunity as well as induction of tolerance and suppression of self-reactivity to overcome autoimmune diseases (29, 38). Thus, we sought to determine whether Ig-MOG given via the oral route would be effective against EAE and to delineate the mechanisms by which oral tolerance restrains myelin-reactive encephalitogenic T cells. IL-10, whether produced by Tregs (30), B cells (39, 40), or DCs (9, 24), usually serves an anti-inflammatory role and contributes to the modulation of autoimmunity. In fact, IL-10 produced by APCs upon intraperitoneal treatment with agg Ig-MOG contributed bystander suppression of autoreactive T cells and modulation of EAE (20, 24). In the oral Ig-MOG model, however, this cytokine did not seem to play a major role in the induction of protection against EAE (Fig. 2). The alternative then was to explore IL-10-independent and other mechanisms for the observed effective suppression of EAE by oral Ig-MOG. The earliest investigations of peripheral T cell tolerance indicated that APCs play a major role in the modulation of self-reactive T cells, particularly through limited costimulation (37). Studies conducted towards the role of APCs in Ig-MOG oral tolerance pointed to lamina propria APCs as major contributors to such tolerance (Fig. 3). Indeed, LP APCs, but not their splenic or lymph node counterparts, were unable to stimulate T cell responses in vitro and in vivo after oral Ig-MOG exposure (Fig. 3 & 4). In fact, these LP APCs displayed tolerogenic function against EAE (Fig. 4). While the observations bode well with previous findings suggesting that mucosal dendritic cells are required for oral tolerance (41), the present study demonstrates that T cells are also required during exposure to oral Ig-MOG for the LP APCs to acquire the tolerogenic functions. This is supported by the findings that, if the exposure to oral Ig-MOG is carried out in Rag2−/ − mice, where T cells are minimal, the LP APCs could not become tolerogenic (Fig. 4). Interestingly, the T cells have to engage the LP APCs through Ag in order for the latter to acquire the tolerogenic functions. This was initially evident when FcγR−/ − mice fed Ig-MOG could not generate tolerogenic LP APCs as T cells are available in this mouse but up-take and presentation of Ig-MOG is not operative due to the deficiency in FcγRs (Fig. 3). A more direct evidence for the requirement of Ag-specific interaction between LP APCs and T cells for the former to acquire tolerogenic functions emanates from the observation that the specificity of naïve T cells had to match the oral chimera for the process to materialize (Fig. 5). This gut-priming induced tolerance, which we believe represents an Ag-specific APC education process, is not unique to Ig-MOG as Ig-OVA was also able to yield tolerogenic LP APCs and also required the presence of OVA-specific OT-II T cells (Fig. 5). As much as we were surprised by the requirement for interaction with T cells for the LP APCs to acquire tolerogenic function, we were puzzled by the findings showing that LP APCs subsequently display broad suppressive functions against diverse T cells regardless of Ag specificity. Notably, LP APCs, which acquired tolerogenic functions by exposure to Ig-MOG, were able to suppress stimulation of OVA-specific OTII T cells and vice versa (Fig. 5). Moreover, LP APCs generated by exposure to Ig-OVA were able to suppress 2D2 T cells in vivo and protect against EAE (Fig. 5). While blockade of MHC molecules and lack of costimulation have previously been shown to suppress the development of autoimmunity (37, 42), neither scenario is likely in this form of suppression. Saturation of MHC II molecules by peptide carry over from exposure to the Ig chimera is not plausible because LP APCs exposed to chimeras that do not match the effector T cells were able to stimulate T cells restricted to the same I-Ab MHC II allele (Fig. S2). In the same line of reasoning, a lack of costimulation is not a likely mechanism for the acquired tolerogenic function because there was no evident down regulation of the key costimulatory molecules (Fig. S3). Since Ag presentation and costimulation capabilities remained intact, it was logical to envision that LP APCs carry out their tolerogenic function through a process of active suppression, especially since the cells displayed up-regulation of PD-L1, a member of the B7 molecule family that is expressed by APCs and functions as an inhibitory receptor ligand for control of self-reactive T cells and modulation of autoimmunity (31, 43–45). In fact, blockade of PD-L1 with an anti-PD-L1 antibody nullifies the tolerogenic functions of LP APCs (Fig. 7). These findings not only explain the lack of oral tolerance in mice deficient for PD-L1 (46, 47) but also bode well with the broad suppressive function observed with LP APCs (Fig. 5) and the inhibition by oral Ig-MOG of CNS homogenate induced EAE which involves multiple epitopes and diverse T cells specificities. Finally, in contrast to the intraperitoneal route, which relied solely on the CD8α−CD4+ splenic DC subset to suppress EAE (20), the oral route extended tolerogenic function to both CD8α−CD4+ and CD8α+ DC subsets, though this was restricted to the lamina propria.
Overall, this study uncovers a novel mechanism by which APCs acquire tolerogenic function upon oral administration of tolerogen. Indeed, we show that the lamina propria is the organized lymphoid tissue uniquely suited to facilitate interaction of resident APCs presenting the oral tolerogen and the cognate T cells. Interestingly, interaction with effector rather than Tregs are responsible for acquisition of tolerogenic function by LP APC (Fig.8). Ultimately, during this antigen-specific contact the APCs are schooled to acquire tolerogenic functions capable of broad suppression against diverse T cell specificities. Specifically, the broad suppression was mediated by the inhibitory PD-L1 molecule which was up-regulated on the LP APCs during oral tolerogen-driven interaction with effector T cells. Interestingly, both CD8α+ and CD8α−CD4+ DC subsets acquired PD-L1-dependent tolerogenic functions and were able to transfer tolerance against myelin-reactive T cells and suppress EAE. Usually DCs promote tolerance by supporting the induction of Tregs (8) or by producing anti-inflammatory cytokines such as TGFβ (48) or IL-10 (9). This study reveals an as of yet unrecognized mechanism whereby DCs acquire active tolerance from T cells and execute suppression through the PD-L1 inhibitory molecule. Although the study raises new questions pertaining to the specific acquisition of tolerogenic function in the LP but not other lymphoid tissues, as well as the mechanism by which effector T cells induce up-regulation of PD-L1, it underscores the usefulness of the oral route as an effective, practical, and attractive approach for amelioration of autoimmunity.
Supplementary Material
Acknowledgments
Funding
This work was supported by grants RO1 NS057194, (to H.Z.) from the National Institutes of Health, and by the J. Lavenia Edwards endowment (to HZ). J.A.C. was supported by Life Sciences Fellowships from the University of Missouri, Columbia.
Footnotes
Abbreviations. agg, aggregated; CNS, central nervous system; DT, diphtheria toxin; DTR, DT receptor; EAE, experimental autoimmune encephalomyelitis; LP, lamina propria; MLN, mesenteric lymph node; MMS, mean maximal score; MOG, myelin oligodendrocyte glycoprotein; PD-L1, programmed death ligand 1. PLN, peripheral lymph node; SP, spleen; STI, soy trypsin inhibitor; Treg, regulatory T cell;
Supplemental Material. The online version of this article contains supplemental material.
References
- 1.Higgins PJ, Weiner HL. Suppression of experimental autoimmune encephalomyelitis by oral administration of myelin basic protein and its fragments. J Immunol. 1988;140:440–445. [PubMed] [Google Scholar]
- 2.Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol. 2003;3:331–341. doi: 10.1038/nri1057. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X, Izikson L, Liu L, Weiner HL. Activation of CD25(+)CD4(+) regulatory T cells by oral antigen administration. J Immunol. 2001;167:4245–4253. doi: 10.4049/jimmunol.167.8.4245. [DOI] [PubMed] [Google Scholar]
- 4.Faria AM, Weiner HL. Oral tolerance and TGF-beta-producing cells. Inflammation & allergy drug targets. 2006;5:179–190. doi: 10.2174/187152806778256034. [DOI] [PubMed] [Google Scholar]
- 5.Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, Muller W, Sparwasser T, Forster R, Pabst O. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity. 2011;34:237–246. doi: 10.1016/j.immuni.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Lelouard H, Fallet M, de Bovis B, Meresse S, Gorvel JP. Peyer’s patch dendritic cells sample antigens by extending dendrites through M cell-specific transcellular pores. Gastroenterology. 2012;142:592–601. e593. doi: 10.1053/j.gastro.2011.11.039. [DOI] [PubMed] [Google Scholar]
- 7.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, Granucci F, Kraehenbuhl JP, Ricciardi-Castagnoli P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2:361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 8.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iwasaki A, Kelsall BL. Freshly isolated Peyer’s patch, but not spleen, dendritic cells produce interleukin 10 and induce the differentiation of T helper type 2 cells. J Exp Med. 1999;190:229–239. doi: 10.1084/jem.190.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204:1765–1774. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 12.Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y, V, Kuchroo K, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science. 1994;265:1237–1240. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- 14.Khoury SJ, Hancock WW, Weiner HL. Oral tolerance to myelin basic protein and natural recovery from experimental autoimmune encephalomyelitis are associated with downregulation of inflammatory cytokines and differential upregulation of transforming growth factor beta, interleukin 4, and prostaglandin E expression in the brain. J Exp Med. 1992;176:1355–1364. doi: 10.1084/jem.176.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thompson HS, Staines NA. Suppression of collagen-induced arthritis with pergastrically or intravenously administered type II collagen. Agents Actions. 1986;19:318–319. doi: 10.1007/BF01971237. [DOI] [PubMed] [Google Scholar]
- 16.Hancock WW, Polanski M, Zhang J, Blogg N, Weiner HL. Suppression of insulitis in non-obese diabetic (NOD) mice by oral insulin administration is associated with selective expression of interleukin-4 and -10, transforming growth factor-beta, and prostaglandin-E. Am J Pathol. 1995;147:1193–1199. [PMC free article] [PubMed] [Google Scholar]
- 17.Ma SW, Zhao DL, Yin ZQ, Mukherjee R, Singh B, Qin HY, Stiller CR, Jevnikar AM. Transgenic plants expressing autoantigens fed to mice to induce oral immune tolerance. Nat Med. 1997;3:793–796. doi: 10.1038/nm0797-793. [DOI] [PubMed] [Google Scholar]
- 18.Zhang ZJ, Davidson L, Eisenbarth G, Weiner HL. Suppression of diabetes in nonobese diabetic mice by oral administration of porcine insulin. Proc Natl Acad Sci U S A. 1991;88:10252–10256. doi: 10.1073/pnas.88.22.10252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neurath MF, Fuss I, Kelsall BL, Presky DH, Waegell W, Strober W. Experimental granulomatous colitis in mice is abrogated by induction of TGF-beta-mediated oral tolerance. J Exp Med. 1996;183:2605–2616. doi: 10.1084/jem.183.6.2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Legge KL, Gregg RK, Maldonado-Lopez R, Li L, Caprio JC, Moser M, Zaghouani H. On the role of dendritic cells in peripheral T cell tolerance and modulation of autoimmunity. J Exp Med. 2002;196:217–227. doi: 10.1084/jem.20011061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med. 2003;197:1073–1081. doi: 10.1084/jem.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 23.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 24.Legge KL, Min B, Bell JJ, Caprio JC, Li L, Gregg RK, Zaghouani H. Coupling of peripheral tolerance to endogenous interleukin 10 promotes effective modulation of myelin-activated T cells and ameliorates experimental allergic encephalomyelitis. J Exp Med. 2000;191:2039–2052. doi: 10.1084/jem.191.12.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li L, Legge KL, Min B, Bell JJ, Gregg R, Caprio J, Zaghouani H. Neonatal immunity develops in a transgenic TCR transfer model and reveals a requirement for elevated cell input to achieve organ-specific responses. J Immunol. 2001;167:2585–2594. doi: 10.4049/jimmunol.167.5.2585. [DOI] [PubMed] [Google Scholar]
- 26.Lee HH, Hoeman CM, Hardaway JC, Guloglu FB, Ellis JS, Jain R, Divekar R, Tartar DM, Haymaker CL, Zaghouani H. Delayed maturation of an IL-12-producing dendritic cell subset explains the early Th2 bias in neonatal immunity. J Exp Med. 2008;205:2269–2280. doi: 10.1084/jem.20071371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lefrancois L, Lycke N. Isolation of mouse small intestinal intraepithelial lymphocytes, Peyer’s patch, and lamina propria cells. Curr Protoc Immunol Chapter. 2001;3(Unit 3):19. doi: 10.1002/0471142735.im0319s17. [DOI] [PubMed] [Google Scholar]
- 28.Romani N, Reider D, Heuer M, Ebner S, Kampgen E, Eibl B, Niederwieser D, Schuler G. Generation of mature dendritic cells from human blood. An improved method with special regard to clinical applicability. J Immunol Methods. 1996;196:137–151. doi: 10.1016/0022-1759(96)00078-6. [DOI] [PubMed] [Google Scholar]
- 29.Weiner HL, da Cunha AP, Quintana F, Wu H. Oral tolerance. Immunol Rev. 2011;241:241–259. doi: 10.1111/j.1600-065X.2011.01017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu P, Gregg RK, Bell JJ, Ellis JS, Divekar R, Lee HH, Jain R, Waldner H, Hardaway JC, Collins M, Kuchroo VK, Zaghouani H. Specific T regulatory cells display broad suppressive functions against experimental allergic encephalomyelitis upon activation with cognate antigen. J Immunol. 2005;174:6772–6780. doi: 10.4049/jimmunol.174.11.6772. [DOI] [PubMed] [Google Scholar]
- 31.Selenko-Gebauer N, Majdic O, Szekeres A, Hofler G, Guthann E, Korthauer U, Zlabinger G, Steinberger P, Pickl WF, Stockinger H, Knapp W, Stockl J. B7-H1 (programmed death-1 ligand) on dendritic cells is involved in the induction and maintenance of T cell anergy. J Immunol. 2003;170:3637–3644. doi: 10.4049/jimmunol.170.7.3637. [DOI] [PubMed] [Google Scholar]
- 32.Gregg RK, Bell JJ, Lee HH, Jain R, Schoenleber SJ, Divekar R, Zaghouani H. IL-10 diminishes CTLA-4 expression on islet-resident T cells and sustains their activation rather than tolerance. J Immunol. 2005;174:662–670. doi: 10.4049/jimmunol.174.2.662. [DOI] [PubMed] [Google Scholar]
- 33.Gregg RK, Jain R, Schoenleber SJ, Divekar R, Bell JJ, Lee HH, Yu P, Zaghouani H. A sudden decline in active membrane-bound TGF-beta impairs both T regulatory cell function and protection against autoimmune diabetes. J Immunol. 2004;173:7308–7316. doi: 10.4049/jimmunol.173.12.7308. [DOI] [PubMed] [Google Scholar]
- 34.Jain R, Tartar DM, Gregg RK, Divekar RD, Bell JJ, Lee HH, Yu P, Ellis JS, Hoeman CM, Franklin CL, Zaghouani H. Innocuous IFNgamma induced by adjuvant-free antigen restores normoglycemia in NOD mice through inhibition of IL-17 production. J Exp Med. 2008;205:207–218. doi: 10.1084/jem.20071878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tartar DM, VanMorlan AM, Wan X, Guloglu FB, Jain R, Haymaker CL, Ellis JS, Hoeman CM, Cascio JA, Dhakal M, Oukka M, Zaghouani H. FoxP3+RORgammat+ T helper intermediates display suppressive function against autoimmune diabetes. J Immunol. 2010;184:3377–3385. doi: 10.4049/jimmunol.0903324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wan X, Guloglu FB, VanMorlan AM, Rowland LM, Jain R, Haymaker CL, Cascio JA, Dhakal M, Hoeman CM, Tartar DM, Zaghouani H. Mechanisms underlying antigen-specific tolerance of stable and convertible Th17 cells during suppression of autoimmune diabetes. Diabetes. 2012;61:2054–2065. doi: 10.2337/db11-1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Legge KL, Bell JJ, Li L, Gregg R, Caprio JC, Zaghouani H. Multimodal antigen specific therapy for autoimmunity. Int Rev Immunol. 2001;20:593–611. doi: 10.3109/08830180109045580. [DOI] [PubMed] [Google Scholar]
- 38.Tsuji NM, Kosaka A. Oral tolerance: intestinal homeostasis and antigen-specific regulatory T cells. Trends Immunol. 2008;29:532–540. doi: 10.1016/j.it.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 39.Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008;28:639–650. doi: 10.1016/j.immuni.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 40.Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3:944–950. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- 41.Viney JL, Mowat AM, O’Malley JM, Williamson E, Fanger NA. Expanding dendritic cells in vivo enhances the induction of oral tolerance. J Immunol. 1998;160:5815–5825. [PubMed] [Google Scholar]
- 42.Adorini L, Valli A, Guery JC. Inhibition of T cell activation by blockade of MHC class II molecules. Seminars in immunology. 1991;3:231–236. [PubMed] [Google Scholar]
- 43.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH, Sharpe AH. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203:883–895. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carter LL, Leach MW, Azoitei ML, Cui J, Pelker JW, Jussif J, Benoit S, Ireland G, Luxenberg D, Askew GR, Milarski KL, Groves C, Brown T, Carito BA, Percival K, Carreno BM, Collins M, Marusic S. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2007;182:124–134. doi: 10.1016/j.jneuroim.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 45.Salama AD, Chitnis T, Imitola J, Ansari MJ, Akiba H, Tushima F, Azuma M, Yagita H, Sayegh MH, Khoury SJ. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med. 2003;198:71–78. doi: 10.1084/jem.20022119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukaya T, Takagi H, Sato Y, Sato K, Eizumi K, Taya H, Shin T, Chen L, Dong C, Azuma M, Yagita H, Malissen B. Crucial roles of B7-H1 and B7-DC expressed on mesenteric lymph node dendritic cells in the generation of antigen-specific CD4+Foxp3+ regulatory T cells in the establishment of oral tolerance. Blood. 2010;116:2266–2276. doi: 10.1182/blood-2009-10-250472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park JJ, Omiya R, Matsumura Y, Sakoda Y, Kuramasu A, Augustine MM, Yao S, Tsushima F, Narazaki H, Anand S, Liu Y, Strome SE, Chen L, Tamada K. B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood. 2010;116:1291–1298. doi: 10.1182/blood-2010-01-265975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weiner HL. The mucosal milieu creates tolerogenic dendritic cells and T(R)1 and T(H)3 regulatory cells. Nature immunology. 2001;2:671–672. doi: 10.1038/90604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.