

Abstract
Mutations in the RNA binding protein FUS (fused in sarcoma) have been linked to a subset of familial amyotrophic lateral sclerosis (ALS) cases. The mutations are clustered in the C-terminal nuclear localization sequence (NLS). Various FUS mutants accumulate in cytoplasm whereas wild-type (WT) FUS is mainly nuclear. Here we investigate the effect of one ALS causing mutant (FUS-ΔNLS, also known as R495X) on gene splicing and expression using genome wide exon-junction arrays. Using a non-neuronal stable cell line with inducible FUS expression, we detected early changes in RNA composition. In particular, mutant FUS-ΔNLS increased calcium/calmodulin-dependent protein kinase II inhibitor 2 (CAMK2N2) at both mRNA and protein levels, whereas WT-FUS had no effect. Chromatin immunoprecipitation experiments showed that FUS-ΔNLS accumulated at the CAMK2N2 promoter region, whereas promoter occupation by WT-FUS remained constant. Given the loss of FUS-ΔNLS in the nucleus through the mutation-induced translocation, this increase of promoter occupancy is surprising. It indicates that, despite the obvious cytoplasmic accumulation, FUS-ΔNLS can act through a nuclear gain of function mechanism.
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the progressive loss of motor neurons in the central nervous system. Recent studies found that mutations in the RNA binding protein FUS (fused in sarcoma) can cause a subset of hereditary forms of ALS in a dominant fashion [1,2]. It has been largely unknown how FUS mutations cause ALS.
FUS, also known as TLS (translocated in sarcoma) and as hnRNP P2, is a nucleic acid binding protein that is mainly nuclear, but can shuttle between cytosol and nucleus. FUS has been implicated in multiple steps of mRNA metabolism, including transcriptional regulation, regulation of RNA processing, and RNA export [3,4,5,6,7]. FUS contains several domains that can bind to nucleic acids including an RNA recognition motif that binds both to DNA and RNA [8], which partially explains the multiple roles in gene expression.
Genomic translocation affecting FUS can result in cancer, especially liposarcomas [9,10]. In these genomic rearrangements, the promoter and N-terminal part of FUS is translocated to the C-terminal domain of various DNA-binding transcription factors, resulting in a fusion protein with a strong transcriptional activation domain (reviewed in [11]). This indicates a role of FUS in gene transcription.
The familial ALS related mutations are clustered in the C-terminal region, which was later determined as a nuclear localization sequence (NLS) [12,13,14]. Mutations in the NLS decrease the nuclear import and cause cytoplasmic accumulation of the mutant FUS protein. In particular, the FUS-ΔNLS mutation lacks the NLS and causes a relatively aggressive ALS clinical phenotype with an early onset [14]. It has been proposed that the loss of a nuclear function or the gain of a cytosolic function or both can be the possible cause for the disease [15,16,17].
Recent studies have reported thousands of RNA targets that can bind to FUS [18,19,20]. The structure of the RNA recognition motif of FUS shows a non-canonical nucleic acid binding site and supports the relatively low specificity binding of RNAs to FUS [8]. To gain insight into how FUS mutations may interfere with mRNA transcription and processing, we performed genome wide exon junction array studies on stably transfected HEK293 cells with inducible FUS expression. We specifically concentrated on early changes in gene expression. Among more than 100 genes showing small changes after 24 hours of FUS expression, calcium/calmodulin-dependent protein kinase II inhibitor 2 (CAMK2N2) showed the most striking difference between wild type and mutant FUS. The change of CAMK2N2 was validated at both RNA and protein levels. Furthermore, chromatin immunoprecipitations experiments showed that FUS-ΔNLS accumulated at the CAMK2N2 promoter region, whereas promoter occupation by WT-FUS remained constant. The results from this study suggest a nuclear gain of function could also be an underlying mechanism contributing to FUS mutation induced alterations.
2. Material and Methods
2.1. Cell culture
HEK293 stable cells with inducible WT GFP-FUS and FUS-ΔNLS expression have been described before. They were created by FRT-mediated recombination is into a single genomic locus. After antibiotic selection, the cells remained pooled to avoid clonal bias [14]. The cells were cultures in DMEM with 10% TET-tested fetal bovine serum (Atlanta Biologicals, s10350H), 2 mM L-glutamine (Gibco, 25030), 15 μg/ml blasticidin (Invitrogen, R210-01), 150 μg/ml hygromycin B (Invitrogen, 10687-010), and 1% penicillin and streptomycin solution. Doxycyline (Clontech, 1 μg/ml) was added to induce the expression of GFP-FUS 6, 24, or 48 hrs before harvest.
2.2. Array analysis
RNA was isolated using Qiagen kits. Its quality was determined by RNA integrity (RIN) number analysis and samples with a RIN >9.5 were used following the Affymetrix labeling procedure.
For the analysis, the signal from Affymetrix human junction arrays (HJAY) was normalized using the “Probe scaling” method. The background was corrected with ProbeEffect from GeneBase [21]. The gene expression index was computed from probes that were selected using ProbeSelect from GeneBase [21]. The gene expression signals were computed using these probes. Genes were considered expressed if the mean intensity was ≥ 500. Genes were considered regulated if 1) they were expressed in at least one condition (i.e., VPA and/or control); 2) the fold-change was greater or equal than 1.5 and; 3) the unpaired t-test p-value between gene intensities was ≤ 0.05. For each probe, a splicing-index was computed. Unpaired t-tests were performed to determine the difference in probe expression between the two samples as described previously [22]. Probe p-values in each probeset were then summarized using Fisher’s method. Using annotation files, splicing patterns (cassette exons, 5’/3’ alternative splice sites and mutually exclusive exons) were tested for a difference between isoforms, selecting the ones with a minimum number of regulated probesets (with a p-value ≤ 0.01) in each competing isoform (at least one third of “exclusion” probesets have to be significant; at least one third of “inclusion” probesets have to be significant and show an opposite regulation for the splicing-index compared to the “exclusion” probesets). For example, for a single cassette exon, the exclusion junction and at least one of the three inclusion probesets (one exon probeset and two inclusion junction probesets) have to be significant and have to show an opposite regulation for the splicing-index.
2.3. RT-PCR
Total RNA from HEK293 cells was extracted using the GenElute Mammalian Total RNA Miniprep Kit (Sigma) according to the manufacturer’s instructions. cDNAs were synthesized from 1 μg of each RNA using SuperScript®; III Reverse Transcriptase (Life Technologies). Reverse transcriptase-polymerase chain reaction (RT-PCR) experiments were carried out using 1 μl of each cDNA as template and specific primers. Products were visualized on gel electrophoresis after ethidium bromide staining. PCR primers were as follows: β-ACTIN_FOR: AGAGCTACGAGCTGCCTGAC; β-ACTIN_REV: GGATGTCCACGTCACACTTC; F_CAMK2N2: ATGTCCGAGATCCTGCCCTA: R_CAMK2N2: ACGTCGTCTATCCGGTCATC; CAMK2N2_cFOR2: ACACAGCGAAAGAGATACGG; CAMK2N2_cREV: AGCAGGAGGGCAAAAGCT
2.4. Chromatin IP
HEK293 cells were lysed and sheared by sonication in a 0,1% NP-40/PBS (Sigma) lysis buffer to generate cellular chromatin fragments of 400–500 bp. The chromatin was immunoprecipitated for 14–16 h at 4°C using antibodies against GFP coupled to sepharose (GFP-Trap_A, Chromotek). After the incubation, chromatin immunoprecipitates were puri ed, then 2 μl of each sample were analyzed by Real – time PCR. The real-time PCR was carried out in the Strategene Mx3005P (Agilent Technologies), using SYBR green reagent (Life Technologies). The relative expression was estimated as follows: 2Ct(reference)−Ct(sample), where Ct (reference) and Ct (sample) were input DNA and specific histone modification chromatin, respectively. For each experiment, at least three immunoprecipitations were analyzed.
2.5. Antisera
Mouse monoclonal anti-FUS antibody (sc-47711) and goat polyclonal anti-β-actin (sc-459) were purchased from Santa Cruz Biotechnology. The CAMK2N2 Antibody (N-term) was a Peptide Affinity Purified Rabbit Polyclonal Antibody (Pab) (AP11140a) from Abgent.
2.6 Transfection
HEK293 cells were seeded in 6 well plates, and were transfected with pEGFP-C3 expression plasmids containing the wild type or ΔNLS FUS cDNA using lipofectamine (Invitrogen) according to the manufacturer’s instructions [13].
2.7 Quantification and statistical analysis
Quantification and statistical analysis were performed using Quantity One® 1-D Analysis Software (Biorad). Results are presented as means ± S.D. Differences between means in two groups were compared using the Student’s t test. A 5 % or 1% significance level and 95 % or 99%, respectively, confidence interval were used in the statistical analysis.
3. Results
3.1. Characterization of stable cell lines with inducible expression of FUS
To determine the molecular role of WT-FUS as well as FUS-ΔNLS, we chose stable HEK293 cell lines with Tet-On inducible expression of either human WT-FUS or FUS-ΔNLS (Figure 1A). Both proteins were GFP-tagged to allow discrimination from endogenous FUS that is expressed in all cells [14]. After six hours of induction, the expression of both WT and FUS-ΔNLS started becoming evident (Figure 1B). The exogenous GFP-FUS expression was comparable to the endogenous FUS expression in the cells.
Figure 1. Expression of GFP-FUS and GFP FUS-ΔNLS in stable cell lines.

A. Domain structure of FUS and FUS-ΔNLS
B. Protein expression after FUS and FUS-ΔNLS induction. HEK293 cells were stably transfected with GFP-FUS and GFP-FUS-ΔNLS expression constructs. The cells were induced with doxycycline for the times indicated. Western blots were performed with an anti FUS antiserum and an anti actin serum as a loading control.
C. Intracellular localization of the FUS variants. The detection of the overexpressed proteins was through their GFP tag. The cells were counterstained with DAPI. The size bar is 10 μm.
The FUS-ΔNLS variant was detected predominantly in the cytoplasm in familial ALS patient tissues. This feature is reflected by the cytosolic accumulation of GFP tagged FUS variants in our cell culture system (Figure 1C). In contrast, WT-FUS is mainly nuclear. We noted that GFP tagged FUS-ΔNLS still showed detectable nuclear staining when it was expressed at the relatively low levels in these cells. This inducible cell culture system offers comparable WT and mutant FUS expression levels, which likely mimics the situation in human ALS patients.
3.2. Genome-wide exon junction array analysis
FUS is part of the hnRNP complex and can bind both to single and double stranded DNA, as well as to RNA [11]. Since significant amount of FUS-ΔNLS leaves the nucleus and accumulates in the cytosol, we hypothesized that the mutation will cause a change in gene expression. We therefore analyzed changes in mRNA expression using Affymetrix genome wide exon junction arrays. In addition to detecting changes in alternative splicing, exon junction arrays are a robust tool to monitor overall gene expression, as each mRNA is detected by multiple probes located in all mRNA regions. To identify only early changes, we compared 24 hours of induction, when both WT-FUS and FUS-ΔNLS started to express. We concentrated on early changes to avoid detection of secondary effects that are not directly caused by FUS-ΔNLS overexpression. To minimize effects caused by different growth conditions, all cells were seeded at the same sub-confluent density prior to doxycyclin induction.
Three datasets were generated for 24 hours of induction. We measured gene expression in cells expressing GFP, WT-FUS and FUS-ΔNLS. These expression levels where then compared and changes larger than a 1.5 fold with a p-value smaller than 0.05 were compared (Figure 2). Comparing GFP overexpression with wild type indicated five changes (Supplementary Table 1). This low number reflects that a modest increase of WT-FUS expression (approximately two fold since the GFP-FUS is comparable to the endogenous FUS, Figure 1B) has no strong effects at the timeframe of the experiment. Comparing FUS-ΔNLS with GFP indicated 156 changes (Supplementary Table 3), which suggested a slightly stronger influence of FUS-ΔNLS on gene expression. Finally, comparing FUS-ΔNLS with FUS-WT showed 43 changes (Supplementary Table 2). This comparison resembles most strongly the situation in humans, where the mutant is expressed together with WT-FUS. There are 23 changes common to the datasets when overexpression of FUS-ΔNLS is compared to WT-FUS and uninduced cells, respectively (Figure 2).
Figure 2. Summary of the array analysis.

The Venn diagram shows pathways most affected by 24 hrs WT-FUS and FUS-ΔNLS overexpression. The changes of the arrays are shown in Supplemental Tables 1–3.
An analysis of KEGG pathways did not indicate pathways that are changed in a statistical significant pathway (p <10−3). The arrays indicated only one change in alternative splicing with high confidence in the comparison of FUS-ΔNLS with GFP, which could however not be validated by RT-PCR (Supplemental Table 3, Pathway analysis tab).
Together, these data suggest that induction of FUS-ΔNLS has a stronger effect on gene expression than expressing WT-FUS. The low number of changes suggests that we monitor only early, direct changes in FUS-ΔNLS dependent modification in gene expression.
3.3. Differential expression of CAMK2N2 by WT and mutant FUS
We next validated the effects of WT-FUS and FUS-ΔNLS overexpression on gene expression using RT-PCR with primers indicated in Figure 3D. We concentrated on the gene with the most pronounced consistent differences during RT-PCR validations, which was CAMK2N2 (calcium/calmodulin-dependent protein kinase II inhibitor 2). CAMK2N2 inhibits calcium/calmodulin-dependent protein kinase II, a major signaling molecule in the brain. It also interacts with the ARHGAP32 protein, also called RICS for Rho GTPase activating protein 32 [23]. RICS promotes GTP hydrolysis on RHOA, CDC42 and RAC1 small GTPases. As shown in Figure 3, WT-FUS down-regulated CAMK2N2 whereas FUS-ΔNLS up-regulated the CAMK2N2 mRNA levels after 6, 24 and 48 hours of FUS expression. These differences between WT and mutant FUS expressing cells were statistically significant (Figure 3C), when normalized to the actin signal that was not influenced by FUS expression.
Figure 3. The CAMK2N2 gene is changed after FUS-ΔNLS expression.

A. RT-PCR detecting CAMK2N2 expression after WT-FUS was induced at the times indicated.
B. RT-PCR detecting CAMK2N2 expression after FUS-ΔNLS was induced at the times indicated.
C. Quantification of the CAMK2N2 signal after normalization to beta actin.
D. Localization of the primer for detection. Primers were F_CAMK2N2 and R_CAMK2N2.
CAMK2N2 changes were highly reproducible in more than four different experiments. Other changes shown in Supplemental Table 1 varied between independent experiments and we did not obtain statistical significant results. These variations between experiments likely reflect the early time points of the analysis that was chosen to detect the early changes caused by the mutation. We therefore identified a change in a mRNA encoding a regulatory protein as a response to the expression of an ALS-causing variant of FUS.
3.4. FUS-ΔNLS upregulates CAMK2N2 protein
To determine whether the changes on the RNA level are reflected on the protein level, we analyzed CAMK2N2 protein levels after WT-FUS and FUS-ΔNLS inductions. As shown in Figure 4, after induction of WT-FUS, CAMK2N2 levels show no statistically significant changes. In contrast, when FUS-ΔNLS is overexpressed, we observe an about 2.5 fold induction on the protein level that reflects the increase on the mRNA level.
Figure 4. FUS-ΔNLS upregulates CAMK2N2 protein levels.
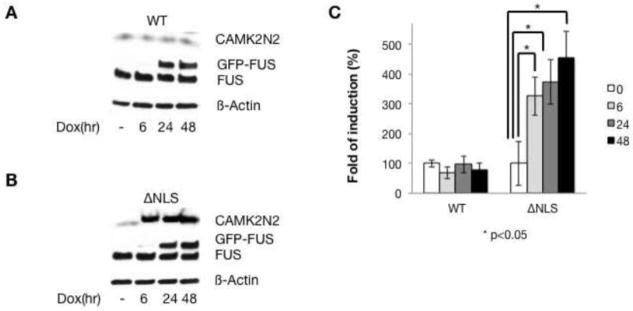
A. After WT-FUS induction, CAMK2N2 expression was detected by Western blot in doxycycline induced cell lines for the times indicated.
B. After FUS-ΔNLS induction, CAMK2N2 expression was detected by Western blot in doxycycline induced cell lines for the times indicated.
C. The quantification of the data is shown on the right.
3.5. FUS-ΔNLS upregulates CAMK2N2 in transient transfections
To rule out the selection of specific clones when making stable cell lines, we tested the effect for FUS-ΔNLS on CAMK2N2 in transient transfection assays. As shown in Figure 5A, similar to the situation is stable cell lines, WT-FUS had no effect on CAMK2N2 mRNA levels. In contrast, we observed a steady increase of CAMK2N2 after transfecting FUS-ΔNLS (Figure 5B). After 48 hrs, CAMK2N2 is upregulated about 2.5 fold, similar to the effect observed in stable cell lines.
Figure 5. The CAMK2N2 gene is changed after FUS-ΔNLS transient expression.

A. RT-PCR detecting CAMK2N2 expression after WT-FUS was transfected at the times indicated. 2μg of expression plasmid per 300,000 cells were used.
B. RT-PCR detecting CAMK2N2 expression after FUS-ΔNLS was transfected at the times indicated. 2μg of expression plasmid per 300,000 cells were used.
C. Quantification of the CAMK2N2 signal after normalization to beta actin.
We next determined the effect of FUS variants on the protein level in transient transfections and observed an about two fold increase of CAMK2N2 with FUS-ΔNLS, but no statistical significant increase with the wild type (Figure 6A–C). The data suggest that FUS-ΔNLS increases both mRNA and protein levels of CAMK2N2, both in stable and transient transfections.
Figure 6. FUS-ΔNLS upregulates CAMK2N2 protein levels after transient transfections.
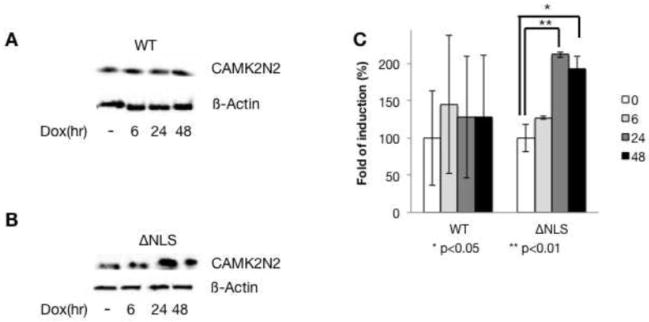
A. After WT-FUS transfection, CAMK2N2 expression was detected by Western blot at the times indicated.
B. After FUS-ΔNLS transfection, CAMK2N2 expression was detected by Western blot at the times indicated.
C. The quantification of the data is shown on the right.
3.6. FUS-ΔNLS binds stronger to the CAMK2N2 promoter than wild-type FUS
FUS has been shown to bind to DNA and to activate transcription [3,4,5]. We therefore measured the binding of WT-FUS and FUS-ΔNLS to the CAMK2N2 promoter. To compare WT and mutant FUS proteins, we immunoprecipitated them using the GFP tag from the stable cell lines. The cells were analyzed at 6 and 48 hrs, to capture the early and strongest changes on the mRNA and protein levels. The levels of CAMK2N2 promoter DNA bound to the immunoprecipitates were determined using real-time PCR. As shown in Figure 7, a 3.5 fold increase of FUS-ΔNLS binding to the CAMK2N2 promoter was observed at 48 hours as compared to WT-FUS. Interestingly, there is a slight decrease after six hrs that precedes the strong increase, suggesting rearrangements at the promoter. In contrast, WT-FUS bound to the promoter remains constant. The data suggest that FUS-ΔNLS accumulates on the CAMK2N2 promoter, which likely causes its induction.
Figure 7. Chromatin immunoprecipitations of CAMK2N2 DNA with FUS and FUS-ΔNLS.

The stable cell lines were induced for six and 48 hours and GFP-tagged FUS-wild type and FUS-ΔNLS were immunoprecipitated using anti-GFP. Chromatin bound to the immunoprecipitates was detected by PCR using the primers located in the promoter regions, as indicated in the cartoon on top. The graph represents three independent experiments. The expression level at time zero was set to one. The differences for FUS-ΔNLS are significant, p<0.05 (*) and p<0.01 (**), as indicated.
4. Discussion
FUS-ΔNLS has been identified as a rare mutation that causes ALS. To gain insight into the molecular mechanism of this mutation, we identified early changes in gene expression caused by FUS-ΔNLS expression. This resembles the disease situation, where FUS-ΔNLS is expressed in addition to WT-FUS. However, in the human system one WT allele is substituted by a mutated allele. Our overexpression system differs from other studies where WT-FUS was knocked out, which resulted in multiple changes in pre-mRNA processing and gene expression [19,20].
We detected only a few changes in gene expression that are small. This is expected, as the disease has an onset of several years and we concentrated only on early changes in gene expression to identify directly regulated genes.
We did not find any changes in pre-mRNA splicing, despite the known involvement of FUS in this process [18]. This is likely due to the early time point of analysis and the fact that we over expressed FUS, which did not drastically change the total amount of WT-FUS.
We found that the FUS-ΔNLS mutant causes an about two-fold increase in CAMK2N2 mRNA and about 3.5 fold increase in protein levels in non-neuronal cells. This was the only change that could be validated in several independent RT-PCR experiments. It therefore validated CAMK2N2 as this first direct target of FUS-ΔNLS.
A deregulation of CAMK2N2 will have consequences for the cell. CAMK2N2 is an inhibitor of Calmodulin dependent kinase 2 (CaMKII) that arrests calmodulin on the kinase, which inhibits it. CAMK2N2 also physically interacts with the Rho GTPase activating protein 32 (RICS), a brain specific regulator for Rho-GAP signaling [23]. Both CAMK2N2 and RICS are involved in signal transduction in the brain. One mode of action for the FUS-ΔNLS variant is the disruption of signaling networks, which could contribute to the disease in vivo. CaMKII is known to phosphorylate AMPA-type glutamate receptors, which are implicated in ALS [24,25]. It is therefore possible that CaMKII-dependent changes in the phosphorylation of AMPA receptors contribute to ALS.
Mechanistically, the change in CAMK2N2 expression is caused by the binding of FUS-ΔNLS protein to the CAMK2N2 promoter. Since WT-FUS binds to the promoter, it is likely that the mutant substitutes endogenous WT-FUS and accumulate over time, which changes the transcription. The molecular reason for this accumulation is unclear, but it could involve an increase of binding to single stranded DNA in the open promoter complex that is aided by proteins that bind to the FUS-ΔNLS mutant stronger than the wild type. Therefore, despite the cytosolic accumulation of FUS-ΔNLS, the protein could act through a nuclear gain of function mechanism.
Together the data indicate that mutant FUS could change the expression of a regulatory protein CAMK2N2 by deregulation of its promoter and that such changes could be involved in the etiology of ALS.
Table 1: Comparison of cells expressing wild type FUS and GFP, each for 24 hours.
Table 2: Comparison of cells expressing FUS-ΔNLS and FUS wild type, each for 24 hours.
Table 3: Comparison of cells expressing FUS-ΔNLS and GFP, each for 24 hours.
Supplementary Material
Highlights.
Mutants of fused in sarcoma (FUS) cause amyotrophic lateral sclerosis (ALS)
CAMK2N2 is the major target in genome wide array analysis of the mutant
FUS-ΔNLS FUS-ΔNLS binds to the promoter of CAMK2N2
FUS-ΔNLS influences transcription and acts through a gain of function mechanism
Acknowledgments
Funding
This work is supported by National Institutes of Health grant R01NS077284 and the ALS Association grant 6SE340 to H. Z. and NIH RO1 GM083187 and 5P20RR020171-08 to S. S.
Footnotes
Array analysis of HEK293 cells stably transfected with constructs expressing GFP, WT-FUS type, FUS-ΔNLS.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 3.Bertolotti A, Lutz Y, Heard DJ, Chambon P, Tora L. hTAF(II)68, a novel RNA/ssDNA-binding protein with homology to the pro-oncoproteins TLS/FUS and EWS is associated with both TFIID and RNA polymerase II. The EMBO journal. 1996;15:5022–5031. [PMC free article] [PubMed] [Google Scholar]
- 4.Bertolotti A, Melot T, Acker J, Vigneron M, Delattre O, Tora L. EWS, but not EWS-FLI-1, is associated with both TFIID and RNA polymerase II: interactions between two members of the TET family, EWS and hTAFII68, and subunits of TFIID and RNA polymerase II complexes. Mol Cell Biol. 1998;18:1489–1497. doi: 10.1128/mcb.18.3.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou Z, Licklider LJ, Gygi SP, Reed R. Comprehensive proteomic analysis of the human spliceosome. Nature. 2002;419:182–185. doi: 10.1038/nature01031. [DOI] [PubMed] [Google Scholar]
- 6.Tan AY, Manley JL. TLS inhibits RNA polymerase III transcription. Mol Cell Biol. 2010;30:186–196. doi: 10.1128/MCB.00884-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron. 2004;43:513–525. doi: 10.1016/j.neuron.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 8.Lui X, Niu C, Ren J, Zhang J, Xie XX, Zhu H, Feng W, Gong W. The RRM domain of human fused in sarcoma protein reveals a non-canonical nucleic acid binding site. Biochem Biophys Acta. 2012 doi: 10.1016/j.bbadis.2012.11.012. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olofsson A, Willen H, Goransson M, Engstrom K, Meis-Kindblom JM, Stenman G, Kindblom LG, Aman P. Abnormal expression of cell cycle regulators in FUS-CHOP carrying liposarcomas. International journal of oncology. 2004;25:1349–1355. [PubMed] [Google Scholar]
- 10.Tan AY, Manley JL. TLS/FUS: a protein in cancer and ALS. Cell cycle. 2012;11:3349–3350. doi: 10.4161/cc.21875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Human molecular genetics. 2010;19:R46–64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, Neumann M, Haass C. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. The EMBO journal. 2010;29:2841–2857. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gal J, Zhang J, Kwinter DM, Zhai J, Jia H, Jia J, Zhu H. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiology of aging. 2011;32:2323, e2327–2340. doi: 10.1016/j.neurobiolaging.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ, Jr, Sapp P, McKenna-Yasek D, Brown RH, Jr, Hayward LJ. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Human molecular genetics. 2010;19:4160–4175. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xia R, Liu Y, Yang L, Gal J, Zhu H, Jia J. Motor neuron apoptosis and neuromuscular junction perturbation are prominent features in a Drosophila model of Fus-mediated ALS. Molecular neurodegeneration. 2012;7:10. doi: 10.1186/1750-1326-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang JW, Brent JR, Tomlinson A, Shneider NA, McCabe BD. The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. The Journal of clinical investigation. 2011;121:4118–4126. doi: 10.1172/JCI57883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Yang M, Deng J, Chen X, Ye Y, Zhu L, Liu J, Ye H, Shen Y, Li Y, Rao EJ, Fushimi K, Zhou X, Bigio EH, Mesulam M, Xu Q, Wu JY. Expression of human FUS protein in Drosophila leads to progressive neurodegeneration. Protein & cell. 2011;2:477–486. doi: 10.1007/s13238-011-1065-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogelj B, Easton LE, Bogu GK, Stanton LW, Rot G, Curk T, Zupan B, Sugimoto Y, Modic M, Haberman N, Tollervey J, Fujii R, Takumi T, Shaw CE, Ule J. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Scientific reports. 2012;2:603. doi: 10.1038/srep00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC, Liang TY, Mazur C, Wancewicz E, Kim AS, Watt A, Freier S, Hicks GG, Donohue JP, Shiue L, Bennett CF, Ravits J, Cleveland DW, Yeo GW. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15:1488–1497. doi: 10.1038/nn.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, Farazi TA, Hafner M, Borkhardt A, Sander C, Tuschl T. RNA targets of wild-type and mutant FET family proteins. Nature structural & molecular biology. 2011;18:1428–1431. doi: 10.1038/nsmb.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kapur K, Jiang H, Xing Y, Wong WH. Cross-hybridization modeling on Affymetrix exon arrays. Bioinformatics. 2008;24:2887–2893. doi: 10.1093/bioinformatics/btn571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen S, Warzecha CC, Carstens RP, Xing Y. MADS+: discovery of differential splicing events from Affymetrix exon junction array data. Bioinformatics. 2010;26:268–269. doi: 10.1093/bioinformatics/btp643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okabe T, Nakamura T, Nishimura YN, Kohu K, Ohwada S, Morishita Y, Akiyama T. RICS, a novel GTPase-activating protein for Cdc42 and Rac1, is involved in the beta-catenin-N-cadherin and N-methyl-D-aspartate receptor signaling. J Biol Chem. 2003;278:9920–9927. doi: 10.1074/jbc.M208872200. [DOI] [PubMed] [Google Scholar]
- 24.Tateno M, Sadakata H, Tanaka M, Itohara S, Shin RM, Miura M, Masuda M, Aosaki T, Urushitani M, Misawa H, Takahashi R. Calcium-permeable AMPA receptors promote misfolding of mutant SOD1 protein and development of amyotrophic lateral sclerosis in a transgenic mouse model. Human molecular genetics. 2004;13:2183–2196. doi: 10.1093/hmg/ddh246. [DOI] [PubMed] [Google Scholar]
- 25.Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A. 1999;96:3239–3244. doi: 10.1073/pnas.96.6.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.