

Abstract
To determine whether a disintegrin and a metalloproteinase-8 (Adam8) regulates allergic airway inflammation (AAI) and airway hyper-responsiveness (AHR), we compared AAI and AHR in wild type (WT) versus Adam8−/− mice in different genetic backgrounds sensitized and challenged with ovalbumin (OVA) or house dust mite protein extract (HDM). OVA- and HDM-treated Adam8−/− mice had higher lung leukocyte counts, more airway mucus metaplasia, greater lung levels of some TH2 cytokines, and higher methacholine-induced increases in central airway resistance than allergen-treated WT mice. Studies of OVA-treated Adam8 bone marrow chimeric mice confirmed that leukocyte-derived Adam8 predominantly mediated Adam8’s anti-inflammatory activities in murine airways. Airway eosinophils and macrophages both expressed Adam8 in WT mice with AAI. Adam8 limited AAI and AHR in mice by reducing leukocyte survival because: 1) Adam8−/− mice with AAI had fewer apoptotic eosinophils and macrophages in their airways than WT mice with AAI; and 2) Adam8−/− macrophages and eosinophils had reduced rates of apoptosis compared with WT leukocytes when the intrinsic (but not the extrinsic) apoptosis pathway was triggered in the cells in vitro. ADAM8 was robustly expressed by airway granulocytes in lung sections from human asthma patients but, surprisingly, airway macrophages had less ADAM8 staining than airway eosinophils. Thus, ADAM8 has anti-inflammatory activities during AAI in mice by activating the intrinsic apoptosis pathway in myeloid leukocytes. Strategies that increase ADAM8 levels in myeloid leukocytes may have therapeutic efficacy in asthma.
Introduction
Asthma is a chronic disease characterized by airway inflammation, airway hyper-reactivity (AHR), and intermittent bronchoconstriction. It affects 300 million people worldwide and causes an estimated 180,000 deaths per year (1). Asthma is caused by complex interactions between environmental and genetic factors, but its pathogenesis is not completely understood.
Matrix metalloproteinases (Mmps¶) regulate AAI and AHR in mice by modulating airway inflammation (2–5), subepithelial fibrosis (6–8), and airway smooth muscle cell proliferation (9). For example, Mmp-2, -7, and -9 regulate AAI in mice by proteolytically cleaving pro-inflammatory mediators to regulate the biologic activities of these mediators (3,5,10). Mmp-8 reduces AAI in mice by increasing granulocyte apoptosis (4). However, little is known about the activities of other metalloproteinase (MP) subfamilies in asthma pathogenesis. The ADAM sub-family of MPs was first linked to asthma in 2002 when ADAM33 was identified as the first asthma susceptibility gene (11). Although Adam33 does not regulate AAI in mice (12), it may promote chronic remodeling processes in asthmatic airways (13). ADAMs are zinc-dependent transmembrane MPs, and are characterized by a multi-domain structure which can include pro, MP, disintegrin, cysteine-rich, transmembrane, and intracellular domains (14). The MP domain proteolytically cleaves and releases signaling molecules and their receptors from cell surfaces to regulate the biological activities of these molecules (15). The disintegrin domain of some ADAMs binds to integrins to regulate cell adhesion and migration (16). The cysteine-rich and EGF-like domains of some ADAMs regulate cell adhesion and cell-cell fusion. The cytoplasmic tail of some ADAMs has the potential to regulate intracellular signaling (17).
ADAM8 (CD156a, MS2) is expressed on the surface of all leukocytes (except T-cells), neurons, microglial cells, and osteoclasts (18). However, there are conflicting reports on whether ADAM8 is expressed by airway epithelium in naïve mice or healthy human subjects (19–21). ADAM8 has the typical domain structure of the ADAM family. Its MP domain cleaves CD23, CD40 ligand, L-selectin, P-selectin, VCAM-1, and pro-TNF-α in vitro (22–24), but it is not clear whether these proteins are substrates for ADAM8 in vivo. Little is known about the functions of the other domains of ADAM8.
ADAM8 expression is upregulated in the lungs of mice with AAI and humans with asthma (19,25,26). However, recent studies of mice over- or under-expressing Adam8 in the ovalbumin (OVA) model of AAI and AHR have reported conflicting results on the activities of Adam8 in regulating AAI and AHR. OVA-sensitized and -challenged transgenic mice that constitutively over-express a soluble form of ADAM8 had decreased peribronchial inflammation when compared with non-transgenic mice (27) suggesting that ADAM8 has antiinflammatory properties in the airways. In contrast, two more recent studies of mice lacking Adam8 both reported that Adam8 promoted OVA-induced AAI in mice (28,29). However, one of these studies reported that Adam8 promoted chemotaxis of T cells, eosinophils, and macrophages (28) whereas the other study reported that Adam8 increased dendritic cell accumulation in the airways of mice (29). Thus, additional studies are needed to better understand the activities of Adam8 in regulating AAI and AHR in mice.
To address the uncertainties about the activities of Adam8 in regulating AAI and AHR, we compared AAI and AHR in Adam8−/− and WT mice in two different genetic backgrounds (mixed SvEv129 X C57BL/6 and BALB/c) sensitized and challenged with two different allergens using different sensitization protocols. We sensitized mice with high vs. low dose ovalbumin (OVA) and an adjuvant by the i.p. route, or with a more clinically-relevant allergen (house dust mite protein extract; HDM) via the respiratory mucosal route in the absence of an adjuvant. We then challenged the mice with the same allergen by the inhaled route. Irrespective of the strain of mouse, allergen, or dose of allergen studied, Adam8−/− mice had greater AAI and AHR than WT mice. Additionally, leukocyte-derived Adam8 reduced AAI by increasing airway myeloid leukocyte cell death rates thereby reducing the accumulation of eosinophils and macrophages in the airways of mice with AAI. Studies of ADAM8 levels in lung sections from human subjects with or without asthma revealed that in asthma patients, airway eosinophils had robust staining for ADAM8, but airway macrophages had minimal ADAM8 staining. Thus, our study identifies leukocyte-derived ADAM8 as a potent anti-inflammatory protein in the airways of mice. Additionally, we report that a member of the ADAM family promotes activation of the intrinsic apoptosis pathway in myeloid leukocytes to thereby limit airway inflammation. Our results identify ADAM8 as a potential therapeutic target for human asthma.
Materials and Methods
Materials
Aluminum hydroxide, Cadenza buffer, and fetal bovine serum were purchased from Thermo Fisher Scientific (Pittsburgh, PA). House dust mite protein extract was obtained from Greer Laboratories Inc. (Lenoir, NC). Recombinant ADAM8, recombinant caspase-3, and murine and goat antibodies to human ADAM8 and Ccr-3, goat anti-murine Tnf-α IgG, and ELISA kits for measuring Mip-1α, Tslp, Ccl17, Ccl22, eotaxin, Tgf-β, and L-selectin were purchased from R & D Systems (Minneapolis, MN). Antibodies to active caspase-3 and isotype-matched control antibodies were obtained from Cell Signaling (Danvers, MA). Rhodamine-conjugated donkeyanti-murine IgG, rhodamine-conjugated donkey anti-rabbit IgG, and fluorescein-conjugated donkey anti-goat IgG were obtained from Jackson ImmunoResearch laboratories Inc. (West grove, PA). The rabbit anti-human CD68 IgG was obtained from Abcam (Cambridge, MA), and the murine anti-human major basic protein IgG was obtained from US Biologicals (Marblehead, MA). The ELISA to murine Adam8 was purchased from Antibodies-Online (Atlanta, GA), and the total protein assay kit was obtained from BioRad (Hercules, CA). All other antibodies were obtained from BD Biosciences (San Jose, CA).
Recombinant cytokines and chemokines were purchased from Peprotech (Rocky Hill, NJ). ELISA kits for measuring lung levels of Il-4, -5, -12, and -13, Tnf-α, Gm-csf, and Il-10 were obtained from eBioscience (San Diego, CA). An ELISA kit for measuring OVA-specific IgE levels in serum samples was purchased from MD Biosciences (Minneapolis, MN). An ELISA kit for quantifying Rantes levels in lung samples was obtained from Antigenix (Huntington Station, NY). The Vector Red staining kit, 3,3′-diaminobenzidine (DAB) staining kit, antigen retrieval solution, peroxidase blocker, and ABC kit were purchased from Vector Labs (Burlingame, CA). Kits for periodic-acid schiff (PAS) and non-specific esterase staining, murine anti-human smooth muscle alpha actin IgG2 antibody, and chicken egg ovalbumin (OVA) were obtained from Sigma-Aldrich. The Mitoprobe® staining kit was obtained from Invitrogen (Carlsbad, CA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Animals
All procedures performed on mice were approved by the Harvard Medical School Institutional Animal Care and Use Committee. Mice were housed in a barrier facility under specific pathogen-free conditions. Adam8−/− mice were generated and initially studied in a mixed SvEv129 X C57BL/6 strain and WT littermate mixed SvEv129 X C57BL/6 strain mice were studied as controls. Adam8−/− mice were obtained from Andrew Docherty, Ph.D. (UBC Cell Tech, Slough, UK) and Carl Blobel, M.D. Ph.D. (Hospital for Special Surgery, New York, NY). Adam8−/− mice have normal lifespan, fertility, and lung development, and no abnormalities have been identified in unchallenged state (20). The Adam8−/− SvEv129 X C57BL/6 strain mice were backcrossed eight generations into a BALB/c genetic background. Initial experiments confirmed that F8 BALB/c wild type (WT) littermate controls had similar responses to allergens as parental WT littermate control mice. Thus, we studied age- and sex-matched F8 BALB/c wild type (WT) parental littermate mice as controls in subsequent experiments. The genotypes of the mice were confirmed using PCR-based protocols performed on genomic DNA extracted from tail biopsies.
OVA-induced AAI using high and low doses of OVA for sensitization
Age- and sex-matched 8–12 week old WT and Adam8−/− mice were used for all studies. Initially, cohorts of SvEv129 X C57BL/6 WT and Adam8−/− mice were sham sensitized using a high dose of OVA (200 μg dissolved in 200 μl of endotoxin-free PBS along with 1 mg alum) on days 0 and 7 by the i.p. route, and then challenged for 20 min with an aerosolized 6% solution of OVA in endotoxin-free PBS on days 14–17. In all subsequent experiments, SvEv129 X C57BL/6 Adam8−/− mice vs. WT littermate SvEv129 X C57BL/6 WT mice and BALB/c Adam8−/− mice vs. BALB/c parental littermate WT mice were sensitized with a low dose (10 μg) of OVA diluted in 200 μl of endotoxin-free PBS along with 1 mg of alum by the i.p. route on days 0 and 7. Mice were then challenged with an aerosolized 6% solution of endotoxin-free OVA in PBS on days 14–17 (30). For all studies, control mice were sensitized with i.p. PBS and challenged with aerosolized PBS. Twenty-four hours after the last OVA or PBS challenge, mice were euthanized, BAL was performed, and total numbers of BAL leukocytes were counted. The percentage of each leukocyte subset in BAL samples was counted on modified Wright-stained cytocentrifuge preparations, and the absolute numbers of each leukocyte subset were calculated (31). Eosinophils were enumerated on Wright Giemsa-stained cytocentrifuge preparations. We also immunostained BAL leukocyte preparations with a phycoerythrin (PE)-conjugated monoclonal antibody to Ccr3 and analyzed the cells by flow cytometry. We gated on the granulocyte population of cells identified by their forward and side scatter characteristics, and quantified the percentage of eosinophils (granulocytes that were Ccr3 positive). In other cohorts of mice, mice were euthanized 24 h after the last OVA challenge, lungs were inflated to 25 cm H2O pressure, fixed in formalin, embedded in paraffin, and mid-sagittal lung sections were stained with hematoxylin and eosin or PAS.
House dust mite protein (HDM)-induced AAI
Mice were sensitized and challenged with HDM protein extract, as described previously (32). Briefly, three doses of HDM protein extract (100 μg in 50 μl of endotoxin-free PBS or PBS alone) were delivered by the intranasal (i.n.) route at weekly intervals, and 72 h after the last HDM challenge, AAI was measured as outlined above. Preliminary experiments confirmed prior reports (32) that 72 h after the last HDM challenge was optimal for measuring AAI and AHR in the mice.
AHR to methacholine challenges
Twenty-four hours after the last OVA-challenge or 72 h after the last HDM challenge, mice were anesthetized with pentobarbital (100 mg/kg) and a tracheostomy was performed. Central airway resistance (Rn) was measured in unchallenged or OVA-treated WT or Adam8−/− mice using a computer-controlled small animal ventilator (Flexivent®, SCIREQ Inc., Montreal, Canada) set at a tidal volume of 10 ml/kg, 120 breaths per min, and 2 cm H2O positive end expiratory pressure. Mice were challenged with PBS alone or a solution of 1–45 mg/ml of methacholine in PBS.
Lung dendritic cell quantification
Lung dendritic cells were quantified as previously described (33). Briefly, lungs were removed from mice, and enzymatically digested using 1 mg/ml collagenase IV and 0.5 mg/ml DNase from bovine pancreas in RPMI 1640 medium. Cells were then stained with allophycocyanin (APC)-conjugated hamster-anti-mouse CD11c IgG or non-immune APC-conjugated hamster IgG as a control. Flow cytometry was performed using a FACS Canto II (Becton Dickson Bioscience, San Jose, CA). Leukocyte subpopulations were identified by their forward and side scatter characteristics and positive immunostaining to quantify auto-fluorescence high CD11cbright cells (macrophages) versus auto-fluorescence low CD11cbright cells (dendritic cells).
Quantification of cytokines and chemokine levels in lung samples from PBS- versus OVA-treated mice
Lungs were harvested 24 h after the last OVA or PBS challenge, homogenized in PBS containing 0.5% (v/v) Triton, 1 mM phenyl-methyl-sulfonyl-fluoride, 1 mM 1,10-o-phenanthroline, Sigma Mammalian Proteinase Inhibitor Cocktail, and 2 mM sodium fluoride. Cytokine levels were analyzed using a cytokine bead array (Pierce Searchlight Technology, Thermo Fisher Scientific, Woburn, MA), or using commercially available ELISA kits.
Measurement of OVA-specific IgE, soluble L-selectin (sL-selectin), and sVcam-1 levels in serum samples from PBS- vs. OVA-treated mice
Serum was obtained from PBS- and OVA-treated WT and Adam8−/− mice by performing right ventricular puncture 24 h after the last allergen challenge. OVA-specific IgE, sL-selectin, and sVcam-1 levels were quantified using commercial ELISA kits.
Generation of Adam8 bone marrow (BM) chimeric mice
Unchallenged BALB/c WT and Adam8−/− mice were lethally irradiated twice, four hours apart, using a radiation dose of 450 centigray using a 137cesium source. Immediately after the second irradiation, mice received two million BM-derived leukocytes intravenously in 200 μl of PBS. We generated and studied four groups of mice by transplanting: 1) WT BM into WT recipients; 2) WT BM into Adam8−/− recipients; 3) Adam8−/− BM into WT recipients; and 4) Adam8−/− BM into Adam8−/− recipients. After 10 weeks of BM engraftment, mice were sensitized and challenged with PBS or OVA (using a low dose of OVA along with alum via the i.p. route for allergen sensitization) as outlined above.
Isolation of bone marrow (BM)-derived murine monocytes
Unchallenged BALB/c strain WT and Adam8−/− mice were euthanized, their femurs and tibias were dissected, and the bone marrow was flushed out by injecting RPMI medium through the marrow cavities. Erythrocytes were removed using a hypotonic lysis step, and cells were cultured for 60 h in RPMI 1640 medium containing 10% fetal calf serum (FCS), 100 U/ml penicillin, 10 μg/ml streptomycin, 250 ng/ml amphotericin B, and 50 ng/ml M-csf. The preparations were 80.5 ± (SEM) 5.2% monocytes as assessed by positive staining for non-specific esterase (a marker of mononuclear phagocytes) and minimal or absent staining for F4/80 (a marker of mature macrophages). Monocytes were 90.2 ± (SEM) 0.7% viable as assessed by lack of staining with propidium iodide using flow cytometry.
Isolation of bone marrow-derived murine eosinophils
Eosinophils were isolated from the bone marrow of unchallenged BALB/c WT and Adam8−/− mice as described previously (34). The preparations were >90% pure eosinophils as assessed by Wright Giemsa staining and immunostaining for Ccr3. Eosinophils were 93.3 ± (SEM) 1.3% viable as assessed by lack of staining with propidium iodide using flow cytometry.
Isolation of murine peritoneal and alveolar macrophages
Whenever possible, we studied alveolar macrophages isolated from BALB/c strain mice for in vitro assays of macrophage function. Alveolar macrophages were isolated by performing bronchoalveolar lavage on mice using 20 × 1 ml aliquots of PBS, followed by adherence of alveolar macrophages to tissue culture plastic. Alveolar macrophages were cultured for 5 days in DMEM medium containing 10% FBS, 100 Units/ml of penicillin, 10 μg/ml of streptomycin and 250 ng/ml of amphotericin B to render them quiescent before assays were initiated. For in vitro assays that required more macrophages than can be obtained by BAL, we studied macrophages isolated from the peritoneal cavities of mice. We delivered 40 mg of thioglycollate in 1 ml of DMEM medium by i.p. injection to BALB/c mice, and peritoneal macrophages were isolated by performing peritoneal lavage four days later. Peritoneal macrophages were isolated by adherence to tissue culture plastic and cultured in DMEM medium containing 10% FBS, 100 Units/ml of penicillin, and 10 μg/ml of streptomycin for 5 days to render them quiescent before in vitro assays were initiated.
Quantification of Adam8 levels in airway eosinophils and macrophages from WT mice with AAI
Macrophages and eosinophils were isolated from the airways of BALB/c WT mice sensitized and challenged with OVA 24 h after the last OVA challenge using BAL. Macrophages were removed by adherence to tissue culture plastic for 1 h at 37°C, and removing non-adherent cells (>90% eosinophils as assessed by Wright-Giemsa staining of cytocentrifuge preparations). Extracts of the eosinophils and macrophages were prepared in RIPA buffer containing 1 mM phenyl-methyl-sulfonyl-fluoride, 1 mM 1,10-o-phenanthroline, and Sigma mammalian protease inhibitor cocktail. Adam8 protein levels and total protein levels were measured in cell extracts using a commercial ELISA kit, and a Bradford dye-binding kit (BioRad, Hercules, CA), respectively. Adam8 levels were normalized to total protein levels in all samples.
Isolation of lung microvascular endothelial cells (LMVECs)
LMVECs were isolated from unchallenged BALB/c WT mice as previously described (35). Lungs were digested in RPMI containing 1 mg/ml collagenase and 0.5 mg/ml DNase at 37°C for 30 min and a positive selection for Pecam-1 was performed using rat anti-murine Pecam-1 antibody, anti-rat microbeads and LS MACS columns (Miltenyi Biotec, Cambridge, MA). LMVECs were cultured in gelatin-coated dishes using the Lonza Bullit® kit growth medium (RPMI with 5% FBS, hydrocortisone, FGF, VEGF, IGF-1, ascorbic acid, EGF, and gentamycin). Media were changed every other day, and cells cultured until they were confluent. Cells were then removed using trypsin, and a positive selection for Icam-2 using rat anti-murine Icam-2 IgG, anti-rat microbeads, and LS MACS columns was performed to isolate LMVECs. LMVECs were cultured until they formed confluent monolayers and then leukocyte adhesion assays were performed.
Leukocyte adhesion assays
Bone marrow-derived monocytes or eosinophils were isolated from unchallenged BALB/c WT and Adam8−/− mice and incubated at 37°C for up to 1 h in triplicate in fibronectin-coated tissue culture wells using 2 × 106 cells/well for all assays (36,37). Monocytes were incubated at 37°C with or without 10 μg/ml of bacterial lipopolysaccharide (LPS) from E. Coli 0111:B4, and eosinophils were incubated with or without 10−7 M LTB4. The percentage of adherent leukocytes was determined by washing and counting non-adherent cells as previously described (36,37). To study monocyte adhesion to LMVECS, LMVECs were pre-incubated at 37°C for 2 h in the presence or absence of 100 ng/ml murine Tnf-α to induce expression of Icam-1. Monocytes were added (106 per well), and cells were co-cultured in duplicate in the presence or absence of 1 μg/ml LPS to activate the monocytes. At intervals for up to 2 h, non-adherent monocytes were removed by washing the monolayers with medium, non-adherent monocytes were counted, and the percentage of adherent monocytes was determined (37). To study eosinophil adhesion to LMVECs, LMVECs were pre-incubated at 37°C for 18 h with or without 10−7 M Il-13 and 10−7 M Il-4 to induce expression of Vcam-1. Eosinophils (106 cells per well in duplicate) were then added to the LMVEC monolayers, and cultures incubated in the presence or absence of 10−7M LTC4 to activate the eosinophils for up to 2 h at 37°C. Adhesion of eosinophils to LMVECs was quantified at intervals by removing the non-adherent eosinophils by washing the LMVEC monolayers with medium, and counting the non-adherent eosinophils (37).
Leukocyte migration assays
We compared the migration of equal numbers eosinophils and monocytes isolated from unchallenged WT versus Adam8−/− mice using Boyden micro-chemotaxis assay chambers (38) or Matrigel™ Invasion chambers (39) using 10−7M murine Mip-1α or 10−7M fMLP as chemoattractants for monocytes, or 100 ng/ml murine eotaxin or 10−7M LTB4 as chemoattractants for eosinophils. Buffer alone in the lower chambers was used as a control for both cell types.
Assessment of the catalytic activity of rhADAM8 against recombinant chemokines and cytokines in vitro
Human pro-ADAM8 (5 μM) was activated with 20 nM thermolysin for 40 min at 37° C in Tris-buffered saline containing 0.05% (vol/vol) Triton (TBST; pH 7.4) and then thermolysin was inactivated by adding 50 μM phosphoramidin. To confirm efficient activation of proADAM8, we incubated 200 nM thermolysin-activated ADAM8 or buffer alone with 10 μM (7-methoxycoumarin-4-yl) acetyl-Pro-Leu-Ala-Gln-Val-(3-[2,4-dinitrophenyl]-L2,3-diaminopropionyl)-Arg-Ser-Ser-Ser-Arg-NH2 (a quenched fluorogenic substrate which is cleaved by ADAM8 (22,23) for up to 18 h at 37°C in 200 μl in TBST, and measured cleavage of the substrate using fluorimetry (Ex λ 320 and Em λ 405). The purified ADAM8 preparation progressively cleaved this fluorogenic substrate over 18 h confirming that the proteinase was active. We then incubated 200 nM active human ADAM8 with or without 0.8 μM human IL-5, 5 μM human IL-4, 5 μM human MIP-1α, 5 μM human RANTES, 5 μM human IP-10, or 1 μM human TNF-α in 25 μl of TBST. Note that preliminary experiments confirmed these concentrations of mediators were optimal for detecting non-cleaved forms of these mediators on silver stained-Tris tricine gels. Reaction products were then separated using 16.5% Tris-Tricine gel electrophoresis and intact mediator and their cleavage products detected using a silver staining kit (31).
Quantification of leukocyte apoptosis in the airways of WT and Adam8−/− mice with AAI
BALB/c WT and BALB/c Adam8−/− mice were sensitized and challenged with OVA (using low-dose OVA and alum delivered by the i.p. route for the allergen sensitization step). Airway leukocytes were harvested from the mice 24 h after the last OVA challenge by performing BAL. BAL leukocytes were fixed in 2% paraformaldehyde in PBS, permeabilized in 100% methanol, and stained with an Alexa 488-conjugated rabbit anti-active caspase-3 (Asp175) IgG or an Alexa 488-conjugated non-immune rabbit IgG (5 μg of antibody/106 cells). Leukocyte subpopulations were identified by their characteristic forward and side scatter using flow cytometry, and intracellular staining for active caspase-3 was quantified by gating on each leukocyte subpopulation. To measure loss of mitochondrial membrane potential in leukocytes, BAL leukocytes were stained with 2 mM 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1, a red fluorophore which accumulates in healthy mitochondria) in the presence and absence of 1 mM carbonylcyanide m-chlorophenylhydrazone (CCCP, a membrane-depolarizing agent which reduces JC-1 accumulation in mitochondria, as a control) using a Mitoprobe® kit. BAL cell subsets were identified by their forward and side scatter characteristics, and data acquired in the FL-1 and FL-2 channels using flow cytometry. Loss of signal in the FL-2 channel (loss of JC-1 accumulation in mitochondria indicative of loss of mitochondrial membrane potential) was quantified for each leukocyte subset.
We also quantified surface levels of apoptosis ligands and their receptors on BAL leukocytes from mice with AAI. Leukocytes were isolated from the airways of BALB/c WT and BALB/c Adam8−/− mice 24 h after the last OVA challenge by performing BAL and removing erythrocytes using a hypotonic lysis buffer step. BAL leukocytes were incubated in 200 μl PBS containing 2% fetal bovine serum, 5 μg/ml of CD16/32 blocking antibody, and 0.1% sodium azide for 30 min at 4°C. Cells were then incubated for 30 min at 4°C with 2 μg/ml hamster anti-murine Fas-ligand (Fas-L) IgG, hamster anti-murine Fas IgG, hamster anti-murine Tnf-r1 IgG, hamster anti-murine Trail IgG, or 0.4 μg/ml of goat anti-murine Tnf-α (or the same concentration of an isotype-matched non-immune primary antibody). Cells were washed twice in PBS, incubated for 20 min at 4°C with an Alexa-488-conjugated rabbit anti-hamster or rabbit anti-goat IgG secondary antibodies, washed twice in PBS, and then fixed using 2% paraformaldehyde in PBS. Cell surface staining was quantified using flow cytometry.
Apoptosis of WT versus Adam8−/− eosinophils and macrophages induced to undergo cell death in vitro
We induced apoptosis of BM-derived eosinophils or macrophages isolated from unchallenged BALB/c WT vs. BALB/c Adam8−/− mice by incubating them at 37°C either: 1) in the absence of serum, agonists, or survival factors for up to 48 h to activate the intrinsic apoptosis pathway; or 2) with 10 μg/ml hamster anti-murine FAS (CD95) IgG2 cross-linking antibody [Clone Jo 2 which activates CD95 (40)] or 10 μg/ml hamster IgG2 antibody (as a control) for up to 24 h to trigger the extrinsic apoptosis pathway. Cells were stained with annexin V conjugated to Alexa-488 and propidium iodide and analyzed using flow cytometry as described previously (31). Surface levels of apoptosis ligands and receptors were quantified on WT and Adam8−/− macrophages and eosinophils using immunostaining and flow cytometry, as described above.
Intracellular active caspase-3, -8, and -9 levels
We measured levels of intracellular active caspases-3, -8, and -9 in macrophages and eosinophils that had been induced to undergo apoptosis as outlined above. Extracts of the cells were all prepared at 5 ×106/ml in caspase lysis buffer [10 mM HEPES containing 0.1% 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate, 2 mM EDTA, 5 μM DTT, 1 mM phenyl-methyl-sulfonyl fluoride, 10 μg/ml pepstatin, 20 μg/ml leupeptin, and 0.1 mM 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride)] as described previously (31). Intracellular levels of active caspase-3 were quantified in macrophage and eosinophil extracts by incubating them in triplicate with a quenched fluorogenic substrate which is specific for active caspase-3 [16 μM Ac-DEVD-7-amino-4-methylcoumarin (AMC)] in the presence and absence of a specific caspase-3 inhibitor (20 μM Z-DEVD-fluoromethyl ketone [FMK]) for 30 min at 37°C protected from light. A standard curve was generated by incubating the substrate with recombinant active caspase-3 assays standards (0–100 ng/ml), and cleavage of the substrate by leukocyte extracts or active caspase-3 assay standards was measured using fluorimetry (F2500 fluorescence spectrophotometer; Hitachi, Tokyo, Japan; Ex λ 400 and Em λ 490). Substrate cleavage in the presence of the caspase-3 inhibitor was subtracted from the substrate cleavage in the absence of the caspase-3 inhibitor (in fluorescence units) for each sample. Caspase-3 activity in cell extracts was determined by interpolation from the standard curve. Similarly, intracellular levels of active caspase-8 and caspase-9 were measured by incubating cell extracts and assay standards of recombinant active caspase-8 or active caspase-9 (both at 0–5 units/ml) in triplicate with a quenched fluorogenic substrate which is specific for either active caspase-8 [14 μM IETD-7-amino-4-trifluoromethylcoumarin (AFC)] or caspase-9 (50 μM LEHD-AFC) both in the presence and absence of an inhibitor of caspase-8 (20 μM Ac-IETD-aldehyde) or caspase-9 (20 μM Z-LEHD-FMK), respectively. Cleavage of the substrates by standards and cell extracts was measured using fluorimetry (Ex 400 λ and Em λ 505). Substrate cleavage in the presence of the caspase-8 inhibitor or caspase-9 inhibitor was subtracted from the substrate cleavage in the absence of the caspase-8 inhibitor or caspase-9 inhibitor for each sample (in fluorescence units). Caspase-8 or caspase-9 activity in cell extracts was determined by interpolation from the standard curve.
Loss of mitochondrial membrane potential
Quiescent alveolar macrophages cultured in chamber-slides or BMderived eosinophils were stained with a Mitoprobe® kit (vide supra), and loss of JC-1 staining was quantified using Metamorph software (Molecular Device, Sunnyvale, CA).
Efferocytosis of apoptotic target cells by macrophages in vitro and in vivo
Peritoneal macrophages isolated from WT and Adam8−/− mice (vide supra) were plated at a density of 2 × 105 cells/ml in 8-well chamber-slides and cultured for 4 days to render them quiescent. Murine PMNs were isolated from the BM of unchallenged BALB/c WT and BALB/c Adam8−/− mice by positive selection using Gr-1 antibody and immunomagnetic beads (39) and incubated in serum-free RPMI 1640 medium at 37°C 5% CO2 for 18 h to induce apoptosis. Apoptotic WT or Adam8−/− PMNs (2 × 106) were added to chamber-slides containing WT or Adam8−/− macrophages, and cells were co-cultured for 1 h at 37°C. PMNs that had not been ingested by macrophages were removed by washing macrophages twice with PBS, the chamber slides stained using Diff Quick™ staining, and macrophage uptake of apoptotic cells quantified by counting apoptotic bodies in 300 macrophages per condition (41). We also quantified apoptotic bodies ingested by 200–300 macrophages isolated from the airways of OVA-treated BALB/c WT and BALB/c Adam8−/− mice 24 h after the last OVA challenge in Diff Quick™-stained cytocentrifuge preparations, as outlined above.
Staining human lung sections for ADAM8
Formalin-fixed paraffin-embedded lung tissue sample from discarded human diagnostic specimens were retrieved from the files of Brigham and Women’s Hospital Department of Pathology. All studies of human subjects were approved by the Partners Healthcare Institutional Review Board. Note that it is very rare for our Pathology Department to receive lung biopsy specimens from adult patients with asthma for an asthma indication alone. Thus, specimens were selected from patients who had undergone surgical resection for an incidental mass (diagnoses included three hamartomas, one metastatic renal cell carcinoma, and five primary lung adenocarcinomas), and lung tissue away from the area of the mass was examined. By history, four patients were current smokers, two were non-smokers, and three were former smokers. Cases were designated by a pathologist (LS) as asthmatic or non-asthmatic based on clinical history and pathologic criteria. Lung sections were de-paraffinized, antigen retrieval was performed using Dako targeted antigen retrieval solution™ and Triton in PBS, endogenous peroxidase activity was blocked using a peroxidase blocker. Non-specific binding of antibody was blocked by incubating slides with 10% rabbit serum at room temperature. Slides were subsequently incubated with primary goat anti-human ADAM8 antibody or non-immune goat IgG (both at 1 μg/ml) in Cadenza buffer containing 10% normal rabbit serum for 18 h at 4°C. Slides were then incubated at 4°C with rabbit anti-goat IgG conjugated to biotin (7.5 μg/ml) and the slides were stained using the Vector ABC kit using DAB as the chromogen and counterstained with 1% methyl green.
Double immuno-fluorescence staining of human lung sections for ADAM8 and markers of eosinophils and macrophages
To identify the leukocyte subsets that express ADAM8 in the airways of patients with asthma, we performed two-color immuno-fluorescence staining of lung sections from three patients with asthma. Briefly, the lung sections were de-paraffinized, and antigen retrieval was performed by heating the slides in a microwave in citrate buffer. The sections were incubated overnight at 4°C for 18 h with either a murine IgG to human major basic protein (MBP; diluted 1:50) as a marker of eosinophils or a rabbit anti-human CD68 IgG (diluted 1:100) as a marker of macrophages. After washing the lung sections with PBS, the sections were incubated at 37°C for 1 h with rhodamine-conjugated donkey-anti-murine IgG (diluted 1:100) or rhodamine-conjugated donkey-anti-rabbit IG (diluted 1:100). Sections were then washed in PBS and incubated at 37°C for 2 h with goat anti-human ADAM8 IgG (diluted 1:100). After washing the lung sections in PBS, fluorescein-conjugated donkey-anti-goat IgG (diluted 1:100) was applied and slides were incubated for an additional 1 h at 37°C. Nuclei were counterstained with 4′-6-diamidino-2-phenylindole (DAPI). Images of the stained lung sections were analyzed using a confocal microscope (Leica Inc., Exton, PA) fitted with air-cooled argon and krypton lasers. Fluorescent confocal micrographs were recorded under dual fluorescent imaging mode in which cells were simultaneously exposed to 488 nm and 568 nm light attenuated by an acusto tunable optical filter. A band pass (530 ± 30 nm) filter was used to select light emitted from the fluorescein-labeled ADAM8 and a long-pass 590 nm filter was used to detect the rhodamine-labeled CD68 or MBP. MetaMorph software was used (39) to measure cell-associated fluorescence (in arbitrary fluorescence units) on images captured under the green filter of cells that were positively stained for major basic protein (eosinophils) or CD68 (macrophages).
Statistics
Statistical analysis was performed using the Sigma Stat™ statistics program. P-values were calculated using paired t-tests if data was normally distributed, and using a Mann-Whitney Rank Sum test if not normally distributed. All data are represented as mean ± SEM unless otherwise indicated. A p ≤ 0.05 was considered significant.
Results
Adam8 limits OVA-induced acute AAI and AHR in mice
Initially, we compared AAI and AHR in SvEv129 X C57BL/6 strain Adam8−/− and WT littermate control mice sham-sensitized with PBS or sensitized with a high dose of OVA (200 μg) along with an adjuvant (alum) via the i.p. route, and then challenged daily with aerosolized PBS or OVA on days 14–17 (42). SvEv129 X C57BL/6 strain Adam8−/− mice had greater OVA-induced AAI, with higher airway macrophage and granulocyte counts (Supplemental Fig. 1A and 1B), and greater AHR to methacholine challenges than SvEv129 X C57BL/6 WT mice (Supplemental Fig. 1C) 24 h after the last OVA challenge. When sensitized with a low dose of OVA (10 μg) along with alum by the i.p. route, and then challenged with aerosolized OVA, SvEv129 X C57BL/6 strain Adam8−/− mice also had greater OVA-induced AAI (Supplemental Fig. 1D) compared with SvEv129 X C57BL/6 strain WT parental littermate control mice.
We then back crossed the mice into the BALB/c background (to the F8 generation), and compared the responses of BALB/c Adam8−/− mice and BALB/c WT parental littermate control mice sensitized with a low dose of OVA (10 μg) along with alum (or sham-sensitized with PBS) via the i.p. route, and then challenged daily on days 14–17 with aerosolized OVA or PBS. OVA sensitization and challenge induced greater airway inflammatory cell infiltrates and more mucus metaplasia in the Adam8−/− mice when compared with WT mice (Figs. 1A and 1B). OVA-treated Adam8−/− mice had 2-fold higher BAL total leukocyte counts, BAL eosinophil counts, BAL macrophage counts, and BAL lymphocyte counts than OVA-treated WT mice 24 h after the last OVA challenge (Fig. 1C). OVA-treated Adam8−/− mice also had significantly greater increases in Rn in response to aerosolized methacholine than OVA-treated WT mice (Fig. 1D).
Fig. 1. Adam8−/− mice have greater OVA-induced AAI than WT mice.

In A–D, BALB/c WT and BALB/c Adam8−/− mice were sham sensitized with PBS or sensitized with a low of dose (10 μg) of OVA along with alum via the i.p. route and then challenged with aerosolized PBS or OVA. In A and B, lungs were inflated and fixed in formalin 24 h after the last PBS or OVA challenge and stained with hematoxylin and eosin (in A, original magnification × 100) or Periodic Acid Schiff (in B, original magnification × 400). The lung sections shown are representative of 6 mice per group. Arrows indicate peri-bronchial and peri-vascular inflammation in A and mucus metaplasia in B. In C, BAL was performed 24 h after the last PBS or OVA challenge and absolute numbers of all BAL leukocytes (All WBCs), eosinophils (Eos), macrophages (Macs), lymphocytes (Lymphs), and PMNs were counted. Data are mean ± SEM; n = 4 mice in the PBS-treated groups and 14–19 mice in the OVA-treated groups; * indicates p ≤ 0.001, and ** p ≤ 0.017 compared with PBS-treated mice belonging to the same genotype. In D, central airway resistance (Rn) to aerosolized methacholine was measured 24 h after the last PBS or OVA challenge. Data are mean ± SEM; n = 5–6 mice/group for PBS-treated mice and n= 14–20 mice/group for OVA-treated mice. Asterisk indicates p ≤ 0.014 vs. OVA-treated WT mice, and ** p 0.021 when compared with PBS-treated mice belonging to the same genotype as the OVA-treated mice. Responses for OVA-treated WT mice ranged from 0.5–1.6 cm H2O/s/ml for 10 mg/ml of methacholine; 0.6–1.9 cm H2O/s/ml for 30 mg/ml of methacholine; and 1.0–1.8 cm H2O/s/ml for 45 mg/ml of methacholine. Responses for OVA treated Adam8−/− mice ranged from 0.7–1.7 cm H2O/s/ml (for 10 mg/ml of methacholine); 1.1–3.4 cm H2O/s/ml (for 30 mg/ml of methacholine); and 1.5–3.4 cm H2O/s/ml (for 45 mg/ml of methacholine).
Adam8 reduces lung levels of some TH2 cytokines and other pro- and anti-inflammatory mediators
Lung levels of Il-4 and Il-5 were significantly higher in the lungs of OVA-treated Adam8−/− mice than OVA-treated WT mice 24 h after the last OVA challenge (Fig. 2A–2B). However, lung levels of Il-13 (Fig. 2C), and thymic stromal lymphopoietin (Tslp; Supplemental Table) did not differ between OVA-treated WT and OVA-treated Adam8−/− mice. We also assessed whether Adam8 limits AAI in mice by reducing lung levels of cytokines and chemokines that promote migration of eosinophils and mononuclear leukocytes into the airways. Lung levels of Rantes (Ccl5), Ip-10 (Cxcl10), and Tnf-α were higher in OVA-treated Adam8−/− than OVA-treated WT mice (Fig. 2D–2F). However, OVA-treated WT vs. Adam8−/− mice did not differ in lung levels of other key chemokines that drive recruitment of eosinophils and mononuclear leukocytes into allergen-exposed airways including eotaxin (Ccl11), Mip-1α (Ccl3), Je (Ccl2), Mdc (Ccl22), or Tarc (Ccl17; Supplemental Table). We also found no differences in lung levels of the anti-inflammatory mediator, transforming growth factor-β (Tgf-β), in OVAtreated WT and Adam8−/− mice (Supplemental Table). However, lung levels of anti-inflammatory Il-10 were higher in OVA-treated Adam8−/− mice compared with OVA-treated WT mice. Thus, OVA-treated Adam8−/− mice had greater AAI and AHR than OVA-treated WT mice despite having higher lung levels of a key antiinflammatory mediator.
Fig. 2. OVA-treated Adam8−/− mice have higher lung levels of TH2 cytokines, Rantes, Ip-10, and Tnf-α compared with OVA-treated WT mice.

In A–F, BALB/c WT and BALB/c Adam8−/− mice were treated with PBS or sensitized with a low dose (10 μg) of OVA along with alum by the i.p. route, and then challenged with aerosolized PBS or OVA. Levels of interleukin-4 (A), interleukin-5 (B), interleukin-13 (C) Rantes (D), interferon-gamma inducible protein-10 (E), and Tnf-α (F) protein were measured in homogenates of lungs isolated from PBS- and OVA-treated BALB/c WT and BALB/c Adam8−/− mice 24 h after the last PBS or OVA challenge using ELISAs. Data are mean ± SEM; n = 4–6 for PBS-treated mice and n = 6–10 for OVA-treated mice. In A–C, * indicates p ≤ 0.046 (and p ≤ 0.003 for OVA- versus PBS-treated mice belonging to the same genotype for each cytokine measured). In D–F, asterisk indicates p = 0.041, and ** indicates p ≤ 0.022 compared with PBS-treated mice belonging to the same genotype as the OVA-treated mice. NS indicates no significant difference between the groups indicated.
Epithelial cell-derived Adam8 does not regulate mucin gene expression in airway epithelial cells
Il-13 is a key cytokine inducing mucus metaplasia during AAI in mice. However, while OVA-treated Adam8−/− mice had greater OVA-induced mucus metaplasia (Fig. 1B) they had similar levels of Il-13 when compared with OVA-treated WT mice (Fig. 2C). Adam8 is expressed by activated airway epithelium as well as leukocytes in mice (19), and other ADAM proteinases expressed by airway epithelial cells regulate mucin production by airway epithelial cells (43). Thus, we assessed whether epithelial-derived Adam8 regulates airway epithelial cell mucus production directly by measuring mucin gene expression in airway epithelial cells isolated from naïve WT vs. Adam8−/− mice and cultured under basal or Il-13 stimulated conditions. Muc5ac transcripts were not detected in epithelial cell cultures from either genotype under either basal or Il-13 stimulated conditions (data not shown). Muc5b expression was ~6-fold higher in unstimulated Adam8−/− epithelial cells when compared with unstimulated WT epithelial cells (Supplemental Fig. 2A). When WT and Adam8−/− epithelial cells were both incubated with Il-13, Muc5b gene expression increased ~11-fold in WT cells when compared with unstimulated WT cells, but Il-13 did not increase Muc5b gene expression in Adam8−/− cultures when compared with expression levels in unstimulated Adam8−/− cells (Supplemental Fig. 2B). When the results for Muc5b gene expression in Il-13-activated epithelial cells were normalized to Muc5b expression in unstimulated WT epithelial cells, Muc5b gene expression increased to a similar extent in WT and Adam8−/− cells. Thus, epithelial cell-derived Adam8 restrains basal Muc5b gene expression in airway epithelial cells. However, under conditions in which Il-13 levels in the airways of mice are high (e.g. during AAI) our in vitro results indicate that Adam8 that is expressed by activated epithelial cells is unlikely to contribute to the Adam8-mediated reduction in mucus metaplasia that we observed in mice with OVA-induced AAI (Fig. 1B).
Adam8 limits house dust mite protein (HDM)-induced acute AAI and AHR in mice
We also compared AAI and AHR in BALB/c WT vs. Adam8−/− mice sensitized and challenged with a second antigen that is a common trigger for human asthma (HDM). HDM-treated Adam8−/− mice had higher BAL leukocyte counts (Fig. 3A) and greater peribronchial inflammation than HDM-treated BALB/c WT mice (Fig. 3B). In addition, HDM-treated Adam8−/− mice had greater AHR to aerosolized methacholine challenges than HDM-treated WT mice (Fig. 3C). Together, these data indicate that Adam8 potently limits AAI and AHR in response to two different allergens in mice sensitized by delivering allergen by two different routes (i.p. for OVA and the respiratory mucosal route for HDM), using either high or low doses of allergen (with or without an adjuvant) for sensitization. In all subsequent experiments, we studied WT and Adam8−/− mice in the BALB/c background in the more commonly-used OVA model of AAI and AHR [using a low dose of OVA 10 μg] along with alum delivered by the i.p. route for the allergen sensitization step.
Fig. 3. HDM-treated Adam8−/− mice have greater AAI and higher AHR than HDM-treated WT mice.

BALB/c WT and BALB/c Adam8−/− mice were sensitized and challenged via the intranasal route with HDM protein extract or sham-treated with PBS as a control. In A, leukocytes were counted in BAL samples 72 h after the last HDM challenge. Data are mean ± SEM; n= 5 PBS-treated mice and n = 10–12 HDM-treated mice. Asterisk indicates p ≤ 0.007 versus PBS-treated mice belonging to the same genotype as the OVA-treated mice, and ** indicates p = 0.017. B shows sections of lungs fixed and inflated in formalin and stained with hematoxylin and eosin. Upper panels in B show greater peribronchial inflammation (arrows) in HDM-treated Adam8−/− mice compared with HDM-treated WT mice. Lower panels in B show that there is predominantly eosinophilic peribronchial inflammation in both genotypes (arrowheads). In C, HMD- or PBS-treated BALB/c WT and BALB/c Adam8−/− mice were exposed to varying concentrations of aerosolized methacholine (or aerosolized PBS) and Rn was measured 72 h after the last HDM challenge. Data are mean ± SEM; n = 5–6 mice/group for PBS-treated mice and n = 7–8 mice per group for HDM-treated mice. Asterisk indicates p < 0.001 when compared with HDM-treated WT mice; and ** indicates p ≤ 0.036 when compared with PBS-treated mice belonging to the same genotype as the OVA-treated mice. Responses for HDM-treated WT mice ranged from 0.17–0.43 cm H2O/s/ml for 1 mg/ml of methacholine; 0.27–0.50 cm H2O/s/ml for 3 mg/ml of methacholine; and 0.40–1.06 cm H2O/s/ml for 10 mg/ml of methacholine; 0.64–1.44 cm H2O/s/ml for 30 mg/ml of methacholine; and 0.66–1.34 cm H2O/s/ml for 45 mg/ml methacholine. Responses for HDM treated Adam8−/− mice ranged from 0.26–0.71 cm H2O/s/ml for 1 mg/ml of methacholine; 0.26–1.64 cm H2O/s/ml for 3 mg/ml of methacholine; 0.48–1.71 cm H2O/s/ml for 10 mg/ml methacholine; 0.86–2.11 cm H2O/s/ml for 30 mg/ml methacholine; and 1.37–1.93 cm H2O/s/ml for 45 mg/ml methacholine.
Leukocytes are the crucial source of anti-inflammatory Adam8 in the airways of mice with AAI
Adam8 is expressed both by leukocytes and activated airway epithelium in mice (19). Thus, we performed bone marrow (BM) transplant experiments to generate Adam8 BM chimeric mice to determine whether leukocyte-derived or lung parenchymal cell-derived Adam8 mediates its anti-inflammatory activities in the airways of mice with AAI. OVA-treated WT recipients transplanted with Adam8−/− BM had greater peribronchial inflammation (Fig. 4A), higher BAL total leukocyte counts (Fig. 4B), greater BAL eosinophil counts (Fig. 4C), and higher BAL macrophage counts (data not shown) than WT recipients transplanted with WT BM. In addition, Adam8−/− recipients transplanted with WT bone marrow had less peribronchial inflammation (Fig. 4A), lower BAL total leukocyte counts (Fig. 4B), lower BAL eosinophil counts (Fig. 4C), and lower BAL macrophage counts (data not shown) than Adam8−/− mice transplanted with Adam8−/− BM. Thus, BM-derived leukocytes are crucial sources of anti-inflammatory Adam8 in mice with AAI. To provide insights into which leukocytes are important sources of Adam8 in the airways of mice with AAI, we isolated eosinophils and macrophages from the airways of OVA-sensitized and -challenged WT mice, and measured Adam8 protein levels in extracts of the cells using an ELISA. Airway eosinophils and macrophages both robustly expressed Adam8 protein and contained similar amounts of Adam8 protein (Table 1; p = 0.802). When we measured Adam8 protein levels in extracts of bone marrow-derived eosinophils versus bone marrow-derived monocytes we detected Adam8 protein in both cell types. However, bone marrow-derived eosinophils contained modestly (1.7-fold) but statistically significantly more Adam8 protein than bone-marrow derived monocytes (mean 1042.3 ± SEM 189.5 versus 605.2 ± SEM 81.5 pg of Adam8 per ng of solubilized protein; n = 6 and 7 preparations, respectively; p = 0.047). These results suggest that eosinophils, monocytes, and macrophages are all important sources of this anti-inflammatory proteinase in the airways of WT mice with AAI.
Fig. 4. Leukocyte-derived Adam8 predominantly mediates Adam8’s anti-inflammatory activities in the airways during AAI.

We generated BALB/c strain Adam8 bone marrow (BM) chimeric mice, as described in Methods. Adam8 BM chimeric mice were sham-sensitized with PBS or sensitized with a low dose (10 μg) of OVA along with alum by the i.p. route and then challenged with aerosolized PBS or OVA. Lungs were inflated and fixed in formalin or BAL was performed. A shows images of hematoxylin and eosin-stained sections of lungs from the mice 24 h after the last OVA challenge (images are representative of 6 mice/group; original magnification × 100). PBS-treated mice had no airway inflammation (data not shown). Twenty four hours after the last OVA or PBS challenge, total leukocytes (B) and granulocytes (>80% eosinophils in C) were counted in BAL samples. Data are mean ± SEM n = 3–4 mice/group for PBS-treated mice and n = 8–22 mice/group for OVA-treated mice. Asterisk indicates p ≤ 0.01 and ** p ≤ 0.023 in both B and C.
Table 1.
Airway eosinophils and macrophages isolated from WT mice with AAI express Adam8 protein
| Adam8 protein levels (pg/μg total protein)** | |
|---|---|
| BAL macrophages* | 236.0 ± 65.7*** |
| BAL eosinophils* | 174.5 ± 24.2 |
BALB/c WT mice were sensitized with 10 μg of OVA by the i.p. route and then challenged with aerosolized OVA. Twenty four h after the last OVA challenge, airway eosinophils and macrophages were isolated, and cell extracts were prepared, as described in Materials and Methods.
Adam8 protein and total protein levels were measured in extracts of BAL macrophages versus BAL eosinophils using an ELISA and a Bradford dye-binding assay kit, respectively. The results are expressed as pg of Adam8 per μg of total protein.
Data are mean ± SEM; n = 16 different paired BAL macrophage and eosinophil preparations (p = 0.802 for the comparison of airway macrophages and eosinophils).
To address the mechanism by which Adam8 reduced AAI and AHR in mice, we tested whether Adam8: 1) inhibited sensitization of mice to allergen; 2) reduced circulating leukocyte counts; 3) inhibited leukocyte adhesion or migration, or cleaved and thereby inactivated pro-inflammatory mediators that promote leukocyte recruitment into the airways; 4) decreased airway leukocyte survival; and/or 5) increased leukocyte clearance from the airways of mice with AAI.
Adam8 does not regulate sensitization of mice to OVA
OVA-treated WT and Adam8−/− mice did not differ in serum levels of OVA-specific IgE (Fig. 5A) indicating that Adam8 did not limit AAI in mice by reducing the sensitization of mice to OVA. Adam8 is expressed by dendritic cells (44) which regulate adaptive immune responses to allergen. Thus, we also quantified dendritic cell numbers in enzymatic lung digests from OVA-sensitized and -challenged WT and Adam8−/− mice. WT and Adam8−/− mice did not differ in their numbers of lung dendritic cells when treated with PBS or OVA (Fig. 5B). Additionally, Adam8 likely did not reduce OVA-induced AAI and AHR via Adam8’s MP domain degrading OVA because we detected no cleavage or degradation of OVA protein when we incubated OVA with 200 nM recombinant active human ADAM8 in vitro (data not shown).
Fig. 5. Adam8 is not required for sensitization of mice to OVA.
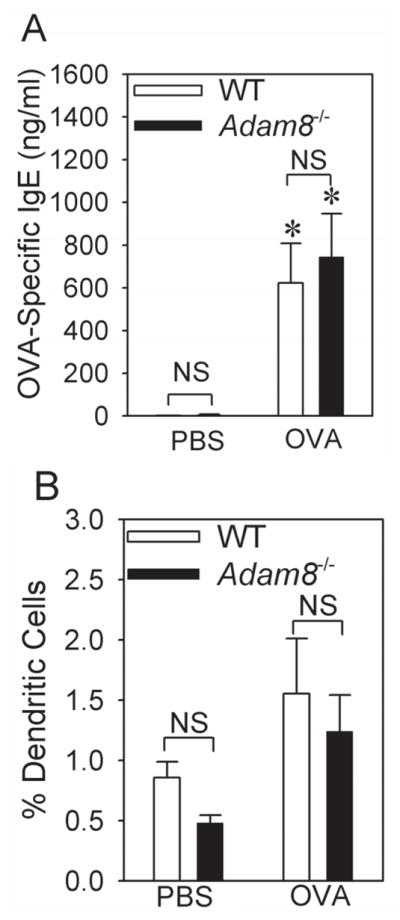
BALB/c WT and BALB/c Adam8−/− mice were sham-sensitized with PBS or sensitized with a low dose (10 μg) of OVA along with alum delivered by the i.p. route, and then challenged with aerosolized PBS or OVA. In A, levels of OVA-specific IgE were measured in serum samples from mice treated with either with PBS (n = 3 mice/group) or OVA (n = 8–9 mice/group). Data are mean ± SEM. Asterisk indicates p < 0.002 when compared with PBS-treated mice of the same genotype. In B, lungs were harvested from the mice 24 h after the last OVA or PBS challenge, and lungs were enzymatically digested, as described in Methods. Dendritic cells were enumerated in single cell suspensions of lung digests by immunostaining and flow cytometry, as described in Methods. Data are mean ± SEM (n = 3 mice/group for PBS-treated mice and n = 5 mice/group for OVA-treated mice).
Adam8 does not regulate circulating leukocyte counts or leukocyte adhesion or migration
Unchallenged WT and Adam8−/− mice do not differ in their peripheral total and differential WBC counts (20). OVA-treated WT and Adam8−/− mice also had similar total and differential leukocyte counts in blood samples (data not shown), indicating that Adam8 did not influence circulating leukocyte counts in mice with AAI.
Next, we tested whether Adam8 restrained AAI and AHR in mice by shedding leukocyte cell adhesion molecules to reduce leukocyte transendothelial migration as ADAM8 sheds L-selectin from the surface of human PMNs activated in vitro (24). We measured circulating leukocyte surface L-selectin levels and levels of soluble L-selectin in serum samples from PBS- or OVA-treated WT vs. Adam8−/− mice. Neither serum sL-selectin levels (data not shown) nor surface L-selectin levels on blood leukocytes (data not shown) differed between PBS- or OVA-challenged WT and Adam8−/− mice. ADAM8 also sheds vascular cell adhesion molecule-1 (Vcam-1) in vitro (27), and Vcam-1 is an important vascular endothelial cell molecule regulating leukocyte migration into the airways during AAI (27). However, WT and Adam8−/− mice with AAI did not differ in serum levels of soluble Vcam-1 (PBS-treated WT mice 106 ± SEM 29 pg/ml; PBS-treated Adam8−/− mice 271 ± 75 pg/ml; OVA-treated WT mice 1581 ± 127 pg/ml; OVA-treated Adam8−/− mice 1894 ± 112 pg/ml; n = 4 PBS-treated mice/group and n= 8 OVA-treated mice/group). Thus, it is unlikely that Adam8 inhibited leukocyte migration into the airways of mice with AAI by shedding leukocyte or endothelial cell adhesion molecules.
Adam8 has a disintegrin domain that binds α9β1 integrin expressed on the surface of osteoclasts (18), and binding of the disintegrin domain of other ADAM family members to integrins regulates cell adhesion and migration (16). Thus, we compared the capacity of WT vs. Adam8−/− leukocytes isolated from unchallenged mice to: 1) adhere to murine lung microvascular endothelial cells (LMVECs) and a representative extracellular matrix (ECM) protein (fibronectin); and 2) migrate in response to chemoattactants in vitro. WT and Adam8−/− eosinophils and monocytes did not differ in their capacity to adhere to murine LMVECs (Fig. 6A–6B) or fibronectin (data not shown) either under basal or stimulated conditions. WT and Adam8−/− eosinophils and monocytes also did not differ in their ability to migrate in response to buffer or chemoattractants in either Boyden microchemotaxis chambers (Fig. 6C and 6D) or Matrigel™ Invasion Chambers (data not shown). Together, these data suggest that Adam8 did not reduce AAI by inhibiting leukocyte transendothelial migration.
Fig. 6. Adam8 is not required for murine eosinophils or monocytes to adhere to lung microvascular endothelial cell (LMVEC) monolayers or migrate in vitro.

In A, equal numbers (106 cells) of bonemarrow-derived monocytes isolated from unchallenged BALB/c WT or BALB/c Adam8−/− mice were incubated at 37°C for 1 h in duplicate with or without 1 μg/ml LPS for 2 h on LMVEC monolayers that had been pre-incubated for 4 h at 37°C with or without 10 ng/ml Tnf-α. In B, equal numbers (106 cells) of bone marrow-derived eosinophils isolated from unchallenged BALB/c WT and BALB/c Adam8−/− mice were incubated in duplicate at 37°C with or without 10−7M LTC4 on LMVEC monolayers that had been pre-incubated for 18 h with or without 10−7M interleukin-4 and 10−7M interleukin-13. In A and B, the percentage of leukocytes adhering to the LMVEC monolayers was determined. Data are mean ± SEM; n= 3–4 experiments. In C and D, we used Boyden micro-chemotaxis assay chambers to compare the migration of bone-marrow derived WT vs. Adam8−/− monocytes to buffer alone, 10−7M fMLP, or 10−7 M Mip-1α in the lower chambers (in C) or the migration of WT vs. Adam8−/− bone marrow derived eosinophils in response to buffer alone, 10−7 M LTB4, or 10−8M murine eotaxin in the lower chambers (in D). Data are expressed as fold increase in cellular migration when compared with cellular migration in response to buffer alone. Data are mean ± SEM from 3–6 wells per experimental condition and 3–6 separate experiments. Asterisk indicates p ≤ 0.031 versus cells in C and p ≤ 0.01 in D when compared with rates of migration of cells of the same genotype in response to buffer.
The MP domain of ADAM8 does not degrade pro-inflammatory mediators
We found higher levels of some TH2 cytokines and mediators of inflammation in the lungs of OVA-treated Adam8−/− mice vs. OVA-treated WT mice and Adam8 has an active MP domain. Thus, we tested whether Adam8 reduced lung levels of Rantes, Ip-10, Tnf-α, Il-4, and/or Il-5 (Fig. 2) by its MP domain degrading these mediators. We studied the activity of recombinant human ADAM8 (rhADAM8) because purified murine Adam8 is not commercially available. First, we confirmed that rhADAM8 ectodomain (which contains the MP domain) is catalytically active by showing that it progressively degraded a quenched fluorogenic peptide substrate which is sensitive to cleavage by ADAM8 [(22); data not shown]. However, active rhADAM8 did not degrade recombinant purified human IL-4, RANTES, TNF-α, or IP-10 (Supplemental Fig. 3 or data not shown) even when high concentrations (200 nM) of hrADAM8 were tested in vitro. Although a lower Mr cleavage product of IL-5 was detected when IL-5 was incubated with 200 nM rhADAM8, most of the IL-5 tested was not degraded (Supplemental Fig. 3). Thus, given the high concentration of ADAM8 tested in vitro and the low amount of the IL-5 cleavage product generated, it is unlikely that ADAM8 reduced lung levels of Il-5 during AAI in mice via its MP domain degrading Il-5. Likely, the higher lung levels of some pro-inflammatory mediators (Fig. 2) and anti-inflammatory Il-10 (Supplemental Table) in Adam8−/− mice with AAI compared with WT mice with AAI were a consequence of the higher leukocyte counts in the airways of Adam8−/− mice with AAI as leukocytes are important cellular sources of these mediators.
Adam8 increases apoptosis of eosinophils and macrophages in the airways of mice with AAI, but has no effect on regulating macrophage uptake of apoptotic leukocytes
To assess whether Adam8 limited AAI by reducing the survival of airway macrophages and eosinophils, we compared rates of apoptosis of macrophages and eosinophils recruited to the airways of WT vs. Adam8−/− mice with AAI. Intracellular levels of active caspase 3 (Fig. 7A) and loss of mitochondrial membrane potential (which is a marker of activation of the intrinsic apoptosis pathway; Fig. 7B) were significantly lower in all Adam8−/− airway leukocytes and also in Adam8−/− airway eosinophils and airway macrophages (but not lymphocytes) than airway leukocytes from WT mice with AAI. This result indicates that Adam8 reduced the survival of airway leukocytes in mice with AAI. Airway macrophages isolated from Adam8−/− mice with AAI also contained fewer apoptotic bodies than airway macrophages from WT mice with AAI (Fig. 7C). We assessed whether the latter finding was due to impaired macrophage uptake of apoptotic cells (efferocytosis) in the absence of Adam8. When WT and Adam8−/− macrophages isolated from unchallenged mice were incubated with equal numbers of granulocytes that had been induced to undergo apoptosis in vitro, the mean number of apoptotic target cells ingested per WT and Adam8−/− macrophage was similar (Fig. 7D). These data indicate that the reduced numbers of apoptotic bodies in airway macrophages from Adam8−/− mice with AAI in Fig. 7C was likely to be due to reduced rates of airway leukocyte apoptosis in mice lacking Adam8 rather than to defects in efferocytosis of airway macrophages lacking Adam8.
Fig. 7. Adam8 increases leukocyte apoptosis but does not regulate clearance of apoptotic leukocytes by macrophages in the airways of mice with AAI.

In A–C, BALB/c strain WT and Adam8−/− mice were shamsensitized with PBS or sensitized with a low dose (10 μg) of OVA along with alum by the i.p. route, and then challenged with aerosolized PBS or OVA. BAL leukocytes were isolated 24 h after the last PBS or OVA challenge. BAL leukocytes were permeabilized and immunostained with an antibody to active caspase-3 or with a non-immune control antibody (in A), or loss of mitochondrial membrane potential was measured in intact cells (in B). Intracellular levels of active caspase-3 and the percentage of cells that had lost mitochondrial membrane potential (apoptotic cells) were quantified in all leukocytes (WBC), granulocytes (Granulo), macrophages (Macs), and lymphocytes (Lymphs) by flow cytometry, as described in Methods. Data are mean ± SEM; n = 16–17 mice/group in A and n = 7–9 mice in B. In A, * indicates p = 0.03 and ** indicates p ≤ 0.006. In B, * indicates p = 0.023 and ** p ≤ 0.014. In C, BAL macrophages were isolated from mice 24 h after the last OVA (n = 11–12 mice/group) or PBS (n = 4 mice/group) challenge. Apoptotic cells that had been ingested by BAL macrophages were counted in 300 macrophages per genotype. Data are mean ± SEM apoptotic cells per BAL macrophage. Asterisk indicates p = 0.042 and ** p ≤ 0.017. In D, macrophages isolated from unchallenged BALB/c WT and BALB/c Adam8−/− mice were incubated for 1 h at 37°C with equal numbers of apoptotic PMNs isolated from unchallenged BALB/c WT or BALB/c Adam8−/− mice. The mean ± SEM number of apoptotic cells ingested per macrophage was counted in 300 macrophages per group (n = 3 separate experiments).
Adam8 does not regulate surface levels of apoptosis ligands or receptors on airway leukocytes during AAI in mice
Adam8 has an active MP domain that has the potential to shed and activate apoptosis ligands from cell surfaces. However, surface levels of Tnf-α, Fas ligand, and Trail and their receptors did not differ on airway eosinophils or macrophages isolated from OVA-treated WT and Adam8−/− mice (data not shown). Thus, it is unlikely that Adam8 reduced survival of airway leukocytes by activating the extrinsic apoptosis pathway in vivo.
Adam8 promotes activation of the intrinsic but not extrinsic apoptosis pathway in myeloid leukocytes in vitro
To determine which apoptosis pathway in myeloid leukocytes is regulated by Adam8, we isolated macrophages or eosinophils from naive WT and Adam8−/− mice and compared their rates of apoptosis after triggering activation of either: 1) the extrinsic apoptosis pathway in eosinophils by ligating FAS; or 2) the intrinsic apoptosis pathway by serum starving the cells. When we activated the extrinsic apoptosis pathway in WT and Adam8−/− macrophages and eosinophils in vitro, we found no differences in their rates of apoptosis, as assessed by measuring intracellular levels of active caspases-3 and -8 (Table 2), and binding of annexin V-Alexa 488 and propidium iodide to the cells (Table 3). WT and Adam8−/− eosinophils and macrophages had similar cell surface levels of apoptosis ligands and their receptors including FAS-ligand, Trail, Tnf-r1, and FAS (data not shown), with the exception of surface Tnf-α levels which were significantly higher on Adam8−/− macrophages compared with WT cells (data not shown). The relevance of the latter in vitro finding to events occurring in the airways of mice with AAI are not clear given that: 1) we found similar levels of Tnf-α on the surface of macrophages from the airways of WT and Adam8−/− mice with AAI (data not shown); and 2) studies of conditional Adam17−/− mice lacking Adam17 only in myeloid leukocytes have confirmed that Adam17 is the main Tnf-α sheddase expressed by murine macrophages in vitro and in vivo (45).
Table 2.
Intracellular levels of active caspase-3 and -8 do not differ in eosinophils undergoing apoptosis induced by activating the extrinsic apoptosis pathway in vitro
| Time point | WT EosinophilsA | Adam8−/− Eosinophils | WT Eosinophils | Adam8−/− Eosinophils |
|---|---|---|---|---|
|
| ||||
| Active caspase-3 in ng/ml Mean (SEM) n |
Active caspase-3 in ng/ml Mean (SEM) n |
Active caspase 8 in units/ml Mean (SEM) n |
Active caspase 8 in units/ml Mean (SEM) n |
|
|
| ||||
| 0 h | 6.4 (8) 4B | 13.4 (14.6) 4 | 0.1 (0.4) 3 | 0.7 (0.4) 3 |
| 2 h | 25.1 (10.2) 4 | 16.1 (13.8) 4 | 0.5 (0.6) 3 | 0.6 (0.5) 3 |
| 4 h | 33.9 (10.6) 4 | 27.8 (13.3) 4 | 1.8 (1.4) 3 | 1.5 (0.9) 3 |
| 6 h | 63.7 (15.9) 4 | 44.8 (18.3) 4 | 2.5 (1.9) 3 | 1.2 (1.2) 3 |
| 8 h | 57.5 (18.1) 4 | 49.1 (23.1) 4 | 2.1 (1.9) 3 | 0.6 (0.9) 3 |
| 20 h | 12.8 (5.9) 4 | 6.6 (2.9) 4 | 0 (0.5) 3 | 0 (0.6) 3 |
Eosinophils were isolated from the bone marrow of unchallenged BALB/c WT and BALB/c Adam8−/− mice, as described in Methods. Macrophages were isolated from the peritoneal cavities of BALB/c WT and BALB/c Adam8−/− mice 4 days after thioglycollate was delivered by the i.p. route, and cells were cultured for 5 days in tissue culture plates to render them quiescent. Equal numbers of eosinophils or macrophages were incubated with a cross-linking antibody to CD95 (FAS) which activates this death domain-containing receptor, or a non-immune isotype control antibody for up to 20 h for eosinophils or 48 h for macrophages. At intervals, cell extracts were prepared at 5 × 106 cells/ml and intracellular levels of active caspase-3 and -8 were measured using specific fluorogenic substrates, as described in Methods.
Data are mean (SEM); n =3–4 mice studied per group.
We were not able to induce significant activation caspase-3 or 8 in either WT or Adam8−/− macrophages by treating cells with the FAS cross-linking antibody for up to 48 h (data not shown).
Table 3.
Binding of annexin-V and propidium iodide to WT vs. Adam8−/− eosinophils do not differ when the extrinsic apoptosis pathway is triggered in vitro
| Time point | WT EosinophilsA | Adam8−/− Eosinophils | WT Eosinophils | Adam8−/− Eosinophils | WT Eosinophils | Adam8−/− Eosinophils |
|---|---|---|---|---|---|---|
|
| ||||||
| % Apoptotic Cells Mean (SEM) n |
% Apoptotic Cells Mean (SEM) n |
% Late Apoptotic Cells Mean (SEM) n |
% Late Apoptotic Cells Mean (SEM) n |
% Viable Cells Mean (SEM) n |
% Viable Cells Mean (SEM) n |
|
|
| ||||||
| 0 h | 0 (2.9) 3B | 0 (3.1) 3 | 0.5 (0.3) 3 | 0 (0.2) 3 | 0.3 (2.7) 3 | 0.2 (3.1) 3 |
| 2 h | 4.1 (6.4) 3 | 8.4 (10.6) 3 | 0.3 (0.1) 3 | 1.2 (0.5) 3 | 3.6 (6.3) 3 | 8.3 (10.7) 3 |
| 4 h | 19 (5.9) 3 | 16.7 (6.1) 3 | 3 (1.3) 3 | 7.3 (0.7) 3 | 18.3 (5) 3 | 19.4 (7) 3 |
| 6 h | 12.2 (9.3) 3 | 9.9 (19.3) 3 | 5.8 (2.5) 3 | 10.9 (8.4) 3 | 16.4 (8.2) 3 | 30.1 (7.8) 3 |
| 8 h | 19.7 (15) 3 | 7.3 (2.2) 3 | 7.5 (1.7) 3 | 8.2 (0.7) 3 | 30.2 (8.9) 3 | 29.5 (6.4) 3 |
| 20 h | 46.2 (10.9) 3 | 40.2 (9.6) 3 | 38.7 (12.5) 3 | 40.6 (8.9) 3 | 57.2 (2.1) 3 | 51.8 (2.6) 3 |
Eosinophils and macrophages were isolated from unchallenged BALB/c WT and BALB/c Adam8−/− mice, as described in Methods. Equal numbers of eosinophils or macrophages were incubated with a cross-linking antibody to CD95 (FAS) which activates this death domain-containing receptor, or a non-immune isotype control antibody for up to 20 h for eosinophils or 48 h for macrophages. At intervals, cells were stained with annexin-V conjugated to Alexa 488 and propidium iodide, and the percentage of cells staining positively and negatively with these reagents was quantified using flow cytometry. The results for cells incubated with a non-immune control antibody were subtracted from those incubated with anti-CD95. We determined the percentages of apoptotic cells (cell staining positively with annexin-V and negatively with propidium iodide), late apoptotic cells (cells staining positively with both annexin-V and propidium iodide), and viable cells (cells not stained with either annexin-V or propidium iodide).
Data are mean ± SEM; n = number of independent experiments.
We were not able to induce significant positive staining for either annexin V or propidium iodide in either WT or Adam8−/− macrophages by treating cells with the FAS cross-linking antibody for up to 48 h.
When we triggered the intrinsic apoptosis pathway in myeloid leukocytes isolated from unchallenged WT and Adam8−/− mice, Adam8−/− eosinophils and/or macrophages had reduced staining for annexin-V and propidium iodide (Fig. 8A) and reduced levels of intracellular active caspase-3 (Fig. 8B and 8C). Additionally, when compared with WT eosinophils and/or macrophages, Adam8−/− cells had reduced levels of two markers of the intrinsic apoptosis pathway activation: 1) loss of mitochondrial membrane potential (Fig. 8D and 8E); and 2) intracellular levels of active caspase-9 (Fig. 8F). Thus, Adam8 triggered activation of the intrinsic (but not the extrinsic) apoptosis pathway in myeloid leukocytes. Likely, this reduced the persistence of leukocytes in the airways (leading to reduced AAI and AHR) because airway macrophages and possibly airway epithelial cells rapidly clear apoptotic leukocytes via efferocytosis (46).
Fig. 8. Adam8 promotes activation of the intrinsic apoptosis in eosinophils and macrophages in vitro.

The intrinsic apoptosis pathway was activated in quiescent peritoneal macrophages (Macs in A, B, F) or quiescent alveolar macrophages (D) or quiescent bone marrow-derived eosinophils (Eos in C and E) isolated from unchallenged BALB/c WT and BALB/c Adam8−/− by incubating cells in the absence of serum, agonists, and survival factors for up to 48 h at 37°C. At intervals, cells were stained with annexin-V conjugated to Alexa-488 and propidium iodide (A) and the percentage of cells staining positively for both annexin-V and propidium iodide was quantified by flow cytometry. Data are mean ± SEM positive cells; n = 3 experiments. Asterisk indicates p ≤ 0.027. Intracellular levels of active caspase-3 (B and C) or active caspase-9 (F) were quantified in macrophage and/or eosinophil cell extracts using quenched fluorogenic substrates that are specific for caspase-3 or caspase-9, respectively, and fluorimetry. Data are mean ± SEM positive cells; n = 3–8 experiments. In B, * indicates p ≤ 0.013, in C, * indicates p ≤ 0.003 vs. Adam8−/− cells studied at the same time point,. In F, * indicates p < 0.029 compared with WT cells studied at the same time point. Loss of mitochondrial membrane potential was measured in serum-starved alveolar macrophages (D) or eosinophils (E) isolated from unchallenged BALB/c WT or BALB/c Adam8−/− mice, as described in Methods. Data are mean ± SEM; n = 4–5 experiments. In D and E, * indicates p ≤ 0.048 and ** p = 0.007.
Adam8 does not regulate lung levels of survival factors other than Il-5 in mice with AAI
Reductions in extracellular levels of survival factors promote activation of the intrinsic apoptosis pathway (47,48). Accordingly, we measured levels of survival factors in lung samples from WT and Adam8−/− mice with AAI. As mentioned above, lung levels of Il-5 (which is an important survival factor for eosinophils) were increased in Adam8−/− mice with AAI compared with WT mice with AAI (Fig. 2B). However, WT and Adam8−/− mice with AAI did not differ in lung levels of M-csf (which is a key survival factor for macrophages) or Gm-csf (which is an important survival signal for both eosinophils and macrophages; Supplemental Table). Thus, it is unlikely that Adam8 promotes apoptosis of macrophages by reducing lung levels of macrophage survival factors.
ADAM8 is robustly expressed in peribronchial granulocytes and airway epithelium, but not in macrophages, in lung sections from human patients with asthma
To gain insights into the cell types that express ADAM8 in asthmatic airways, we immuno-stained lung sections from asthma patients and control subjects without asthma for ADAM8. ADAM8 was expressed to a similar extent in bronchial epithelium and type II pneumocytes in the lungs of patients with and without asthma (Fig. 9A). The bronchial epithelium had membranous and cytoplasmic staining patterns, with an apical accentuation in both asthmatics and non-asthmatics. There was some suggestion of a more diffuse cytoplasmic reactivity in the asthmatic cases but no significant difference in the degree of positive staining for ADAM8 in bronchial epithelium in lung sections from asthmatic versus non-asthmatic subjects (Figs. 9A and 9B). Eosinophils in this study set were found exclusively in the asthmatic tissues and predominantly within the airway walls, and were strongly positive for ADAM8 (Fig. 9A and black arrows in Fig. 9B). In contrast, all macrophages identified in lung sections (by LS) from all five asthmatic subjects studied had similar minimal staining for ADAM8 as macrophages in control lung sections (Fig. 9B; arrowheads). As expected, there was no staining for ADAM8 in predominantly lymphocytic peri-bronchial infiltration in a control subject with chronic bronchitis (top panel in Fig. 9B, red arrows). ADAM8 was first cloned from macrophages and is highly expressed by activated macrophages (49–51). Thus, it was surprising that we detected only low levels of ADAM8 in macrophages in asthmatic airways which we expect to be activated state. To confirm this finding, we double immunostained lung sections from three patients with asthma with a red fluorophore for a marker of macrophages (CD68) or eosinophils (major basic protein) and with a green fluorophore for ADAM8, and quantified the staining in each cell type. These results (which are representative of ADAM8 staining in lung sections from 3 asthma subjects) confirmed that airway eosinophils stained robustly for ADAM8 in asthmatic subjects whereas macrophages had relatively low-level ADAM8 staining (Fig. 10). Peripheral blood eosinophils or monocytes from healthy human subjects expressed minimal quantities of ADAM8 on their surface under basal conditions, but incubating the cells with a pro-inflammatory mediator (10−7 M leukotriene B4) for 30 min at 37°C induced 2–3 fold increases in surface ADAM8 protein levels on monocytes and eosinophils (data not shown). Thus, surprisingly ADAM8 (which has anti-inflammatory actions in the airways of mice with AAI) is expressed at only low levels in airway macrophages in human asthma patients.
Fig. 9. ADAM8 expression in the airways of asthma patients versus control subjects.

A. Lung sections from human patients without (top panels) or with (bottom panels) asthma were stained with either hematoxylin and eosin (left hand panels), or an isotype control antibody (middle panels), or an anti-ADAM8 antibody (right hand panels; original magnification × 100). B. High magnification images (original magnification × 400) of lung sections from patients with and without asthma showing intense staining for ADAM8 in peribronchial granulocytes (eosinophils; black arrows) in the asthma patients, minimal staining for ADAM8 in macrophages in either control subjects or patients with asthma (black arrowheads), and a lack of staining for ADAM8 in the predominantly lymphocytic peribronchial infiltration on a patient with chronic bronchitis (top right, red arrows). The images shown in A and B are representative of lungs sections from five asthmatic subjects and four control subjects without asthma.
Fig. 10. ADAM8 is expressed at higher levels in eosinophils than macrophages in the lungs of patients with asthma.

Lung sections from an asthmatic subject were double immuno-stained with FITC for ADAM8 (left panels in A and B) and with rhodamine for a marker of eosinophils [major basic protein (MBP) middle panels in A), or with rhodamine for a marker of macrophages (CD68; middle panels in B) and examined using a confocal microscope (original magnification × 600). Merged images are shown in the right panels for A and B. The images shown are representative of immunostained lung sections from three asthmatic subjects. Lung sections stained with non-immune control primary antibodies showed no staining (data not shown). In C, ADAM8 staining in cells identified as eosinophils versus macrophages (by their positive staining with rhodamine using the markers listed above) was quantified using MetaMorph image analysis software. The lines in the boxes in the box plots show the 25th percentiles, medians, and 75th percentiles and the error bars represent the 10th and 90th percentiles. Data are mean ± SEM (n = 18 eosinophils and 9 macrophages). Asterisk indicates p = 0.025.
Discussion
We report consistent and robust anti-inflammatory actions for leukocyte-derived Adam8 in murine AAI by studying WT vs. Adam8−/− mice in two genetic backgrounds exposed to different allergens and different sensitization protocols. Our results showed that airway eosinophils and macrophages robustly expressed Adam8 in WT mice with AAI and both cell types are likely to be crucial sources of anti-inflammatory Adam8 in mice with AAI. We also report that an Adam family member promotes activation of the intrinsic apoptosis pathway in both macrophages and eosinophils thereby restraining allergen-induced AAI and AHR in mice.
Until now, ADAM family members have been thought to regulate cellular apoptosis via their MP domains shedding apoptosis ligands and/or receptors (14). However, we found no evidence that Adam8 sheds apoptosis ligands or their receptors from the surface of myeloid leukocytes or activates the extrinsic apoptosis pathway in airway myeloid leukocytes in mice with AAI. Apoptosis promotes resolution of inflammatory responses, and non-phlogistic removal of apoptotic cells is associated with the release of anti-inflammatory mediators by macrophages (52). Asthma has been strongly linked to reduced rates of apoptosis of leukocytes in peripheral blood and airways (53–58) including two leukocyte subsets that express ADAM8 [eosinophils and macrophages (54,59–62)]. There is evidence that prolonged survival of airway eosinophils and macrophages contributes to airway inflammation in asthma (47,54,63,64). However, little is known about the pathways that regulate leukocyte apoptosis in asthma. Our study has identified Adam8 as a key molecule that promotes leukocyte apoptosis in the airways of mice with AAI. We speculate that the cytoplasmic tail of Adam8 binds to intracellular signaling molecules to trigger activation of the intrinsic pathway in myeloid leukocytes, and our future studies will investigate this possibility. Alternatively, Adam8 reduced the levels or activity of pro-survival factors in the airways of mice with AAI. Consistent with this, we found higher lung levels of the eosinophil pro-survival signal, Il-5 (47), in Adam8−/− mice with AAI compared with WT mice with AAI (Fig. 2B), which was unlikely to be due to the MP domain of Adam8 degrading Il-5 (Supplemental Fig. 3). Likely, the higher lung levels of Il-5 reflected the greater number of Il-5 producing cells in the airways of Adam8−/− mice with AAI when compared to WT mice with AAI. Although we found no evidence (Supplemental Table) that Adam8 regulated lung levels of M-csf or Gm-csf (key survival factors for myeloid leukocytes), it is possible that the MP domain of Adam8 cleaved and thereby regulated lung levels of other yet-to-be identified eosinophil or macrophage pro-survival factors in mice with AAI.
Although lung levels of some TH2 cytokines and pro-inflammatory mediators were increased in Adam8−/− mice compared with WT mice with AAI, the MP domain of rADAM8 did not degrade these mediators in vitro (Supplemental Fig. 3 or not shown). Adam8−/− mice had greater allergen-induced AAI and AHR than WT mice despite having higher levels of the anti-inflammatory cytokine, Il-10 (Supplemental Table). Likely, the higher lung levels of pro-inflammatory mediators (and Il-10) reflected the increased numbers of leukocytes resistant to apoptosis in the airways of Adam8−/− mice with AAI because leukocytes are key sources of these mediators. Adam8 may have reduced AAI in mice by polarizing airway macrophages from an alternatively activated (M2) phenotype [which other studies have reported to be increased in the airways of mice with AAI and linked to disease severity (65)] toward a classically activated (M1) phenotype. However, this is unlikely because when compared with WT mice with AAI, Adam8−/− mice with AAI had higher lung levels of some markers of M1 macrophages (Il-12, Tnf-α, and Ccl-5) along with higher levels of a classic M2 marker (Il-10), but similar lung levels of other M1 (Ccl2) and M2 (Tgf-β) markers (Fig. 2 and Supplemental Table).
An interesting finding in our paper was that while lung levels of Il-4 and -5 levels were higher in OVA-treated Adam8−/− mice than OVA-treated WT mice, lung levels of Il-13 were similar in OVA-treated WT and Adam8−/− mice. Likely, CD4+ TH2 T cell counts were increased in OVA-treated Adam8−/− mice compared to OVA-treated WT mice leading to the higher lung levels of Il-4 and Il-5 in the Adam8−/− mice. One explanation for the similar lung levels of Il-13 in OVA-treated WT and Adam8−/− mice is that Il-13 is produced by cells other than CD4+ TH2 including smooth muscle cells (66) and nuocytes which are innate immune cells that produce early bursts of Il-13 in tissues with allergic inflammation (67). It is possible that in our study, Adam8 reduced TH2 T cell counts but increased the activation or numbers of these other Il-13-producing cells during AAI in mice leading to overall similar lung levels of Il-13 in OVA-treated WT and Adam8−/− mice. These possibilities will be the focus of future studies in our laboratory.
Another interesting finding in our paper was that OVA-treated Adam8−/− mice had greater mucus metaplasia in their airways than OVA-treated WT mice despite having similar lung levels of Il-13, an important cytokine driving mucus metaplasia in the airways of allergen-treated mice (68). Although epithelial cell-derived Adam-10 and -17 regulate mucin gene expression in epithelial cells by activating the EGF receptor (43), we found no evidence that epithelial-derived Adam8 reduced mucus production by Il-13-activated airway epithelium (Supplemental Fig. 2). One possible explanation for the increased mucus metaplasia in the airways of OVA-treated Adam8−/− vs. WT mice in the presence of similar lung levels of Il-13 is that OVA-treated Adam8−/− mice have higher airway macrophages and eosinophil counts (Fig. 1C) and these cells and their products (including TNF-α) increase mucin gene expression by airway epithelium (69,70). It is noteworthy that we detected higher lung levels of TNF-α in OVA-treated Adam8−/− mice (Fig. 2F). Studies of the regulation of mucus metaplasia in airway epithelial cells by Adam8 are beyond the scope of this study but will be the focus of our future studies.
Prior studies of mice over-expressing or lacking Adam8 have reported opposite results on the activities of Adam8 in regulating AAI and AHR in mice. Matsuno et al reported that OVA-treated C57BL/6 strain transgenic mice that constitutively over-expressed a soluble Adam8 (sAdam8) lacking the transmembrane domain in the liver had reduced peri-bronchial inflammation, but similar BAL leukocyte counts and BALF levels of Il-4 and Il-5 (27) when compared with OVA-treated non-transgenic mice. While Matsuno et al reported that sAdam8 sheds Vcam-1 from endothelial cells in vitro (27), sAdam8 was only a modest Vcam-1 sheddase in vitro, and sVcam-1 levels were not measured in Adam8 transgenic vs. non-transgenic mice (27). We did not find differences in sVcam-1 levels in serum samples from OVA-treated WT and OVA-treated Adam8−/− mice which suggests that Adam8 is not a significant Vcam-1 sheddase during AAI in mice. It is noteworthy that Matsuno et al did not evaluate airway leukocyte apoptosis in sAdam8 transgenic mice.
In contrast, two other groups studied C57BL/6 strain WT vs. Adam8−/− mice in OVA-induced AAI and reported that Adam8 promoted AAI and AHR in C57BL/6 mice but by different mechanisms (28,29). Paulissen et al reported that Adam8 increased AAI in mice by increasing lung dendritic cell counts and lung levels of eotaxin and Ccl22 (Mdc), but the underlying mechanisms were not elucidated (29). Naus et al reported that Adam8 was essential for the migration of T-cells, macrophages, eosinophils, and dendritic cells because, unlike WT leukocytes, Adam8−/− leukocytes failed to migrate in response to agonists in vitro (28). Unlike Paulissen et al, we did not find any differences in lung dendritic cell counts or lung levels of Ccl22 or eotaxin in WT vs. Adam8−/− mice with AAI. Unlike Naus et al, we did not find any defects in the capacity of Adam8−/− eosinophils or mononuclear phagocytes to migrate in vitro using both Boyden chamber (Fig. 6) and Matrigel invasion assays (data not shown). Naus et al and our study both tested eotaxin as a chemoattractant for eosinophils. However, we tested fMLP and MIP-1α as monocyte chemoattractants, whereas Naus et al used stromal cell-derived factor-1 to stimulate dendritic cell migration and LPS and TNF-α to stimulate macrophage migration in vitro. It is possible that differences in the stimuli used for mononuclear phagocyte migration assays may explain the differences in the results obtained. It is interesting to note that while Naus et al reported that T-lymphocytes from mice with AAI expressed Adam8 and T-cell-derived Adam8 was essential for Adam8’s pro-inflammatory actions in murine airways, another group found that T lymphocytes do not express ADAM8 (44). Overall, our results are more consistent with those of Matsuno et al on sAdam8 transgenic mice with OVA-induced AAI.
Possible explanations for the conflicting results of our study versus those of Naus and coworkers and Paulissen et al on WT vs. Adam8−/− mice with AAI include the strain of mouse studied, and the sensitization dose and route used to deliver the allergen to mice. Most of our studies were conducted on Adam8−/− mice in the BALB/c strain whereas Naus and Paulissen studied Adam8−/− C57BL/6 strain mice. These strain differences [reviewed elegantly in (65)] have produced divergent results in other studies of mice with AAI (70–72). However, it is unlikely that strain differences explain the divergent results for Adam8−/− mice with AAI because we found similar anti-inflammatory activities for Adam8 when we studied Adam8−/− and WT mice in a mixed SvEv129 X C57BL/6 strain and the BALB/c strain. The sensitization dose of OVA used by Naus et al was significantly higher than the dose that we used in most of our studies [2 mg/ml vs. 50 μg/ml (or 10 μg/mouse), respectively]. Sensitization of C57BL/6 strain mice with high rather than low doses of OVA leads to a TH1-type airway immune response (73) and a reduction in allergic phenotype due to the recruitment of anti-inflammatory effector CD8+ T cells into the airways (74). However, it is unlikely that the allergen sensitization dose explains the divergent results obtained for Adam8−/− mice with AAI in our study vs. the studies of Naus and Paulissen and coworkers because: 1) Paulissen et al used the same low dose of OVA in C57BL/6 mice that we used in BALB/c and mixed SvEv129 X C57BL/6 strain mice; and 2) when we sensitized Adam8−/− vs. WT mice with a low vs. high sensitization dose of OVA, Adam8−/− mice had greater AAI than WT mice using both protocols (Supplemental Fig. 1). Both Naus et al and Paulissen et al used lower OVA concentrations for challenging mice (than we used (0.2%, 1%, and 6%, respectively). Thus, it is possible that Adam8 has different activities in regulating mild versus severe AAI in mice. Future studies will assess the effects of varying concentrations of aerosolized OVA on the airway responses of OVA-sensitized WT vs. Adam8−/− mice.
Our studies of ADAM8 expression in lung sections from asthmatic patients showed that ADAM8 was robustly expressed in airway epithelium and sub-epithelial granulocytes which two pathologists (LS and LK) identified as being mainly eosinophils. These results are consistent with those from a previous study of ADAM8 expression in bronchial biopsies from patients with asthma (26). In contrast to previous reports, we found that ADAM8 was expressed by lung epithelial cells of human subjects without pathological features of asthma including epithelial cells in the small airways and type II pneumocytes (Fig. 9A), and that ADAM8 expression in airway epithelial cells was similar in subjects with and without asthma. Overall, ADAM8 expression was increased in the airways of asthmatic patients due to robust expression of ADAM8 in granulocytes (but staining for ADAM8 in macrophages was weak). Double immuno-fluorescence using markers of eosinophils vs. macrophages confirmed robust expression of ADAM8 in airway eosinophils but relatively low-level expression of ADAM8 in airway macrophages. ADAM8 was first cloned from macrophages and is expressed at high levels in activated macrophages (49–51) including macrophages present in other inflamed tissues (51,75,76). We detected robust levels of Adam8 protein in airway macrophages which were similar to levels detected in airway eosinophils from WT mice with AAI (Table 1). Surface ADAM8 levels also increased on both monocytes and eosinophils from healthy human subjects that were activated in vitro with a pro-inflammatory mediator (leukotriene B4). Thus, it was surprising that we detected only low-level staining for ADAM8 in peri-bronchial macrophages in lung sections from all five asthmatic subjects studied because we expected peri-bronchial macrophages to be activated due to the inflammation present in the asthmatic airways. It is noteworthy that in our OVA model of AAI in mice, Adam8 was robustly expressed by airway macrophages and airway inflammation resolves when we stopped challenging the mice with allergen. However asthma tends to be a chronic disease in human subjects. Given the potent anti-inflammatory activity of Adam8 in mice with AAI, our results suggest that the lack of robust up-regulation of ADAM8 in airway macrophages in human asthmatic subjects could contribute to the severity or persistence of airway inflammation in asthma patients. This possibility will be the focus of our future studies.
The mechanism underlying the lack of robust up-regulation of ADAM8 in peri-bronchial macrophages in asthmatic subjects is not clear, but could be due to reduced synthesis of ADAM8 or increased removal of ADAM8 from the surface of airway macrophages in patients with asthma. In this respect it is noteworthy that single nucleotide polymorphisms (SNPs) in the ADAM8 locus are associated with increased plasma levels of a soluble form of ADAM8 and increased risk of myocardial infarction (77). However, SNPs in the ADAM8 locus were not found to be significantly associated with asthma or atopy in one cohort of subjects (78). Additional longitudinal studies are warranted in larger cohorts of patients with asthma to determine whether SNPs in the ADAM8 gene are associated with asthma risk or severity. However, if sADAM8 is found to have anti-inflammatory activities in human subjects as well as mice, reduced plasma or lung levels of sADAM8 in patients with severe asthma could contribute to their more severe airway inflammation.
In summary, we report that leukocyte-derived Adam8 has potent anti-inflammatory activity during in AAI in mice, and this is due to leukocyte-derived Adam8 increasing eosinophil and macrophage apoptosis in the airways of mice with AAI. We now show that an ADAM family member limits inflammatory responses in vivo by activating the intrinsic apoptosis pathway. Furthermore, human asthma patients appear to have defective up-regulation of ADAM8 in their airway macrophages which could contribute to the persistent inflammation in their airways by increasing the survival of these cells. Together, our results suggest that strategies that increase ADAM8 levels on leukocyte cell surfaces in human patients with asthma (or sADAM8 levels in extracellular fluids) may represent novel therapeutic approaches to limit the severity of airway inflammation, and thereby reduce the morbidity and mortality associated with asthma.
Supplementary Material
Acknowledgments
We thank Carl Blobel, MD, PhD (Hospital for Special Surgery, New York, NY) and Andrew Docherty (UBC Cell Tech, Slough, UK) for providing us with Adam8−/− mice.
Footnotes
Abbreviations used: AAI, allergic airway inflammation; ADAM, a proteinase with a disintegrin and a metalloproteinase domain; AFC, 7-amino-4-trifluoro-methylcoumarin; AHR, airway hyper-responsiveness; BAL, bronchoalveolar lavage; BM, bone marrow; CCCP, carbonylcyanide m-chlorophenylhydrazone; DAB, 3,3′-diaminobenzidine; ECM, extracellular matrix; eos, eosinophils; FEV1, forced expiratory volume in one second; FAS-L, FAS-ligand; granulo, granulocyte; HDM, house dust mite protein extract; IL; interleukin; ICS, inhaled corticosteroids; i.n, intranasal; JC-1, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide; LMVECs, lung microvascular endothelial cells; lymphs, lymphocytes; Mmp, matrix metalloproteinase; MP, metalloproteinase; OVA, ovalbumin; PMN, polymorphonuclear neutrophil; RN, central airway resistance; SNP, single nucleotide polymorphisms; sADAM8, soluble ADAM8; WT, wild type.
References
- 1.Braman SS. The global burden of asthma. Chest. 2006;130:4S–12S. doi: 10.1378/chest.130.1_suppl.4S. [DOI] [PubMed] [Google Scholar]
- 2.Corry DB, Rishi K, Kanellis J, Kiss A, Song LZ, Xu J, Feng L, Werb Z, Kheradmand F. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2-deficiency. Nat Immunol. 2002;3:347–353. doi: 10.1038/ni773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McMillan SJ, Kearley J, Campbell JD, Zhu XW, Larbi KY, Shipley JM, Senior RM, Nourshargh S, Lloyd CM. Matrix metalloproteinase-9 deficiency results in enhanced allergen-induced airway inflammation. J Immunol. 2004;172:2586–2594. doi: 10.4049/jimmunol.172.4.2586. [DOI] [PubMed] [Google Scholar]
- 4.Gueders MM, Balbin M, Rocks N, Foidart JM, Gosset P, Louis R, Shapiro S, Lopez-Otin C, Noel A, Cataldo DD. Matrix metalloproteinase-8 deficiency promotes granulocytic allergen-induced airway inflammation. J Immunol. 2005;175:2589–2597. doi: 10.4049/jimmunol.175.4.2589. [DOI] [PubMed] [Google Scholar]
- 5.Goswami S, Angkasekwinai P, Shan M, Greenlee KJ, Barranco WT, Polikepahad S, Seryshev A, Song LZ, Redding D, Singh B, Sur S, Woodruff P, Dong C, Corry DB, Kheradmand F. Divergent functions for airway epithelial matrix metalloproteinase 7 and retinoic acid in experimental asthma. Nat Immunol. 2009;10:496–503. doi: 10.1038/ni.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoshino M, Nakamura Y, Sim J, Shimojo J, Isogai S. Bronchial subepithelial fibrosis and expression of matrix metalloproteinase-9 in asthmatic airway inflammation. J Allergy Clin Immunol. 1998;102:783–788. doi: 10.1016/s0091-6749(98)70018-1. [DOI] [PubMed] [Google Scholar]
- 7.Xu J, Benyon RC, Leir SH, Zhang S, Holgate ST, Lackie PM. Matrix metalloproteinase-2 from bronchial epithelial cells induces the proliferation of subepithelial fibroblasts. Clin Exp Allergy. 2002;32:881–888. doi: 10.1046/j.1365-2745.2002.01386.x. [DOI] [PubMed] [Google Scholar]
- 8.Corbel M, Caulet-Maugendre S, Germain N, Lagente V, Boichot E. Enhancement of gelatinase activity during development of subepithelial fibrosis in a murine model of asthma. Clin Exp Allergy. 2003;33:696–704. doi: 10.1046/j.1365-2222.2003.01581.x. [DOI] [PubMed] [Google Scholar]
- 9.Johnson S, Knox A. Autocrine production of matrix metalloproteinase-2 is required for human airway smooth muscle proliferation. Am J Physiol. 1999;277:L1109–L1117. doi: 10.1152/ajplung.1999.277.6.L1109. [DOI] [PubMed] [Google Scholar]
- 10.Corry DB, Kiss A, Song LZ, Song L, Xu J, Lee SH, Werb Z, Kheradmand F. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. FASEB J. 2004;18:995–997. doi: 10.1096/fj.03-1412fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Eerdewegh P, Little RD, Dupuis J, Del Mastro RG, Falls K, Simon J, Torrey D, Pandit S, McKenny J, Braunschweiger K, Walsh A, Liu Z, Hayward B, Folz C, Manning SP, Bawa A, Saracino L, Thackston M, Benchekroun Y, Capparell N, Wang M, Adair R, Feng Y, Dubois J, FitzGerald MG, Huang H, Gibson R, Allen KM, Pedan A, Danzig MR, Umland SP, Egan RW, Cuss FM, Rorke S, Clough JB, Holloway JW, Holgate ST, Keith TP. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature. 2002;418:426–430. doi: 10.1038/nature00878. [DOI] [PubMed] [Google Scholar]
- 12.Chen C, Huang X, Sheppard D. ADAM33 is not essential for growth and development and does not modulate allergic asthma in mice. Mol Cell Biol. 2006;26:6950–6956. doi: 10.1128/MCB.00646-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Puxeddu I, Pang YY, Harvey A, Haitchi HM, Nicholas B, Yoshisue H, Ribatti D, Clough G, Powell RM, Murphy G, Hanley NA, Wilson DI, Howarth PH, Holgate ST, Davies DE. The soluble form of a disintegrin and metalloprotease 33 promotes angiogenesis: implications for airway remodeling in asthma. J Allergy Clin Immunol. 2008;121:1400–6. 1406. doi: 10.1016/j.jaci.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 2003;17:7–30. doi: 10.1101/gad.1039703. [DOI] [PubMed] [Google Scholar]
- 15.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 16.Herren B, Garton KJ, Coats S, Bowen-Pope DF, Ross R, Raines EW. ADAM15 overexpression in NIH3T3 cells enhances cell-cell interactions. Exp Cell Res. 2001;271:152–160. doi: 10.1006/excr.2001.5353. [DOI] [PubMed] [Google Scholar]
- 17.Tousseyn T, Jorissen E, Reiss K, Hartmann D. (Make) stick and cut loose--disintegrin metalloproteases in development and disease. Birth Defects Res C Embryo Today. 2006;78:24–46. doi: 10.1002/bdrc.20066. [DOI] [PubMed] [Google Scholar]
- 18.Roodman GD. Regulation of osteoclast differentiation. Ann N Y Acad Sci. 2006;1068:100–109. doi: 10.1196/annals.1346.013. [DOI] [PubMed] [Google Scholar]
- 19.King NE, Zimmermann N, Pope SM, Fulkerson PC, Nikolaidis NM, Mishra A, Witte DP, Rothenberg ME. Expression and regulation of a disintegrin and metalloproteinase (ADAM) 8 in experimental asthma. Am J Respir Cell Mol Biol. 2004;31:257–265. doi: 10.1165/rcmb.2004-0026OC. [DOI] [PubMed] [Google Scholar]
- 20.Kelly K, Hutchinson G, Nebenius-Oosthuizen D, Smith AJ, Bartsch JW, Horiuchi K, Rittger A, Manova K, Docherty AJ, Blobel CP. Metalloprotease-disintegrin ADAM8: expression analysis and targeted deletion in mice. Dev Dyn. 2005;232:221–231. doi: 10.1002/dvdy.20221. [DOI] [PubMed] [Google Scholar]
- 21.Dijkstra A, Postma DS, Noordhoek JA, Lodewijk ME, Kauffman HF, ten Hacken NH, Timens W. Expression of ADAMs (“a disintegrin and metalloprotease”) in the human lung. Virchows Arch. 2009 doi: 10.1007/s00428-009-0748-4. [DOI] [PubMed] [Google Scholar]
- 22.Amour A, Knight CG, English WR, Webster A, Slocombe PM, Knauper V, Docherty AJ, Becherer JD, Blobel CP, Murphy G. The enzymatic activity of ADAM8 and ADAM9 is not regulated by TIMPs. FEBS Lett. 2002;524:154–158. doi: 10.1016/s0014-5793(02)03047-8. [DOI] [PubMed] [Google Scholar]
- 23.Naus S, Reipschlager S, Wildeboer D, Lichtenthaler SF, Mitterreiter S, Guan Z, Moss ML, Bartsch JW. Identification of candidate substrates for ectodomain shedding by the metalloprotease-disintegrin ADAM8. Biol Chem. 2006;387:337–346. doi: 10.1515/BC.2006.045. [DOI] [PubMed] [Google Scholar]
- 24.Gomez-Gaviro M, Dominguez-Luis M, Canchado J, Calafat J, Janssen H, Lara-Pezzi E, Fourie A, Tugores A, Valenzuela-Fernandez A, Mollinedo F, Sanchez-Madrid F, Diaz-Gonzalez F. Expression and regulation of the metalloproteinase ADAM-8 during human neutrophil pathophysiological activation and its catalytic activity on L-selectin shedding. J Immunol. 2007;178:8053–8063. doi: 10.4049/jimmunol.178.12.8053. [DOI] [PubMed] [Google Scholar]
- 25.Paulissen G, Rocks N, Quesada-Calvo F, Gosset P, Foidart JM, Noel A, Louis R, Cataldo DD. Expression of ADAMs and their inhibitors in sputum from patients with asthma. Mol Med. 2006;12:171–179. doi: 10.2119/2006-00028.Paulissen. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foley SC, Mogas AK, Olivenstein R, Fiset PO, Chakir J, Bourbeau J, Ernst P, Lemiere C, Martin JG, Hamid Q. Increased expression of ADAM33 and ADAM8 with disease progression in asthma. J Allergy Clin Immunol. 2007;119:863–871. doi: 10.1016/j.jaci.2006.12.665. [DOI] [PubMed] [Google Scholar]
- 27.Matsuno O, Miyazaki E, Nureki S, Ueno T, Kumamoto T, Higuchi Y. Role of ADAM8 in experimental asthma. Immunol Lett. 2006;102:67–73. doi: 10.1016/j.imlet.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 28.Naus S, Blanchet MR, Gossens K, Zaph C, Bartsch JW, McNagny KM, Ziltener HJ. The metalloprotease-disintegrin ADAM8 is essential for the development of experimental asthma. Am J Respir Crit Care Med. 2010;181:1318–1328. doi: 10.1164/rccm.200909-1396OC. [DOI] [PubMed] [Google Scholar]
- 29.Paulissen G, Rocks N, Gueders MM, Bedoret D, Crahay C, Quesada-Calvo F, Hacha J, Bekaert S, Desmet C, Foidart JM, Bureau F, Noel A, Cataldo DD. ADAM-8, a metalloproteinase, drives acute allergen-induced airway inflammation. Eur J Immunol. 2011;41:380–391. doi: 10.1002/eji.200940286. [DOI] [PubMed] [Google Scholar]
- 30.Levy BD, De Sanctis GT, Devchand PR, Kim E, Ackerman K, Schmidt BA, Szczeklik W, Drazen JM, Serhan CN. Multi-pronged inhibition of airway hyper-responsiveness and inflammation by lipoxin A(4) Nat Med. 2002;8:1018–1023. doi: 10.1038/nm748. [DOI] [PubMed] [Google Scholar]
- 31.Quintero PA, Knolle MD, Cala LF, Zhuang Y, Owen CA. Matrix metalloproteinase-8 inactivates macrophage inflammatory protein-1 alpha to reduce acute lung inflammation and injury in mice. J Immunol. 2010;184:1575–1588. doi: 10.4049/jimmunol.0900290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang X, I, Lewkowich P, Kohl G, Clark JR, Wills-Karp M, Kohl J. A protective role for C5a in the development of allergic asthma associated with altered levels of B7-H1 and B7-DC on plasmacytoid dendritic cells. J Immunol. 2009;182:5123–5130. doi: 10.4049/jimmunol.0804276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vermaelen K, Pauwels R. Accurate and simple discrimination of mouse pulmonary dendritic cell and macrophage populations by flow cytometry: methodology and new insights. Cytometry A. 2004;61:170–177. doi: 10.1002/cyto.a.20064. [DOI] [PubMed] [Google Scholar]
- 34.Dyer KD, Moser JM, Czapiga M, Siegel SJ, Percopo CM, Rosenberg HF. Functionally competent eosinophils differentiated ex vivo in high purity from normal mouse bone marrow. J Immunol. 2008;181:4004–4009. doi: 10.4049/jimmunol.181.6.4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kisseleva T, Song L, Vorontchikhina M, Feirt N, Kitajewski J, Schindler C. NF-kappaB regulation of endothelial cell function during LPS-induced toxemia and cancer. J Clin Invest. 2006;116:2955–2963. doi: 10.1172/JCI27392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Owen CA, Campbell MA, Boukedes SS, Stockley RA, Campbell EJ. A discrete subpopulation of human monocytes expresses a neutrophil-like pro-inflammatory (P) phenotype. Am J Physiol (Lung Cell Mol Physiol 11) 1994;267:L775–L785. doi: 10.1152/ajplung.1994.267.6.L775. [DOI] [PubMed] [Google Scholar]
- 37.Owen CA, Campbell EJ, Hill SL, Stockley RA. Increased adherence of monocytes to fibronectin in bronchiectasis: Regulatory effects of bacterial lipopolysaccharide and role of CD11/CD18 integrins. Am Rev Respir Dis. 1992;145:626–631. doi: 10.1164/ajrccm/145.3.626. [DOI] [PubMed] [Google Scholar]
- 38.Owen CA, Campbell MA, Boukedes SS, Campbell EJ. Monocytes recruited to sites of inflammation express a distinctive pro-inflammatory (P) phenotype. Am J Physiol (Lung Cell Mol Physiol 11) 1994;267:L786–L796. doi: 10.1152/ajplung.1994.267.6.L786. [DOI] [PubMed] [Google Scholar]
- 39.Owen CA, Hu Z, Lopez-Otin C, Shapiro SD. Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinase. J Immunol. 2004;172:7791–7803. doi: 10.4049/jimmunol.172.12.7791. [DOI] [PubMed] [Google Scholar]
- 40.Enari M, Hug H, Nagata S. Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature. 1995;375:78–81. doi: 10.1038/375078a0. [DOI] [PubMed] [Google Scholar]
- 41.El KD, Jozsef L, Pan W, Wang L, Petasis NA, Serhan CN, Filep JG. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am J Respir Crit Care Med. 2009;180:311–319. doi: 10.1164/rccm.200810-1601OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haworth O, Cernadas M, Yang R, Serhan CN, Levy BD. Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat Immunol. 2008;9:873–879. doi: 10.1038/ni.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Basbaum C, Li D, Gensch E, Gallup M, Lemjabbar H. Mechanisms by which gram-positive bacteria and tobacco smoke stimulate mucin induction through the epidermal growth factor receptor (EGFR) Novartis Found Symp. 2002;248:171–176. [PubMed] [Google Scholar]
- 44.Richens J, Fairclough L, Ghaemmaghami AM, Mahdavi J, Shakib F, Sewell HF. The detection of ADAM8 protein on cells of the human immune system and the demonstration of its expression on peripheral blood B cells, dendritic cells and monocyte subsets. Immunobiology. 2007;212:29–38. doi: 10.1016/j.imbio.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 45.Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y, Blobel CP. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol. 2007;179:2686–2689. doi: 10.4049/jimmunol.179.5.2686. [DOI] [PubMed] [Google Scholar]
- 46.Vandivier RW, Richens TR, Horstmann SA, deCathelineau AM, Ghosh M, Reynolds SD, Xiao YQ, Riches DW, Plumb J, Vachon E, Downey GP, Henson PM. Dysfunctional cystic fibrosis transmembrane conductance regulator inhibits phagocytosis of apoptotic cells with proinflammatory consequences. Am J Physiol Lung Cell Mol Physiol. 2009;297:L677–L686. doi: 10.1152/ajplung.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang CD, Wang CH, Liu CY, Lin SM, Chou CL, Liu WT, Lin HC, Kuo HP. Eosinophils from asthmatics release IL-5 in an autocrine fashion to prevent apoptosis through upregulation of Bcl-2 expression. J Asthma. 2005;42:395–403. doi: 10.1081/JAS-63001. [DOI] [PubMed] [Google Scholar]
- 48.Comalada M, Xaus J, Sanchez E, Valledor AF, Celada A. Macrophage colony-stimulating factor-, granulocyte-macrophage colony-stimulating factor-, or IL-3-dependent survival of macrophages, but not proliferation, requires the expression of p21(Waf1) through the phosphatidylinositol 3-kinase/Akt pathway. Eur J Immunol. 2004;34:2257–2267. doi: 10.1002/eji.200425110. [DOI] [PubMed] [Google Scholar]
- 49.Yoshida S, Setoguchi M, Higuchi Y, Akizuki S, Yamamoto S. Molecular cloning of cDNA encoding MS2 antigen, a novel cell surface antigen strongly expressed in murine monocytic lineage. Int Immunol. 1990;2:585–591. doi: 10.1093/intimm/2.6.585. [DOI] [PubMed] [Google Scholar]
- 50.Yoshiyama K, Higuchi Y, Kataoka M, Matsuura K, Yamamoto S. CD156 (human ADAM8): expression, primary amino acid sequence, and gene location. Genomics. 1997;41:56–62. doi: 10.1006/geno.1997.4607. [DOI] [PubMed] [Google Scholar]
- 51.Dehmel T, Janke A, Hartung HP, Goebel HH, Wiendl H, Kieseier BC. The cell-specific expression of metalloproteinase-disintegrins (ADAMs) in inflammatory myopathies. Neurobiol Dis. 2007;25:665–674. doi: 10.1016/j.nbd.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 52.Fadok VA, Bratton DL, Henson PM. Phagocyte receptors for apoptotic cells: recognition, uptake, and consequences. J Clin Invest. 2001;108:957–962. doi: 10.1172/JCI14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Druilhe A, Wallaert B, Tsicopoulos Lapa A, Silva E, Tillie-Leblond JRI, Tonnel AB, Pretolani M. Apoptosis, proliferation, and expression of Bcl-2, Fas, and Fas ligand in bronchial biopsies from asthmatics. Am J Respir Cell Mol Biol. 1998;19:747–757. doi: 10.1165/ajrcmb.19.5.3166. [DOI] [PubMed] [Google Scholar]
- 54.Vignola AM, Chanez P, Chiappara G, Siena L, Merendino A, Reina C, Gagliardo R, Profita M, Bousquet J, Bonsignore G. Evaluation of apoptosis of eosinophils, macrophages, and T lymphocytes in mucosal biopsy specimens of patients with asthma and chronic bronchitis. J Allergy Clin Immunol. 1999;103:563–573. doi: 10.1016/s0091-6749(99)70225-3. [DOI] [PubMed] [Google Scholar]
- 55.Lamb JP, James A, Carroll N, Siena L, Elliot J, Vignola AM. Reduced apoptosis of memory T-cells in the inner airway wall of mild and severe asthma. Eur Respir J. 2005;26:265–270. doi: 10.1183/09031936.05.00144304. [DOI] [PubMed] [Google Scholar]
- 56.Kankaanranta H, Lindsay MA, Giembycz MA, Zhang X, Moilanen E, Barnes PJ. Delayed eosinophil apoptosis in asthma. J Allergy Clin Immunol. 2000;106:77–83. doi: 10.1067/mai.2000.107038. [DOI] [PubMed] [Google Scholar]
- 57.Lee E, Robertson T, Smith J, Kilfeather S. Leukotriene receptor antagonists and synthesis inhibitors reverse survival in eosinophils of asthmatic individuals. Am J Respir Crit Care Med. 2000;161:1881–1886. doi: 10.1164/ajrccm.161.6.9907054. [DOI] [PubMed] [Google Scholar]
- 58.Maa SH, Wang CH, Liu CY, Lin HC, Huang KH, Kuo HP. Endogenous nitric oxide downregulates the Bcl-2 expression of eosinophils through mitogen-activated protein kinase in bronchial asthma. J Allergy Clin Immunol. 2003;112:761–767. doi: 10.1016/s0091-6749(03)02009-8. [DOI] [PubMed] [Google Scholar]
- 59.Kankaanranta H, Lindsay MA, Giembycz MA, Zhang X, Moilanen E, Barnes PJ. Delayed eosinophil apoptosis in asthma. J Allergy Clin Immunol. 2000;106:77–83. doi: 10.1067/mai.2000.107038. [DOI] [PubMed] [Google Scholar]
- 60.Duncan CJ, Lawrie A, Blaylock MG, Douglas JG, Walsh GM. Reduced eosinophil apoptosis in induced sputum correlates with asthma severity. Eur Respir J. 2003;22:484–490. doi: 10.1183/09031936.03.00109803a. [DOI] [PubMed] [Google Scholar]
- 61.Jang AS, I, Choi S, Lee S, Seo JP, Yang SW, Park CS. Bcl-2 expression in sputum eosinophils in patients with acute asthma. Thorax. 2000;55:370–374. doi: 10.1136/thorax.55.5.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bureau F, Seumois G, Jaspar F, Vanderplasschen A, Detry B, Pastoret PP, Louis R, Lekeux P. CD40 engagement enhances eosinophil survival through induction of cellular inhibitor of apoptosis protein 2 expression: Possible involvement in allergic inflammation. J Allergy Clin Immunol. 2002;110:443–449. doi: 10.1067/mai.2002.126781. [DOI] [PubMed] [Google Scholar]
- 63.Woolley KL, Gibson PG, Carty K, Wilson AJ, Twaddell SH, Woolley MJ. Eosinophil apoptosis and the resolution of airway inflammation in asthma. Am J Respir Crit Care Med. 1996;154:237–243. doi: 10.1164/ajrccm.154.1.8680686. [DOI] [PubMed] [Google Scholar]
- 64.Walsh GM. Eosinophil apoptosis: mechanisms and clinical relevance in asthmatic and allergic inflammation. Br J Haematol. 2000;111:61–67. [PubMed] [Google Scholar]
- 65.Byers DE, Holtzman MJ. Alternatively activated macrophages and airway disease. Chest. 2011;140:768–774. doi: 10.1378/chest.10-2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bansal G, Wong CM, Liu L, Suzuki YJ. Oxidant signaling for interleukin-13 gene expression in lung smooth muscle cells. Free Radic Biol Med. 2012;52:1552–1559. doi: 10.1016/j.freeradbiomed.2012.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, McKenzie AN. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol. 2012;129:191–198. doi: 10.1016/j.jaci.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 68.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 69.Silva MA, Bercik P. Macrophages are related to goblet cell hyperplasia and induce MUC5B but not MUC5AC in human bronchus epithelial cells. Lab Invest. 2012;92:937–948. doi: 10.1038/labinvest.2012.15. [DOI] [PubMed] [Google Scholar]
- 70.Lee JJ, Dimina D, Macias MP, Ochkur SI, McGarry MP, O’Neill KR, Protheroe C, Pero R, Nguyen T, Cormier SA, Lenkiewicz E, Colbert D, Rinaldi L, Ackerman SJ, Irvin CG, Lee NA. Defining a link with asthma in mice congenitally deficient in eosinophils. Science. 2004;305:1773–1776. doi: 10.1126/science.1099472. [DOI] [PubMed] [Google Scholar]
- 71.Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE, Ghiran S, Gerard NP, Yu C, Orkin SH, Gerard C. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305:1776–1779. doi: 10.1126/science.1100283. [DOI] [PubMed] [Google Scholar]
- 72.Wills-Karp M, Karp CL. Biomedicine. Eosinophils in asthma: remodeling a tangled tale. Science. 2004;305:1726–1729. doi: 10.1126/science.1104134. [DOI] [PubMed] [Google Scholar]
- 73.Morokata T, Ishikawa J, Yamada T. Antigen dose defines T helper 1 and T helper 2 responses in the lungs of C57BL/6 and BALB/c mice independently of splenic responses. Immunol Lett. 2000;72:119–126. doi: 10.1016/s0165-2478(00)00188-7. [DOI] [PubMed] [Google Scholar]
- 74.Aguilar-Pimentel JA, Alessandrini F, Huster KM, Jakob T, Schulz H, Behrendt H, Ring J, de Angelis MH, Busch DH, Mempel M, Ollert M. Specific CD8 T cells in IgE-mediated allergy correlate with allergen dose and allergic phenotype. Am J Respir Crit Care Med. 2010;181:7–16. doi: 10.1164/rccm.200902-0190OC. [DOI] [PubMed] [Google Scholar]
- 75.Schlomann U, Rathke-Hartlieb S, Yamamoto S, Jockusch H, Bartsch JW. Tumor necrosis factor alpha induces a metalloprotease-disintegrin, ADAM8 (CD 156): implications for neuronglia interactions during neurodegeneration. J Neurosci. 2000;20:7964–7971. doi: 10.1523/JNEUROSCI.20-21-07964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chiba Y, Onoda S, Hattori Y, Maitani Y, Sakai H, Misawa M. Upregulation of ADAM8 in the airways of mice with allergic bronchial asthma. Lung. 2009;187:179–185. doi: 10.1007/s00408-009-9145-7. [DOI] [PubMed] [Google Scholar]
- 77.Raitoharju E, Seppala I, Levula M, Kuukasjarvi P, Laurikka J, Nikus K, Huovila AP, Oksala N, Klopp N, Illig T, Laaksonen R, Karhunen PJ, Viik J, Lehtinen R, Pelto-Huikko M, Tarkka M, Kahonen M, Lehtimaki T. Common variation in the ADAM8 gene affects serum sADAM8 concentrations and the risk of myocardial infarction in two independent cohorts. Atherosclerosis. 2011;218:127–133. doi: 10.1016/j.atherosclerosis.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 78.Tremblay K, Lemire M, Potvin C, Tremblay A, Hunninghake GM, Raby BA, Hudson TJ, Perez-Iratxeta C, Andrade-Navarro MA, Laprise C. Genes to diseases (G2D) computational method to identify asthma candidate genes. PLoS ONE. 2008;3:e2907. doi: 10.1371/journal.pone.0002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.