

Abstract
Adenoviruses are employed in the study of cellular processes and as expression vectors used in gene therapy. The success and reproducibility of these studies is dependent in part on having accurate and meaningful titers of replication competent and helper-dependent adenovirus stocks, which is problematic due to the use of varied and divergent titration protocols. Physical titration methods, which quantify the total number of viral particles, are used by many, but are poor at estimating activity. Biological titration methods, such as plaque assays, are more biologically relevant, but are time consuming and not applicable to helper-dependent gene therapy vectors. To address this, a protocol was developed called “infectious genome titration” in which viral DNA is isolated from the nuclei of cells ~3 h post-infection, and then quantified by Q-PCR. This approach ensures that only biologically active virions are counted as part of the titer determination. This approach is rapid, robust, sensitive, reproducible, and applicable to all forms of adenovirus. Unlike other Q-PCR-based methods, titers determined by this protocol are well correlated with biological activity.
Keywords: adenovirus, gene therapy vectors, titration, Q-PCR
1. Introduction
1.1 Adenovirus
Since their discovery in 1953, adenoviruses have been used as a model to elucidate fundamental aspects of virology and cellular biology, and as a vector for the delivery of transgenes into cultured cells and directly into the tissues of live animals (Fields et al., 2007). Studies using adenovirus have led to the discovery of intron splicing in mRNA (Berget et al., 1977; Chow et al., 1977) and have aided in our understanding of gene expression and cell cycle control (Berk, 2005). Additionally, adenovirus is used frequently in the field of gene therapy (Ghosh et al., 2006; Brunetti-Pierri and Ng, 2008; Raty et al., 2008; Pesonen et al., 2010). Adenovirus-based gene delivery vectors have many benefits when compared to other viral and non-viral vectors. They can be produced in large quantities, have a relatively large transgene-carrying capacity, and they can be used to transduce a wide variety of cells (Parks and Graham, 1997; Harui et al., 1999; Palmer and Ng, 2003; Rots et al., 2003). Given these advantages, adenovirus-based vectors have been used in ~23 % of gene therapy clinical trials as of 2012 (http://www.abedia.com/wiley/vectors.php, 02/13/2013).
In the first attempts to use adenovirus as a gene delivery vehicle, the viral early genes E1A, E1B, and often E3 were deleted from the 36 kb, double stranded DNA genome of adenovirus type 5 or 2 (Rosenfeld et al., 1992; Rich et al., 1993). The deletions in these so called “first generation” adenovirus vectors (FG-Ad) were designed to make the vector incompetent for replication in targeted cells and to increase their cloning capacity for transgenes. In time, it was observed that basal expression from the remaining viral genes in FG-Ad vectors could be both cytotoxic and immunogenic (Dai et al., 1995; Lieber et al., 1996; Tan et al., 1999). In some instances, these properties were exploited. For example, adenovirus-based oncolytic vectors have been used to target and kill tumor cells (Pesonen et al., 2010). Additionally, FG-Ad vectors have been used specifically for their immunogenicity in the development of vaccines to diseases such as HIV and H1N1 influenza (Prevec et al., 1991; Steitz et al., 2010). However, when adenovirus vectors are used to deliver a therapeutic transgene, the cytotoxicity and immunogenicity observed with FG-Ad can be problematic. To alleviate this, a new generation of adenovirus vectors was developed in which all viral open reading frames were excised. These vectors are known as helper-dependent adenovirus (HD-Ad) vectors because they can only be propagated in the presence of a helper adenovirus that provides necessary viral proteins in trans (Parks et al., 1996).
Outside of gene therapy, adenovirus continues to be a useful tool for elucidating important cellular processes such as transcriptional regulation and oncogenesis (Berk, 2005). As a tumor virus, adenovirus has been studied to understand the steps involved in oncogenic transformation (Endter and Dobner, 2004). In particular, adenoviral proteins have been shown to manipulate p53, Rb, the ubiquitin ligase pathway, and a host of chromatin remodeling proteins in a well orchestrated reprogramming of infected cells (Weitzman and Ornelles, 2005; DeCaprio, 2009; Ferrari et al., 2009; Levine, 2009).
1.2 The Importance of Titer
It is important that experiments performed with adenovirus and adenovirus-based vectors are conducted on stocks that are well characterized and quantified. Having accurate titers for stocks of adenovirus is necessary to allow comparisons between results from different laboratories, and for the comparison of different vectors. Accurate titers can also facilitate the rational design of experiments, and help to ensure the reproducibility of results. The importance of titer is especially critical in gene therapy experiments where the correct dose can mean the difference between therapeutic benefit and pathological consequences (Raper et al., 2003).
In the case of HD-Ad vectors, the problem of having accurate titers is compounded by the fact that these stocks contain some percentage of contaminating helper virus. Despite creative efforts to exclude the helper virus from manufactured stocks of HD-Ad vectors, it has not been possible to eliminate a low level of helper virus contamination (Parks et al., 1996; Ng et al., 2002; Alba et al., 2007). As the helper virus contains the same cytotoxic and immunogenic viral genes as FG-Ad vectors, too much helper virus contamination can adversely affect the therapeutic benefit of the HD-Ad vector. Therefore, it is important that HD-Ad stocks are titered not just for the total number of vector particles, but also to determine the relative proportions of HD-Ad and helper virus.
1.3 Titration Methods
Adenovirus titration methods can be divided into two categories: physical titration methods and biological titration methods. The physical titration methods are ones in which the total number of viral particles is determined, typically by quantifying the DNA content of the stock. These approaches have an advantage in that they are extremely rapid and easy to perform. The most common of these is a protocol in which the viral DNA from a stock that has been highly purified by CsCl buoyant density centrifugation is quantified by its optical absorbance at 260 nm (OD260) (Maizel et al., 1968). A conversion factor is then used that converts the optical absorbance to a titer that is measured in terms of viral particles per milliliter (VP/mL).
In contrast, the biological titration methods, also known as functional titration methods, assign potency to a stock of virus based on its ability to cause a biological effect in transduced cells. The classic example of this is the plaque assay in which patches of lysed cells in a monolayer are counted to determine the titer of the virus in terms of plaque forming units per milliliter (PFU/mL) (Green et al., 1967). A variation on this technique, known as tissue culture infectious dose at 50% (TCID50), is performed in a 96-well tissue culture plate of permissive cells (Grabow et al., 1992). Each row of wells receives a different dose of virus in an eight-log dilution series. After a period of time, each row is scored for the number of infected wells as determined by the observance of cytopathic effect. The dilution of the row in which 50% of the wells are scored as positive for infection is then used to calculate the original titer of the stock. This is reported in terms of infectious untis per milliliter (IU/mL).
Biological potency can also be assigned based on the expression of an easily assayed reporter protein such as β-galactosidase or green fluorescent protein (GFP) (Philipson, 1961; Gueret et al., 2002). For example, the number of GFP expressing cells in a monolayer can be used to determine the titer of a GFP-expressing virus in terms of green transducing units per milliliter (GTU/mL). Similarly, β-galactosidase expressing vectors can be titered in terms of blue forming units per milliliter (BFU/mL). Such protocols are relatively rapid and easy to perform, but they necessitate that the vector being titered express an easily assayed gene product. Unfortunately, easily quantifiable reporter genes such as GFP are often themselves immunogenic (Stripecke et al., 1999). This makes inclusion of a reporter gene for the purposes of titration undesirable in gene therapy vectors (Gil et al., 2010).
Lastly, FG-Ad and wild type stocks of adenovirus may be titered by immuno-staining infected cells for the expression of viral proteins. This approach has been commercialized in kits such as the Adeno-X Rapid Titer kit (Clontech, Mountain View, CA, USA), which uses antibodies against the adenovirus hexon protein to identify infected cells in monolayer of permissive cells.
1.4 Extreme Variation Between Titers by Different Methods
Given the wide array of potential vector titration methods available to researchers, it is possible to titer a single adenovirus stock in terms of VP, PFU, IU, GTU, and more. But how do these different values compare? In short, they do not. This group and others have observed that there are often log-scale differences in the titer of a single stock when it is titered by different methods (Nyberg-Hoffman et al., 1997; Ma et al., 2001; Kreppel et al., 2002). Physical titers, like VP, are typically much higher than biological titers like GTU. PFU titers are typically lower than GTU and TCID50 IU titers (Gueret et al., 2002). Thus, it is not atypical to see a 100-fold difference or more between a PFU titer and a VP titer of the same stock. This makes intuitive sense when one considers that each method is assaying a different property of the adenovirus stock. A VP titer is typically higher than a PFU titer because only a fraction of virions in an infection is successful in initiating the multiple rounds of infection necessary to form a plaque (Mittereder et al., 1996). Additionally, defective virions will contribute their DNA to the optical absorbance used in the OD260 assay, but they will not produce additional infected wells in a TCID50 assay. This problem is exacerbated by the fact that the ratios of PFU to GTU or VP to IU vary from stock to stock (Kreppel et al., 2002; Crettaz et al., 2008). The fact that the ratio of physical titer to biological titer is variable is recognized by the US government. The USFDA requires that the ratio of VP to IU be determined for all adenovirus vectors used in human gene therapy, and that this ratio be less than 30:1 (Nyberg-Hoffman et al., 1997; Murphy, 1998).
The lack of agreement on a single method for titering adenovirus or adenovirus vector stocks between different laboratories is problematic. For example, an adenovirus experiment conducted by one investigator using a multiplicity of infection of five as determined by VP would likely yield different results from another investigator that uses a multiplicity of infection of five as determined by PFU. This problem has an additional caveat in the case of HD-Ad vector stocks. In many published reports, the HD-Ad vector and the helper virus are assayed by different methods (Parks et al., 1996; Umana et al., 2001; Zhou et al., 2002). When the helper virus is assayed in terms of PFU, and the HD-Ad vector is assayed in terms of BFU, there is liable to be a significant underestimation of the helper virus contamination.
1.5 Biological Significance
Given that different titration methods can yield different results, this raises the question of which method is best? Certainly, this depends on what experiment is being conducted. In the case of administering a gene therapy vector into an experimental animal or human patient, the total number of VP is important because too many VPs can be pathogenic (Raper et al., 2003). However, if 90% of those viral particles are functionally inert, the dose in terms of VP will be poorly predictive of the biological effect under study.
In most cases that involve the use of adenovirus, including all experiments in which adenovirus is used as an expression vector, arguably the most relevant approach is to use a biological titration method. Titers derived by biological means, such as by quantifying cells transduced by a GFP-expressing vector, may be the most useful for determining the dose of adenovirus vector required to observe a certain biological effect. Also, biological titration methods are less likely to be skewed by an excess of functionally inert defective virions or unpackaged DNA. Put another way, titration methods based on the activity of the virus have more biological relevance.
1.6 Q-PCR for Adenovirus Titration
In the effort to find a rapid all-purpose titration protocol, perhaps the most promising approaches involve the use of Q-PCR ( Ma et al., 2001; Wang et al., 2005; Puntel et al., 2006; Crettaz et al., 2008). Q-PCR as a technique is rapid, sensitive, easy to perform, and capable of assaying many samples simultaneously (Ginzinger, 2002). One particularly useful advantage of Q-PCR is that amplification is sequence specific. This means that, unlike the OD260 approach, the vector DNA can be quantified uniquely regardless of the presence of other sources of DNA. In the case of HD-Ad vector stocks, this specificity means that the HD-Ad vector and the helper virus can be assayed independently and simultaneously (Puntel et al., 2006).
Frequently, Q-PCR is used to quantify all of the vector DNA in a given stock. In some published reports, the capsids in a given vector stock are disrupted by means of a detergent, and the resulting solution of naked DNA is assayed (Sandig et al., 2000; Wang et al., 2006). Results obtained in this fashion are therefore functionally equivalent to results obtained by OD260. In other words, they lack biological significance. In a more common variation on this approach, unpackaged DNA is eliminated by treatment with DNase I prior to disruption of the viral capsids (Ma et al., 2001; Puntel et al., 2006; Segura et al., 2010). Vector DNA packaged in protein capsids is resistant to DNase I. Theoretically, this means that only packaged vector DNA is quantified in the subsequent Q-PCR. Unfortunately, titers determined in this fashion are still not biologically relevant (Ma et al., 2001; and this manuscript).
1.7 The Infectious Genome Q-PCR Approach
Given the limitations of available titration protocols described above, a hybrid assay, presented here, was developed that combines the best elements of physical titrations methods (i.e. ease and speed) with the best elements of biological titration methods (i.e. the ability to assay biological potency). In this approach, Q-PCR is used to quantify the amount of vector DNA that is purified from the nuclei of an infected monolayer of HeLa cells. By including this infection step, only vector genomes that are part of functional virions capable of delivering packaged DNA to the cell nucleus are assayed. We call this approach infectious genome quantification, and the results are reported in terms of infectious genome units per milliliter (IGU/mL). Similar approaches have been reported earlier (Crettaz et al., 2008), but those protocols are dependent on replication of the virus which can be highly variable between different viruses (Zhou et al., 2002). This is problematic in a titration assay. Here we present a protocol that is universally applicable to all adenovirus and adenovirus vectors regardless of their replication potential. This technique is rapid and easy to perform, and includes internal controls that reduce variation and validate the assay results. Additionally, we demonstrate that this approach produces titers that are highly predictive of biological activity following infection of mouse hepatocytes by tail vein injection.
2. Methods and Materials
2.1 Virus Stocks and Tissue Culture
All tissue culture was performed in either HeLa cell monolayers or 293HEK cell monolayers (ATCC, Manassas, VA, USA) grown in high-glucose Dulbecco’s Modification of Eagle Medium (Invitrogen, Grand Island, NY, USA) supplemented with 10% bovine calf serum. Vector stocks were prepared as described previously (Dorigo et al., 2004; Gallaher et al., 2009), except for the Adenovirus Reference Material (ARM) which was purchased from the ATCC.
For stocks described as “crude lysate”, infected cells were harvested in 10% of the media used to culture cells during virus or vector production, lysed by three successive rounds of freezing, thawing and vortexing, and then clarified by centrifugation for 30 minutes at 3000 g. Stocks described as “CsCl purified” were taken through the additional step of loading onto CsCl buoyant density gradients to separate viral particles from cellular debris and empty capsids (Vellekamp et al., 2001). Finally, these stocks were dialyzed to remove the CsCl as described previously (Gallaher et al., 2009).
2.2. Infectious Genome Protocol (IGU)
2.2.1 HeLa Cell Infection
Approximately 2×106 HeLa cells per well were seeded onto 6-well tissue culture plates one day before infection. Once the cells were 90-100% confluent, wells were infected, in duplicate, with 5 μL of vector stock diluted in 500 μL of serum-free medium. Plates were allowed to incubate from three to six hours. Media was removed, and cells were washed twice with 1 mL of PBS (Invitrogen, Grand Island, NY, USA) per well. Cell lysis was performed by addition of 500 μL of freshly prepared NP-40 lysis buffer containing 0.65% NP-40 substitute (Calbiochem, Billerica, MA, USA), 150 mM NaCl, 10 mM Tris, pH 8.0. Cells were incubated for 5 to 10 minutes at room temperature to allow cell lysis to occur. The lysate was pipetted up and down 10-15 times to aid in lysis, and then transferred to Eppendorf tubes. Nuclei were pelleted by centrifugation for three minutes at 2000 g in a microfuge. Supernatant was decanted, and an additional 1 mL of NP-40 lysis buffer was added. The pellet of nuclei in fresh buffer was vortexed briefly before an additional three-minute centrifugation at 2000 g. Supernatant was decanted, and nuclei were suspended in 200 μL PBS. Samples were either purified immediately as described in section 2.2.2, or were stored at 4°C for up to one day.
2.2.2 DNA Isolation and Purification
Total DNA was isolated and purified by means of the DNeasy Blood & Tissue kit (QIAgen, Valencia, CA, USA) using the protocol for “Purification of Total DNA from Animal blood or Cells”. Briefly, the pelleted nuclei were treated with 20 μL of 15 mg/mL proteinase K and 200 μL of QIAgen buffer AL for 20 minutes at 55°C. 200 μL of ethanol were added, and DNA was immobilized on a DNeasy column by centrifugation. After two wash steps with QIAgen buffers AW1 and AW2, the DNA was eluted with two successive elutions of QIAgen AE buffer (200 μL each) and pooled. Purified DNA was either assayed immediately as described in 2.2.3, or was stored at 4°C for later analysis.
2.2.3 Q-PCR Conditions
All PCR was performed in optical-grade, 96-well plates (Applied Biosystems, Carlsbad, CA, USA). Each 25 μL reaction contained 12.5 μL of TaqMan Universal Master Mix (Applied Biosystems, Carlsbad, CA, USA), 5 μL of purified DNA template, primers (900 nM final concentration), and probe (250 nM final concentration). PCR runs were performed on an ABI 7500 Real Time Thermocycler using SDS software v.1.3 (Applied Biosystems, Carlsbad, CA, USA). Reaction conditions were as follows: 10 min at 94°C followed by 40 cycles of 10 seconds at 94°C and 30 seconds at 60°C.
2.2.4 Standard Curves
Plasmids containing the relevant template DNA were constructed for use in standard curves. PCR amplicons of 61 and 74 bp were generated using the L2 and BGH primer sets (see section 2.4 for sequences) and appropriate template DNA using the conditions described in section 2.2.3. The resulting amplicons were purified using QIAquick PCR cleanup kits (QIAgen, Valencia, CA, USA) and ligated into pPCR-Script TA cloning plasmids (Stratagene, Santa Clara, CA, USA). The resulting plasmids, pPCR-L2 and pPCR-BGH, were used to transform DH5α Escherichia coli (Invitrogen, Grand Island, NY, USA), grown in 300 mL TYE cultures, and purified by Plasmid Maxi Prep kits (QIAgen, Valencia, CA, USA). The concentrations of the resulting plasmid stocks were determined by optical absorbance at 260 nm. This was used to calculate the number of DNA molecules per μL given that each standard plasmid is 3.03 kb in size (1 mg of 1 kb DNA = 9.5×1011 molecules). Plasmids were serially diluted in TE to 1×109 through 1×102 copies per PCR.
2.3 Vector Genome Protocol (VG)
DNA for Q-PCR was prepared as follows. 5 μL of vector stock was treated with 40,000 U of DNase I (Invitrogen, Grand Island, NY, USA) in buffer containing 5.6 mM MgCl2, 500 mM KCl, and 200 mM Tris pH 8.0 for 30 minutes at 37°C. The reaction was stopped by adding EDTA to a final concentration of 50 mM, and then diluting with 200 μL of PBS. Virions were lysed and DNA was purified following the procedure described in section 2.2.2 and used as template DNA as described in section 2.2.3.
2.4 Primer Design
Primers and probes were designed to be as universal as possible in order to make comparisons between stocks of different vectors and viruses highly comparable. The “L2” primer/probe set was designed to hybridize to a region of the wild type adenovirus serotype 5 genome in the L2 gene region. This region was chosen because it is present in most non-HD-Ad adenoviruses; including FG-Ad vectors, helper viruses, and wild type human adenovirus 5. For titering HD-Ad vectors which lack the L2 region, we designed a second set of primers and probes, the “BGH” set, to target the bovine growth hormone poly-adenylation site that is included in all of the HD-Ad vectors used in these studies. All PCR primers and probes were designed using Primer Express Software v.2.0 (Applied Biosystems, Carlsbad, CA, USA). Primers and probes were custom manufactured (Operon, Petaluma, CA, USA). Primers were ordered using the 50 nM scale, desalted. Probes were ordered using the 250 nM scale, HPLC purified. Fluorophores and their appropriate Black Hole Quenchers (BHQ) are indicated below. Primers and probes were reconstituted in TE and their concentration measured by optical absorbance at 260 nm.
BGH Forward Primer: AATGCGATGCAATTTCCTCAT
BGH Reverse Primer: TGCCTTCCTTGACCCTGG
BGH Probe: [HEX]-TTAGGAAAGGACAGTGGGAGTGGCACC-[BHQ1A]
L2 Forward Primer: TTGTGGTTCTTGCAGATATGGC
L2 Reverse Primer: TCGGAATCCCGGCACC
L2 Probe: [Cy5]-CTCACCTGCCGCCTCCGTTTCC-[BHQ2A]
2.5 Data Analysis
Each Q-PCR run was analyzed by SDS software v.1.3 (Applied Biosystems, Carlsbad, CA, USA). Runs in which the R2 value of the linearity of the standard curve fell bellow 0.99 were rejected. The slope of the line comparing the cycle threshold number to the log of the starting titer was used to calculate the efficiency of the PCR according to the formula:
(Ginzinger, 2002). PCR runs where the efficiency was below 90% or above 100% were rejected.
Data that satisfied these criteria were transferred to Excel 2004 (Microsoft, Redmond, WA, USA) for further analysis. For each vector stock, the replicate determinations were averaged and the standard deviation was calculated. Three additional stocks of adenovirus were quantified in parallel in each assay (see sections 4.1.1 and 4.1.2). One of these, identified as the “reference stock”, was titered repeatedly by TCID50 or by GFP transduction assay. Deviation of the observed value for the reference stock in the IGU assay relative to its previously established biological titer was used to adjust the value of all stocks in that assay. The other two stocks, labeled “validation stocks”, were titered repeatedly by the IGU assay. The observed titers for these stocks in any given IGU assay were compared to the previously established titers and standard deviations. If the observed adjusted titer of either validation stock was greater than or less than two standard deviations from its established mean, then the run was rejected.
2.6 Statistical Analysis
Statistical analysis was made using Prism v5.0 software (GraphPad, La Jolla, CA, USA) using either the Student’s t-test or Bonferroni’s multiple comparison test as a post-ANOVA analysis. The variance component analysis was performed using SPSS Statistics 19 software (IBM, Armonk, NY, USA) with a restricted maximum likelihood (REML) fitting of variance components. In all cases P values less than 0.05 were considered significant.
2.6 Other Titration Methods
2.6.1 GFP Transduction Assay
One day prior to infection, 48-well tissue culture plates were seeded with 5×105 HEK293 (ATCC, Manassas, VA, USA) cells per well. The next day, the virus stock to be titered was serially diluted through five 10x dilutions. 100 μL of each dilution was used to infect wells in duplicate for 1 hour at 37°C followed by the addition of 400 μL of media. Plates were incubated for two to three days at 37°C. Observation with a fluorescence microscope was used to identify the wells with 10–30% of cells expressing GFP. These wells were washed with PBS, and gently trypsinized. Cells were pelleted in a microfuge at 2000 g, and resuspended in 100 μL of media before being transferred to a hemocytometer for counting on a fluorescence microscope. Titers were calculated using the relevant dilution factor and were expressed in terms of GTU/mL.
2.6.2 TCID50
TCID50 assays were performed as described in the AdEasy Vector System Application Manual v1.4 (Qbiogene, Solon, OH, USA) except that the number of infected wells was scored on multiple occasions between 3 days and 20 days as indicated.
2.6.3 OD260
Titrations by optical absorbance at 260 nm were performed as described (Mittereder et al., 1996).
2.7 Mouse Work
In vivo experiments were described in detail in Gallaher et al. (2009). Briefly, several different stocks of Renilla luciferase-expressing HD-Ad vectors were injected via the tail vein into adult, female NU-Foxn1nu nude mice (Charles River, Wilmington, MA, USA). One week post infection, 20 μg of Renilla luciferase substrate, coelenterazine (NanoLight Technology, Pinetop, AZ, USA), was injected IV and imaged by bioluminescent imaging (IVIS, Hopkinton, MA, USA). Data was analyzed by Living Image software v.2.20 (Xenogen, Hopkinton, MA, USA) and was used to assay luciferase expression in terms of maximum photons per second in the region of interest.
3. Results
3.1 Titrations by TCID50
The TCID50 protocol is used by many for determining the infectious potency of adenovirus stocks. In fact, a variation on the protocol was chosen by the Adenovirus Reference Material Working Group (ARMWG) to assign an infectious potency to the ARM. However, we observed that the titer determinations by TCID50 were poorly predictive of the biological activity of viral stocks. This was particularly obvious for stocks of adenovirus vectors that express a readily detectible reporter gene such as GFP. In certain stocks, the IU/mL titer by TCID50 significantly under-estimated the biological activity of the vector.
To address this shortcoming of the TCID50 titration method, we examined the various elements of the protocol to identify potential sources of variation. In most published examples of the protocol, including that of the ARMWG, infected wells are scored as positive or negative after 10 days of infection (Qbiogene; Li et al., 2009; Segura et al., 2010). To determine if this duration is sufficient for detecting all infected wells, a time course experiment was conducted. Several different stocks of FG-Ad vectors and the ARM were assayed. Each was scored for the number of positive and negative wells at multiple time points ranging from 3 to 20 days post infection. The same infected plates were evaluated repeatedly.
It was observed that in all but one case, the titer, as determined by the percentage of positive wells, continued to increase after 10 days of infection (Figure 1). In the case of the ARM, the titer increased by a modest 58%. However, the titer of several other stocks increased dramatically. One stock, Ad-FL helper adenovirus (Umana et al., 2001) increased 31 times between day 10 and day 17. In each case, the increase in observed titer reached a plateau by day 20.
Figure 1. Duration of TCID50 Assay Affects Result.
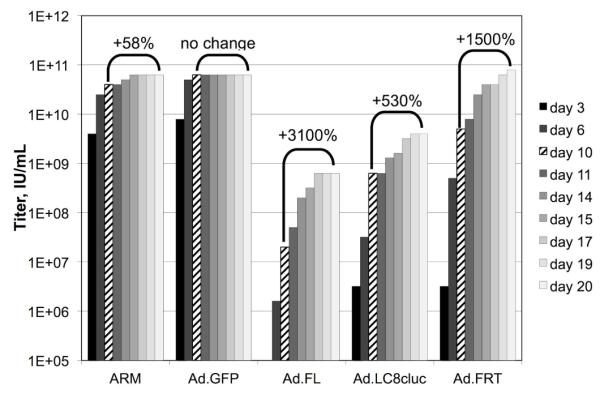
Five different stocks of adenovirus were titered according to a standard TCID50 protocol. At multiple points over 20 days, infected wells were identified by the observance of cytopathic effects and used to calculate the titer of each stock. 10 days (indicated by diagonal hash marks) is the standard duration for most protocols. The brackets indicate the change in observed titer between 10 days and 20 days post-infection.
3.2 Development of the Infectious Genome Titration Protocol
While the TCID50 titration protocol is both repeatable and biologically predictive, its utility is greatly reduced by the fact that it takes between 10 and 20 days to complete, and cannot be used for the titration of HD-Ad vectors. In order to address both of these limitations, this group began to use Q-PCR-based protocols for the titration of adenovirus-based vectors. A protocol was developed in which the vector stock is infected onto HeLa monolayers. Following a three-hour infection, nuclear DNA was isolated, purified, and quantified by Q-PCR (Figure 2). Isolation of nuclei was included to exclude biologically inactive viral particles that would otherwise artificially inflate Q-PCR based titers. Consequently, the infectious genome protocol (IGU/mL) should be more predictive of the biological activity of a given stock than titers assigned by total vector genomes (VG/mL).
Figure 2. Overview of the Infectious Genome Protocol.

Each stock of adenovirus to be titered was used to infect replicate wells of HeLa cell monolayers in 6-well tissue culture plates. 5 μL of vector stock (crude or purified) was diluted into 500 μL of serum-free media and overlaid on the monolayer for three to six hours at 37°C. The inoculant was aspirated, and monolayers were washed twice with PBS. Cell membranes, but not nuclear membranes, were lysed in place by the addition of NP-40 detergent. The nuclei were pelleted, and used to prepare total DNA by means of a commercial kit. The resulting DNA was then used as template in Q-PCR to quantify the number of vector genomes that had translocated to the nucleus during the incubation period. The titer of each stock was then determined relative to a reference standard stock of pre-established titer that was processed in parallel. Additionally, two validation stocks of pre-established titer were processed in parallel in order to identify errors during processing.
Evaluation of this Q-PCR titration protocol began with an examination of the standard curve used in the PCR to quantify the vector DNA. A plasmid bearing the L2 amplicon was diluted in 10-fold steps to give between 1×109 through 1×102 copies per PCR. This dilution series was assayed in triplicate by a standard Q-PCR with the L2 primer/probe set. Next, the log of the initial number of plasmid copies was plotted against the PCR cycle number (Ct) at which the exponential growth curve of each reaction crossed an arbitrary threshold (Supplementary Figure S1A). Linear regression of this analysis showed that there was a high degree of linearity between Ct and the log of the initial copy number over seven logs (R2=0.9997). The slope of this line was then used to calculate the efficiency of the PCR using the equation:
In this instance, the PCR using the L2 primer/probe set was found to be 97% efficient. PCR efficiencies in the range of 95% - 100% were consistently observed with both the L2 and the BGH primer/probe sets (data not shown).
Given the observed seven logs of linearity in the PCR portion of the titration protocol, the linear range of the IG approach overall can be predicted. In the protocol as presented here, each copy detected in the PCR should correspond to 2×104 IGU/mL in the virus stock being titered. Therefore, the linear range of the PCR (between 102 and 109 copies/reaction) should allow the detection of adenovirus stocks whose potencies range from 2×106 to 2×1013 IGU/mL.
In order to test this hypothetical linear range empirically, a stock of HD-Ad vector with a reasonable titer, 3×1010 IGU/mL, was subjected to three serial 10-fold dilutions. Each of these was used to infect duplicate wells of HeLa cells and titered following the standard protocol (Supplemental Figure S1B). When the expected titer was plotted versus the observed titer, there was a high degree of linearity (R2=0.9997). This suggests the protocol as written has a linear range of at least 3×1010 to 3×107 IGU/mL. Slight variations in the protocol should allow this range to be extended up or down as needed.
3.3 Comparison of Q-PCR Titration Methods
In order to evaluate the proposed infectious genome approach, titrations of two FG-Ad stocks, a CsCl purified stock and a crude lysate stock, were performed using either the infectious genome approach described above (IGU protocol) or by Q-PCR of viral DNA following DNase I treatment of the viral stocks (VG protocol). Additionally, each stock was analyzed by a third protocol in which no effort was made to remove unpackaged DNA or defective viral particles (Total Vector DNA). To assay the effectiveness of the VG and IGU protocols in eliminating biologically inactive DNA, an unrelated plasmid was spiked into each viral stock at comparable copy numbers to the viral DNA in the stocks. This plasmid was independently assayed using a different primer/probe set.
While the spiked plasmid was readily detected in the untreated viral stocks, it was decreased to below the limit of detection by both the VG and IGU protocols; a decrease of over four logs (Figure 3). This suggests that either method is highly effective in eliminating naked vector DNA.
Figure 3. Comparison of Q-PCR-based Titration Protocols.
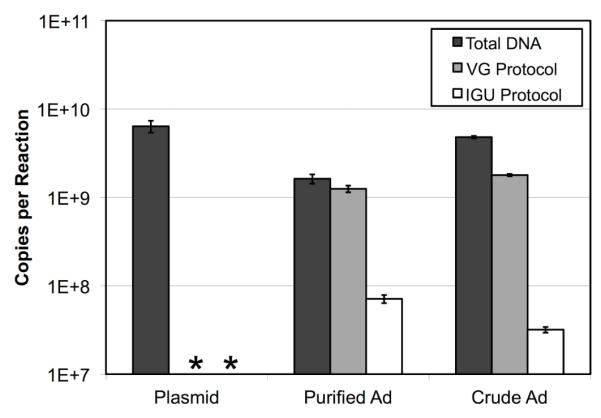
Two stocks of a FG-Ad vector, one crude lysate and one purified by CsCl gradient, were subjected to three different protocols designed to determine the vector’s titer. In the IGU protocol, non-infectious and naked DNA were eliminated by selectively harvesting vector DNA capable of infecting HeLa cell nuclei in culture (Figure 2). In the VG protocol, naked DNA was removed by incubating the stocks for 30 minutes at 37°C with DNase I. No effort was made to remove naked DNA in the Total DNA protocol. In all three cases, the vector DNA was liberated from capsids and purified by means of a commercial DNA cleanup kit, and then quantified by Q-PCR. As a control, an unrelated plasmid with an independently assayable template was spiked into the purified FG-Ad stock and processed in parallel. * indicates the result was below the limit of detection (< 5×104 copies). Bars indicate the mean of four replicates ± standard deviation.
The VG protocol measured similar amounts of vector DNA to the Total Vector DNA protocol without treatment with DNase I. In contrast, there was a dramatic decrease in titers determined by the IGU protocol. Specifically, 77% of the copies of vector DNA in the purified stock were found to be DNase I resistant, while 4.4% of the total copies were infectious as assayed by the IGU protocol. In the case of the crude stock, 37% of the total vector copies were DNase I resistant, and 0.66% were found to be infectious. These results suggest that the VG protocol and IGU protocol are assaying very different properties of a given vector stock. This raised the question of which value is more useful as a predictor of biological activity.
3.4 Comparison of Q-PCR to Other Physical and Biological Titration Methods
A single stock of CsCl purified FG-Ad expressing GFP was titered by a variety of physical and biological titration methods (Figure 4). There were significant differences between the titers determined by each of the methods with the OD260 being the highest at 1.1×1012 VP/mL and TCID50 being the lowest at 6.1×1010 IU/mL. Consistent with the comparison of VG to IGU titers presented in section 3.3, a dramatic difference was observed between the two methods (2.0×1012 VG/mL versus 1.3×1011 IGU/mL).
Figure 4. Comparison of Physical and Biological Titration Methods.
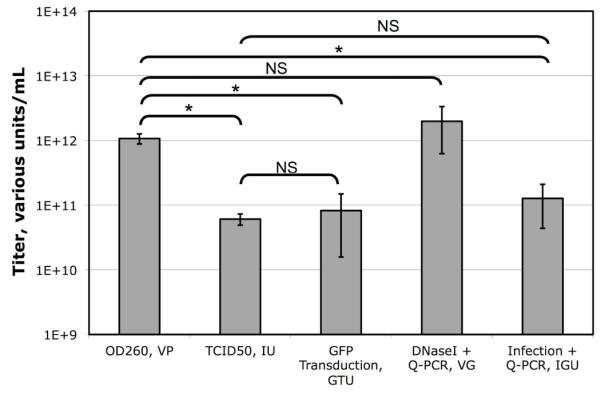
A single stock of purified GFP-expressing FG-Ad vector was titered by five different methods. Each bar indicates the mean of multiple assays ± standard deviation (OD260 N=6, TCID50 N=5, GFP Transduction N=5, VG N=8, IGU N=27). Statistical significance was determined by post-ANOVA Bonferroni’s multiple comparison test. * indicates statistical significance with P < 0.05.
To determine which of these approaches are comparable, a series of Student T-tests was performed. The concentration of vector molecules determined by OD260 of detergent lysed virions was not statistically different from the VG titer determined by Q-PCR of DNase I resistant DNA. However, the concentration of infectious DNA molecules determined by TCID50, GFP transducing activity or the IGU protocol presented here was only 3-6% of the total vector DNA molecules in the stock. Significantly, the titer of infectious genomes/mL determined by the rapid IGU protocol was not statistically different from that determined by the slower GFP-transducing assay, or the much slower TCID50 assay. This analysis suggests that the IGU titration protocol is a good proxy for biological titration methods such as TCID50 and reporter gene expression, but that the VG protocol is not.
3.5 Biological Predictiveness of Titers Assigned by the Infectious Genome Protocol
The results of the preceding section suggested that infectious potencies determined by qPCR of nuclei-transduced vector DNA as in the IGU protocol are comparable to potencies determined by other biological titration methods. This suggests that IGU titers should correlate more highly with the biological activity of a given vector stock than VG titers determined by Q-PCR of DNase I treated vector stocks. To test this directly, we examined five different CsCl purified stocks of luciferase-expressing HD-Ad vectors. Each stock was titered by Q-PCR using both the VG and IGU protocols (Figure 5A). Once again, there were large discrepancies in the titers of each stock when comparing the two methods, ranging from 10-fold up to 3600-fold higher for the VG titer compared to the IGU titer. Next, these five different stocks were injected into the tail veins of mice in a series of in vivo expression experiments. In each case, a different stock and/or a different volume was used. One week post injection, the mice were assayed for luciferase expression by in vivo bioluminescence imaging over the liver. If IGU titers are more biologically relevant than VG titers, then luciferase expression should correlate more highly with the number of IGU injected per mouse than the number of VG injected. To evaluate this hypothesis, both IGU and VG versus maximum luciferase activity were plotted for each mouse (Figure 5B). A correlation coefficient was calculated for both sets of data and used to calculate two-tailed P-values. The P-value for the relationship between VG titers and activity was 0.944. This indicates that the VG titers did not correlate at all with luciferase expression. In contrast, the P-value for IGU titers relative to luciferase was 0.0053, which is highly significant. This demonstrates how IGU titers, but not VG titers, are predictive of biological activity.
Figure 5. Relationship of Q-PCR Titers to Biological Activity.

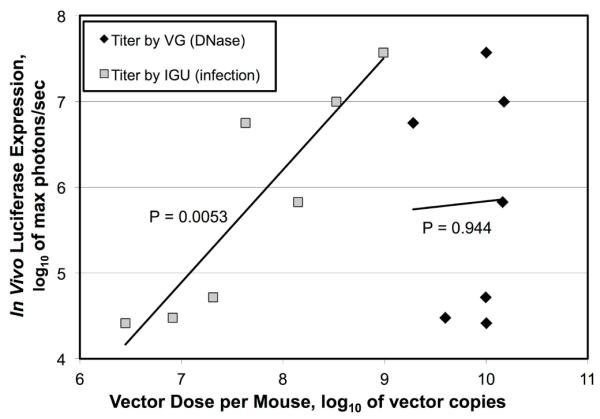
(A) Five different stocks of luciferase expressing HD-Ad vectors were titered by Q-PCR using two different protocols; one including an infection step (IGU/mL) and one that included only a DNase I treatment (VG/mL). Brackets indicate the ratio of VG to IGU. (B) In a series of in vivo experiments, each stock was injected into mice and assayed by bioluminescent imaging. This figure plots the log of the maximum level of luciferase expression against the log of the dose injected as determined by each titration method. Each set of seven data points was used to calculate a curve and to calculate a correlation coefficient. The correlation coefficient was used to calculate two-tailed P-values with five degrees of freedom.
3.6 Ability to Simultaneously and Independently Titer Two Vectors in a Mixed Population
One advantage of Q-PCR technology is its ability to independently quantify two or more amplicons in the same reaction (duplexing or multiplexing, respectively). This trait makes Q-PCR ideal for mixed populations of adenovirus. For example, HD-Ad vectors, which can only be propagated in the presence of a helper adenovirus, always contain some percentage of contaminating helper virus. For purposes of gene therapy, it is important to know the titer of the both.
To determine if the IGU protocol works for titering mixed populations of virus in a duplexing assay, a stock of an HD-Ad with contaminating helper virus was titered. The resulting purified nuclear DNA was assayed in two separate series of Q-PCR. In one, both the BGH primer/probe, set which detects the HD-Ad, and the L2 primer/probe set, which detects helper virus, were used. In the second series of Q-PCR, only the BGH primer/probe set was used (Supplemental Figure S2). There was no statistical difference in the titer determinations between the duplex and the singleplex reactions. Likewise in titrations of the helper virus alone, titering it with just the L2 primer/probe set or with both sets produced the same result. This indicates that amplification by one of these primer/probe set did not influence amplification by the other primer/probe set.
3.7 Effect of Extraneous Non-template DNA
In the IGU protocol, total nuclear DNA is harvested from infected HeLa cells. This means that all of the cellular DNA, as well as cellular RNA, is present in the Q-PCR in excess relative to the vector DNA being titered. To determine if this “non-template” DNA can affect the quantitation of the vector, a series of quantifications on a plasmid with the L2 amplicon were performed. Three 3-fold dilutions of this plasmid were prepared. Each dilution was split into two aliquots, and 500 ng of purified HeLa total cellular DNA, equivalent to 2×107 cells, was then spiked into one of each pair of aliquots. Lastly, each of these was quantitated by Q-PCR and plotted relative to the predicted copy number (Supplemental Figure S3).
At each of the three dilutions (1×107, 3×106, 1×106 copies), there was no statistical difference between the vector copy in the presence or in the absence of 500 ng HeLa DNA. This represents a 10,000-fold excess of HeLa DNA by mass at the highest plasmid copy number. The relative number of copies of HeLa cell DNA and vector were chosen to be far in excess of the ratios that would be encountered in the IGU protocol.
3.8 Analysis of the Precision of the Infectious Genome Protocol and Sources of Variance
An important aspect of any titration protocol is its precision; will a given titration result be repeatable from sample-to-sample and from day-to-day. Three stocks were chosen for this analysis: a FG-Ad, a HD-Ad, and the ARM. Each stock was titered using duplicate or triplicate determinations of each DNA preparation. Each preparation was performed in duplicate or triplicate on any given assay day. Lastly, each of the three stocks was assayed on separate days. Over 600 Q-PCR determinations were included in this analysis. For each stock, the mean and coefficient of variation (CV) are presented in Table 1.
Table 1.
Variance Component Analysis of Replicates
| Stock | Titer | CV | Day to Day | Prep to Prep | Determination to Determination | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| IGU/mL | Variation | N | % | Variation | N | % | Variation | N | % | ||
| FG-Ad | 1.3E+11 | 0.61 | 3.5E+21 | 24 | 52% | 3.0E+21 | 102 | 45% | 2.0E+20 | 264 | 3.0% |
| HD-Ad | 6.3E+09 | 0.33 | 1.1E+18 | 4 | 22% | 3.5E+18 | 18 | 70% | 4.3E+17 | 54 | 8.6% |
| ARM | 3.7E+10 | 0.53 | 2.4E+20 | 44 | 52% | 2.1E+20 | 140 | 46% | 8.9E+18 | 300 | 1.9% |
In order to determine the relative contribution of determination-to-determination variance, preparation-to-preparation variance, and day-to-day variance to the overall variance, the data were subjected to variance component analysis. For each of the three stocks examined, the contribution of determination-to-determination variance was the least significant source of variance. This indicates that the difference between replicate PCRs performed on a single preparation of DNA was relatively small. In contrast, the day-to-day and preparation-to-preparation variances were more significant. Interestingly, the relative percentages of the sources of variance for the two largest datasets (FG-Ad and ARM) were nearly identical: 52% versus 52% for the day-to-day variance and 45% vs 46% for the prep-to-prep variance.
3.9 Analysis of the Adenovirus Reference Material
The ARM is a single stock of wild type adenovirus that has been titered independently by the various labs of the ARMWG. It was designed to be a common frame of reference for different investigators to compare their titration protocols (Palmer and Ng, 2004). Two different titers are provided for the ARM: a physical titer by OD260 and a biological titer by TCID50.
An aliquot of the ARM was obtained and quantified by OD260, TCID50, and by the IGU protocol (Figure 6A). The number of viral particles as determined by OD260 was 4.3 ± 1.6 ×1011 VP/mL (N=4). This is not statistically different from the published titer of 5.8 × 1011 VP/mL. Likewise, the TCID50 determination of 4.2 ± 1.8 ×1010 IU/mL (N=3) was statistically the same as the published titer of 7×1010 normalized adjusted standard IU/mL.
Figure 6. Titrations of the Adenovirus Reference Material (ARM).

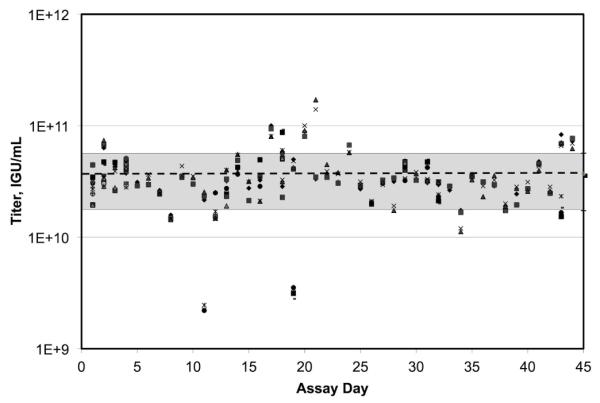
(A) The ARM was titered by OD260 (N=4) and by TCID50 (N=3). The results are presented relative to results published by the ARM working group. Bars equal the mean ± the standard deviation. Comparisons between determinations were made by Student’s t-test. * indicates significance with P < 0.05. (B) The ARM was titered by the IGU protocol using duplicate or triplicate Q-PCR determinations of 140 DNA preparations performed on 44 assay days over the course of a year. Each of 300 data points is plotted. The dotted line indicates the mean. The grey area indicates ± one standard deviation.
Lastly, the ARM was titrated repeatedly over the course of a year by our IGU protocol (Figure 6B). The mean result of 44 separate assay days was 3.7 ± 2.0 ×1010 IGU/mL (mean ± SD). Interestingly, this titer was not statistically different from the observed TCID50 titer of the ARM (P=0.689). Consistent with the observations reported in section 3.4, the IGU titer was in better agreement with the biological TCID50 titer than the physical OD260 titer.
4. Discussion
4.1 Development of the IGU Protocol
4.1.1 Use of a Reference Standard
Q-PCR technology is a powerful tool for comparing the relative quantity of two or more samples. However, unlike TCID50 assays and plaque assays, Q-PCR technology is inherently unable to provide absolute determinations of the quantity of a given sample (Ginzinger, 2002). This limitation is circumvented by comparing all unknown quantities to a standard curve of known quantity. This means that any determination of a vector’s potency by Q-PCR is only as good as the standard curve.
In most published examples that use Q-PCR to quantify adenovirus, the standard curve is generated by serially diluting a plasmid of known copy number, as determined by optical absorbance, which shares the same template region with the vector being quantified. One advantage of this approach is that is possible to generate a curve that is linear over a large range. We have observed a high degree of linearity over seven logs (Supplemental Figure S1A). This large range ensures that vectors can be assayed without foreknowledge of the expected titer.
However, this begs the question of whether or not a plasmid standard provides a good approximation for a vector genome in absolute numbers? In Wang et al. (2006), they determined that plasmid standards do differ from vector standards by a significant degree. We have had similar findings in our own experiments (data not shown). The researchers surmised that this might be due to slight differences in the initial amplification of a supercoiled plasmid relative to a linear adenoviral genome. This affect skews the absolute quantity determinations, but not the relative results because those are based on the comparative amplification during exponential growth phase of the PCR. By this point in the reaction, the polymerase is mostly utilizing newly synthesized amplicon as its template, which is the same across all reactions.
To sidestep this potential source of inaccuracy, we use a “reference standard” in all Q-PCR titrations. This is a stock whose infectious potency has been well established by biological assays (either TCID50 or GFP transduction) and is assayed in parallel with the unknown vector stocks (Figure 2). In the Q-PCR step, the plasmid standard, the reference standards, and any number of unknown vector stocks are all assayed in parallel. By analysis of the results from the plasmid standard curve, it can be determined how much starting material was present for each unknown stock relative to the reference standard. These relative numbers are then used to assign infectious potency to each unknown vector stock relative to the previously determined biological titer of the reference stock.
In our protocol, the reference stock is assayed in parallel with the unknown stocks through all stages of the protocol. An additional benefit of this approach is that any systematic errors that may have occurred during the infection step or the DNA purification step should be cancelled out. This should also mitigate any effects due to differences in the infectivity of the HeLa cells used from one titration to another. Lastly, by adjusting the absolute value of the standard curve relative to the reference stock, the titers of all unknown stocks are effectively tethered to a biological titer. This helps ensure that titers determined by the IGU protocol are predictive of biological activity.
4.1.2 Use of a Validation Standard
No matter how careful a laboratory technician is, variations and mistakes in processing inevitably occur that can skew results. As such, we have found that it is beneficial to include one or two additional stocks with well-established infectious potencies that are assayed in parallel to the reference standard and the unknown vector stocks. These vector stocks, which we term “validation stocks”, are treated in the analysis as unknowns. Once the data analysis is complete, the observed titer is compared to the previously established titer. Significant deviations between the known and observed titer for the validation stocks alert the operator that errors may have occurred during the assay.
4.1.2 Choice of the Number of Replicates
The deviation between an observed titer and the “true” titer of a given stock is due to both random fluctuations and to systematic error (skew). By pinning the titer of an unknown stock to the reference standard assayed in parallel, we hope to minimize the contribution of systematic error. However, random error must also be dealt with. The most obvious approach is to take the mean of several replicate determinations. But how many replicates are necessary to bring the mean assayed result in line with the true results? We performed an extensive variance component analysis to address this. In this analysis, we observed that the variance between multiple Q-PCR determinations of a single preparation of DNA were minimal compared with the variance between multiple preparations of DNA from vector infected HeLa monolayers. From this we conclude that it is preferable to perform replicate parallel infections of a given vector stock than to perform additional Q-PCR determinations from a single infection. Since the day-to-day variation is also significant, we find that assaying a given stock over several days is preferable to preparing all replicates at one time.
4.2 Comparison of VG, IGU and Total Vector DNA
Most previously published examples of using Q-PCR to assign potencies to adenovirus stocks do so either by quantifying all of the vector DNA, or by using the VG approach in which DNase I is used to remove any unpackaged vector DNA (Ma et al., 2001). Not surprisingly, we find that the first method, quantifying total vector DNA, is comparable to OD260 and thus does a poor job of predicting the functional potency of a given stock. More surprisingly, we find that the second method of quantifying only DNase I-resistant vector DNA to be not much better (Figure 5B).
To examine this phenomenon more closely, we quantified the amount of vector DNA by three different Q-PCR-based methods: total DNA, DNase I-resistant DNA (VG), and infectious DNA (IGU) as presented in Figure 3. A control plasmid was included in the analysis to determine if the DNase I and HeLa-infection approaches were capable of removing naked DNA from the analysis. In this analysis both methods were completely effective at removing the naked plasmid DNA.
In the case of the crude lysate, 63% of the total DNA was susceptible to DNase I digestion. This is not surprising since crude lysate is expected to contain a great deal of vector DNA that had been replicated, but not packaged, during the propagation. More surprising was the result in the case of the CsCl-purified stock. The purification process is designed to remove unpackaged DNA. Nevertheless, a significant amount, 23% of the total DNA, was susceptible to DNase I. It is not clear if this fraction represents contaminating naked DNA or severely compromised virions.
Of the fraction of total vector DNA that was found to be DNase I-resistant (i.e. believed to be packaged into virions), only 6% was found to be infectious in the case of the purified stock, and 2% in the case of the crude lysate. This large discrepancy helps to explain why titers derived from the IGU protocol are so different than titers from the VG protocol. The low fraction of virions that successfully deliver their packaged DNA into the nucleus of HeLa cells may be due to a low probability of correctly completing all of the steps of the complex infection process. These include binding of the fiber knob to the CAR receptor, penton base association with integrins and the resulting induction of endocytosis, escape from the endosome, association of partially uncoated particles with microtubules via dynein motor proteins, intracellular transport on microtubules to the microtubule organizing center, association with nuclear pore complexes, and active import of the packaged DNA in complex with virion protein VII through nuclear pore complexes into the nucleus (reviewed in Fields et al. (2007)).
4.3 Comparison to Other Titration Methods
4.3.1 Comparison to Physical Titration Methods
Physical titration methods, such as OD260 and total DNA Q-PCR, are used frequently in adenovirus studies due to their ease and rapidity. An OD260 determination of a virus stock can be completed in a manner of minutes. By assaying the number of copies of viral DNA, these approaches provide a convenient way of quantifying an adenovirus stock. However, we have found that their is variation over two logs between the ability of different CsCl density gradient purified adenovirus vectors to deliver their packaged DNA into nuclei (Figure 5B). This makes simple determination of the total concentration of vector genomes in a CsCl purified stock of little value in predicting the concentration of infectious units. In the design of experiments and in the reporting of results, it is typically more important to know the biological potency of virus stock under investigation rather than the number of viral particles.
This problem would be ameliorated if there were a consistent relationship between infectious potencies and particle number. However, we have observed repeatedly that this is not the case. For example, the ratio of VG to IGU ranged from 10:1 to 3600:1 across the five stocks assayed in Figure 5A. In Figure 3, the ratios were 18:1 and 56:1 for the two stocks evaluated in those experiments. This variation may be due to damage to virion particles during CsCl equilibrium buoyant density centrifugation or during dialysis to remove CsCl. However, once a stock is prepared and stored at −70°C, we observed variations of IGU/mL of only ~2 fold or less when the same vector stock was assayed on separate days (Table 1). Given these results, we feel that it is incumbent on investigators to report the titers of their viral stocks in terms of biological units.
4.3.2 Comparison to Biological Titration Methods
The plaque assay has been a commonly used titration method for adenovirus for decades (Green et al., 1967). Clearly, there are a great many advantages to this protocol and to the closely related TCID50 approach. They are well established, and they assign potencies to adenovirus stocks with a high degree of biological relevance. However, these approaches have two major shortcomings: the low throughput of the approach and the length of time required to perform the assay. A standard plaque assay takes 7 to 14 days (Mittereder et al., 1996). It was observed that a TCID50 titration can take up to 20 days (Figure 1). This delay can be a major impediment to the advancement of an adenovirus research project. In contrast, the IGU protocol presented here is complete in a day or two. Additionally, the IGU protocol can easily be performed on multiple vector stocks in parallel. Six to ten stocks are assayed routinely at a time.
Another major limitation of plaque assays and TCID50 assays is that they are dependent on the ability of the virus being titered to undergo a productive infection. A great many adenovirus-based vectors are either unable or only weakly able to form a plaque. It has been observed that a number of commonly used viruses, such as AdLC8clucif (Parks et al., 1996), are somewhat compromised in their ability to replicate, and therefore are difficult to titer by plaque assay (Zhou et al., 2002). Viruses that are difficult to plaque also tend to take longer in the TCID50 assay to reach their full titer (Figure 1). We have also observed this with adenovirus-based expression vectors that express weekly cytotoxic transgenes. However, each of these can be readily titered by the IGU protocol.
This limitation of plaque assays and TCID50 assays is even more of an issue in the case of HD-Ad vectors, which are completely incapable of inducing a productive infection. Additionally, we (Supplemental Figure S2), and others (Sandig et al., 2000; Puntel et al., 2006), have shown that a Q-PCR-based approach is ideal for HD-Ad vectors because it allows for the simultaneous determination of the titers of both the HD-Ad vector and any contaminating helper virus. It is not surprising then, that some form of Q-PCR is used so often in the titration of HD-Ad vectors.
Some groups have developed adenovirus titration assays that are abased on the hybridization of labeled probes to the vector DNA (Kreppel et al., 2002; Palmer and Ng, 2004). These approaches share many of the same advantages and disadvantages of the Q-PCR-based approach described above. In fact, one group has found that results from Q-PCR and from labeled-oligo hybridization are effectively equivalent (Crettaz et al., 2008). The one factor that tips in the favor of Q-PCR is the wide dynamic range. It was shown (Supplemental Figure S1A and B) that this approach is capable of accurately quantifying vector stocks in a range of four to seven logs compared to 2.5 logs in the slot blot assay (Kreppel et al., 2002; Crettaz et al., 2008).
5. Conclusion
The IGU titration protocol presented here is rapid, scalable, reproducible, sensitive, and is applicable to adenovirus and all types of adenovirus-based expression vectors. Most importantly, it produces titers that reflect the biological activity of the virus stock.
Supplementary Material
Highlights.
We present a universal method for titering adenovirus, recombinant adenovirus and helper dependent adenovirus vectors.
This method is superior to other commonly used methods because it is rapid, reproducible, and sensitive.
Titers determined by this method are superior at predicting biological activity than other Q-PCR based methods.
Acknowledgements
The authors thank Carol Eng for her excellent technical assistance and Michael Balamotis and Ahmed Yousef for their advice. This work was supported by grant R37-CA025235 and T32-GM07104 from the NCI, NIH, USA. Additional funding was provided by the Warsaw Family Fellowship.
Abbreviations
- Q-PCR
Quantitative Polymerase Chain Reaction
- TCID50
Tissue culture infectious dose at 50%
- IU/mL
Infectious Units per milliliter as determined by TCID50 assay
- VG/mL
Vector Genomes per milliliter as determined by Q-PCR protocol
- IGU/mL
Infectious Genome Units per milliliter as determined by the protocol described here
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References
- Alba R, Hearing P, Bosch A, Chillon M. Differential amplification of adenovirus vectors by flanking the packaging signal with attB/attP-PhiC31 sequences: implications for helper-dependent adenovirus production. Virology. 2007;367:51–8. doi: 10.1016/j.virol.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Berget SM, Moore C, Sharp PA. Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc Natl Acad Sci U S A. 1977;74:3171–5. doi: 10.1073/pnas.74.8.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk AJ. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene. 2005;24:7673–85. doi: 10.1038/sj.onc.1209040. [DOI] [PubMed] [Google Scholar]
- Brunetti-Pierri N, Ng P. Progress and prospects: gene therapy for genetic diseases with helper-dependent adenoviral vectors. Gene Ther. 2008;15:553–60. doi: 10.1038/gt.2008.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow LT, Gelinas RE, Broker TR, Roberts RJ. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell. 1977;12:1–8. doi: 10.1016/0092-8674(77)90180-5. [DOI] [PubMed] [Google Scholar]
- Crettaz J, Olague C, Vales A, Aurrekoetxea I, Berraondo P, Otano I, Kochanek S, Prieto J, Gonzalez-Aseguinolaza G. Characterization of high-capacity adenovirus production by the quantitative real-time polymerase chain reaction: a comparative study of different titration methods. J Gene Med. 2008;10:1092–101. doi: 10.1002/jgm.1236. [DOI] [PubMed] [Google Scholar]
- Dai Y, Schwarz EM, Gu D, Zhang WW, Sarvetnick N, Verma IM. Cellular and humoral immune responses to adenoviral vectors containing factor IX gene: tolerization of factor IX and vector antigens allows for long-term expression. Proc Natl Acad Sci U S A. 1995;92:1401–5. doi: 10.1073/pnas.92.5.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaprio JA. How the Rb tumor suppressor structure and function was revealed by the study of Adenovirus and SV40. Virology. 2009;384:274–84. doi: 10.1016/j.virol.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Dorigo O, Gil JS, Gallaher SD, Tan BT, Castro MG, Lowenstein PR, Calos MP, Berk AJ. Development of a novel helper-dependent adenovirus-Epstein-Barr virus hybrid system for the stable transformation of mammalian cells. J Virol. 2004;78:6556–66. doi: 10.1128/JVI.78.12.6556-6566.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endter C, Dobner T. Cell transformation by human adenoviruses. Curr Top Microbiol Immunol. 2004;273:163–214. doi: 10.1007/978-3-662-05599-1_6. [DOI] [PubMed] [Google Scholar]
- Ferrari R, Berk AJ, Kurdistani SK. Viral manipulation of the host epigenome for oncogenic transformation. Nat Rev Genet. 2009;10:290–4. doi: 10.1038/nrg2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields BN, Knipe DM, Howley PM. Fields’ virology. Wolters Kluwer Health/Lippincott Williams & Wilkins; Philadelphia: 2007. [Google Scholar]
- Gallaher SD, Gil JS, Dorigo O, Berk AJ. Robust in vivo transduction of a genetically stable Epstein-Barr virus episome to hepatocytes in mice by a hybrid viral vector. J Virol. 2009;83:3249–57. doi: 10.1128/JVI.01721-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh SS, Gopinath P, Ramesh A. Adenoviral vectors: a promising tool for gene therapy. Appl Biochem Biotechnol. 2006;133:9–29. doi: 10.1385/abab:133:1:9. [DOI] [PubMed] [Google Scholar]
- Gil JS, Gallaher SD, Berk AJ. Delivery of an EBV episome by a self-circularizing helper-dependent adenovirus: long-term transgene expression in immunocompetent mice. Gene Ther. 2010;17:1288–93. doi: 10.1038/gt.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginzinger DG. Gene quantification using real-time quantitative PCR: an emerging technology hits the mainstream. Exp Hematol. 2002;30:503–12. doi: 10.1016/s0301-472x(02)00806-8. [DOI] [PubMed] [Google Scholar]
- Grabow WO, Puttergill DL, Bosch A. Propagation of adenovirus types 40 and 41 in the PLC/PRF/5 primary liver carcinoma cell line. J Virol Methods. 1992;37:201–7. doi: 10.1016/0166-0934(92)90047-h. [DOI] [PubMed] [Google Scholar]
- Green M, Pina M, Kimes RC. Biochemical studies on adenovirus multiplication. XII. Plaquing efficiencies of purified human adenoviruses. Virology. 1967;31:562–5. doi: 10.1016/0042-6822(67)90241-3. [DOI] [PubMed] [Google Scholar]
- Gueret V, Negrete-Virgen JA, Lyddiatt A, Al-Rubeai M. Rapid titration of adenoviral infectivity by flow cytometry in batch culture of infected HEK293 cells. Cytotechnology. 2002;38:87–97. doi: 10.1023/A:1021106116887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harui A, Suzuki S, Kochanek S, Mitani K. Frequency and stability of chromosomal integration of adenovirus vectors. J Virol. 1999;73:6141–6. doi: 10.1128/jvi.73.7.6141-6146.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreppel F, Biermann V, Kochanek S, Schiedner G. A DNA-based method to assay total and infectious particle contents and helper virus contamination in high-capacity adenoviral vector preparations. Hum Gene Ther. 2002;13:1151–6. doi: 10.1089/104303402320138934. [DOI] [PubMed] [Google Scholar]
- Levine AJ. The common mechanisms of transformation by the small DNA tumor viruses: The inactivation of tumor suppressor gene products: p53. Virology. 2009;384:285–93. doi: 10.1016/j.virol.2008.09.034. [DOI] [PubMed] [Google Scholar]
- Li F, Feng L, Liu Y, Zhen X, Chen L. An integrated cell culture and quantitative polymerase chain reaction technique for determining titers of functional and infectious adenoviruses. Anal Biochem. 2009;391:157–9. doi: 10.1016/j.ab.2009.05.017. [DOI] [PubMed] [Google Scholar]
- Lieber A, He CY, Kirillova I, Kay MA. Recombinant adenoviruses with large deletions generated by Cre-mediated excision exhibit different biological properties compared with first-generation vectors in vitro and in vivo. J Virol. 1996;70:8944–60. doi: 10.1128/jvi.70.12.8944-8960.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Bluyssen HA, De Raeymaeker M, Laurysens V, van der Beek N, Pavliska H, van Zonneveld AJ, Tomme P, van Es HH. Rapid determination of adenoviral vector titers by quantitative real-time PCR. J Virol Methods. 2001;93:181–8. doi: 10.1016/s0166-0934(01)00257-9. [DOI] [PubMed] [Google Scholar]
- Maizel JV, Jr., White DO, Scharff MD. The polypeptides of adenovirus. I. Evidence for multiple protein components in the virion and a comparison of types 2, 7A, and 12. Virology. 1968;36:115–25. doi: 10.1016/0042-6822(68)90121-9. [DOI] [PubMed] [Google Scholar]
- Mittereder N, March KL, Trapnell BC. Evaluation of the concentration and bioactivity of adenovirus vectors for gene therapy. J Virol. 1996;70:7498–509. doi: 10.1128/jvi.70.11.7498-7509.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DB. In: Guidance for Industry: Guidance for Human Somatic Cell Therapy and Gene Therapy. F.a.D.A. U.S. Department of Health and Human Services, editor. U.S. Food and Drug Administration; Rockville, MD: 1998. p. 30. [Google Scholar]
- Ng P, Evelegh C, Cummings D, Graham FL. Cre levels limit packaging signal excision efficiency in the Cre/loxP helper-dependent adenoviral vector system. J Virol. 2002;76:4181–9. doi: 10.1128/JVI.76.9.4181-4189.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyberg-Hoffman C, Shabram P, Li W, Giroux D, Aguilar-Cordova E. Sensitivity and reproducibility in adenoviral infectious titer determination. Nat Med. 1997;3:808–11. doi: 10.1038/nm0797-808. [DOI] [PubMed] [Google Scholar]
- Palmer D, Ng P. Improved system for helper-dependent adenoviral vector production. Mol Ther. 2003;8:846–52. doi: 10.1016/j.ymthe.2003.08.014. [DOI] [PubMed] [Google Scholar]
- Palmer DJ, Ng P. Physical and infectious titers of helper-dependent adenoviral vectors: a method of direct comparison to the adenovirus reference material. Mol Ther. 2004;10:792–8. doi: 10.1016/j.ymthe.2004.06.1013. [DOI] [PubMed] [Google Scholar]
- Parks RJ, Chen L, Anton M, Sankar U, Rudnicki MA, Graham FL. A helper-dependent adenovirus vector system: removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc Natl Acad Sci U S A. 1996;93:13565–70. doi: 10.1073/pnas.93.24.13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks RJ, Graham FL. A helper-dependent system for adenovirus vector production helps define a lower limit for efficient DNA packaging. J Virol. 1997;71:3293–8. doi: 10.1128/jvi.71.4.3293-3298.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesonen S, Kangasniemi L, Hemminki A. Oncolytic adenoviruses for the treatment of human cancer: focus on translational and clinical data. Mol Pharm. 2010;8:12–28. doi: 10.1021/mp100219n. [DOI] [PubMed] [Google Scholar]
- Philipson L. Adenovirus assay by the fluorescent cell-counting procedure. Virology. 1961;15:263–8. doi: 10.1016/0042-6822(61)90357-9. [DOI] [PubMed] [Google Scholar]
- Prevec L, Christie BS, Laurie KE, Bailey MM, Graham FL, Rosenthal KL. Immune response to HIV-1 gag antigens induced by recombinant adenovirus vectors in mice and rhesus macaque monkeys. J Acquir Immune Defic Syndr. 1991;4:568–76. [PubMed] [Google Scholar]
- Puntel M, Curtin JF, Zirger JM, Muhammad AK, Xiong W, Liu C, Hu J, Kroeger KM, Czer P, Sciascia S, Mondkar S, Lowenstein PR, Castro MG. Quantification of high-capacity helper-dependent adenoviral vector genomes in vitro and in vivo, using quantitative TaqMan real-time polymerase chain reaction. Hum Gene Ther. 2006;17:531–44. doi: 10.1089/hum.2006.17.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qbiogene . In: AdEasy Vector System Application Manual. Qbiogene, editor. Carlsbad, CA: [Google Scholar]
- Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP, Wilson JM, Batshaw ML. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. 2003;80:148–58. doi: 10.1016/j.ymgme.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Raty JK, Pikkarainen JT, Wirth T, Yla-Herttuala S. Gene therapy: the first approved gene-based medicines, molecular mechanisms and clinical indications. Curr Mol Pharmacol. 2008;1:13–23. doi: 10.2174/1874467210801010013. [DOI] [PubMed] [Google Scholar]
- Rich DP, Couture LA, Cardoza LM, Guiggio VM, Armentano D, Espino PC, Hehir K, Welsh MJ, Smith AE, yGregory RJ. Development and analysis of recombinant adenoviruses for gene therapy of cystic fibrosis. Hum Gene Ther. 1993;4:461–76. doi: 10.1089/hum.1993.4.4-461. [DOI] [PubMed] [Google Scholar]
- Rosenfeld MA, Yoshimura K, Trapnell BC, Yoneyama K, Rosenthal ER, Dalemans W, Fukayama M, Bargon J, Stier LE, Stratford-Perricaudet L, et al. In vivo transfer of the human cystic fibrosis transmembrane conductance regulator gene to the airway epithelium. Cell. 1992;68:143–55. doi: 10.1016/0092-8674(92)90213-v. [DOI] [PubMed] [Google Scholar]
- Rots MG, Curiel DT, Gerritsen WR, Haisma HJ. Targeted cancer gene therapy: the flexibility of adenoviral gene therapy vectors. J Control Release. 2003;87:159–65. doi: 10.1016/s0168-3659(02)00360-7. [DOI] [PubMed] [Google Scholar]
- Sandig V, Youil R, Bett AJ, Franlin LL, Oshima M, Maione D, Wang F, Metzker ML, Savino R, Caskey CT. Optimization of the helper-dependent adenovirus system for production and potency in vivo. Proc Natl Acad Sci U S A. 2000;97:1002–7. doi: 10.1073/pnas.97.3.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura MM, Monfar M, Puig M, Mennechet F, Ibanes S, Chillon M. A real-time PCR assay for quantification of canine adenoviral vectors. J Virol Methods. 2010;163:129–36. doi: 10.1016/j.jviromet.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Steitz J, Barlow PG, Hossain J, Kim E, Okada K, Kenniston T, Rea S, Donis RO, Gambotto A. A candidate H1N1 pandemic influenza vaccine elicits protective immunity in mice. PLoS One. 2010;5:e10492. doi: 10.1371/journal.pone.0010492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stripecke R, Carmen Villacres M, Skelton D, Satake N, Halene S, Kohn D. Immune response to green fluorescent protein: implications for gene therapy. Gene Ther. 1999;6:1305–12. doi: 10.1038/sj.gt.3300951. [DOI] [PubMed] [Google Scholar]
- Tan BT, Wu L, Berk AJ. An adenovirus-Epstein-Barr virus hybrid vector that stably transforms cultured cells with high efficiency. J Virol. 1999;73:7582–9. doi: 10.1128/jvi.73.9.7582-7589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umana P, Gerdes CA, Stone D, Davis JR, Ward D, Castro MG, Lowenstein PR. Efficient FLPe recombinase enables scalable production of helper-dependent adenoviral vectors with negligible helper-virus contamination. Nat Biotechnol. 2001;19:582–5. doi: 10.1038/89349. [DOI] [PubMed] [Google Scholar]
- Vellekamp G, Porter FW, Sutjipto S, Cutler C, Bondoc L, Liu YH, Wylie D, Cannon-Carlson S, Tang JT, Frei A, Voloch M, Zhuang S. Empty capsids in column-purified recombinant adenovirus preparations. Hum Gene Ther. 2001;12:1923–36. doi: 10.1089/104303401753153974. [DOI] [PubMed] [Google Scholar]
- Wang F, Puddy AC, Mathis BC, Montalvo AG, Louis AA, McMackin JL, Xu J, Zhang Y, Tan CY, Schofield TL, Wolf JJ, Lewis JA. Using QPCR to assign infectious potencies to adenovirus based vaccines and vectors for gene therapy: toward a universal method for the facile quantitation of virus and vector potency. Vaccine. 2005;23:4500–8. doi: 10.1016/j.vaccine.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Wang L, Wang CJ, Tan CY, Hsu D, Hennessey JP. A robust approach for the quantitation of viral concentration in an adenoviral vector-based human immunodeficiency virus vaccine by real-time quantitative polymerase chain reaction. Hum Gene Ther. 2006;17:728–40. doi: 10.1089/hum.2006.17.728. [DOI] [PubMed] [Google Scholar]
- Weitzman MD, Ornelles DA. Inactivating intracellular antiviral responses during adenovirus infection. Oncogene. 2005;24:7686–96. doi: 10.1038/sj.onc.1209063. [DOI] [PubMed] [Google Scholar]
- Zhou HS, Zhao T, Rao XM, Beaudet AL. Production of helper-dependent adenovirus vector relies on helper virus structure and complementing. J Gene Med. 2002;4:498–509. doi: 10.1002/jgm.301. [DOI] [PubMed] [Google Scholar]
8. Web References
- Journal of Gene Medicine 02/22/2012. http://www.abedia.com/wiley/vectors.php.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.