

Abstract
The ‘activating' E2fs (E2f1-3) are transcription factors that potently induce quiescent cells to divide. Work on cultured fibroblasts suggested they were essential for division, but in vivo analysis in the developing retina and other tissues disproved this notion. The retina, therefore, is an ideal location to assess other in vivo adenovirus E2 promoter binding factor (E2f) functions. It is thought that E2f1 directly induces apoptosis, whereas other activating E2fs only induce death indirectly by upregulating E2f1 expression. Indeed, mouse retinoblastoma (Rb)-null retinal neuron death requires E2f1, but not E2f2 or E2f3. However, we report an entirely distinct mechanism in dying cone photoreceptors. These neurons survive Rb loss, but undergo apoptosis in the cancer-prone retina lacking both Rb and its relative p107. We show that while E2f1 killed Rb/p107 null rod, bipolar and ganglion neurons, E2f2 was required and sufficient for cone death, independent of E2f1 and E2f3. Moreover, whereas E2f1-dependent apoptosis was p53 and p73-independent, E2f2 caused p53-dependent cone death. Our in vivo analysis of cone photoreceptors provides unequivocal proof that E2f-induces apoptosis independent of E2f1, and reveals distinct E2f1- and E2f2-activated death pathways in response to a single tumorigenic insult.
Keywords: retinoblastoma, E2f, p53, cone photoreceptor, retina
Retinoblastoma protein (Rb), the prototype tumor suppressor, was discovered because of its role in childhood retinal cancer.1 It is related to p107 (Rbl1) and p130 (Rbl2) and these proteins form repressive transcriptional complexes with a subset of E2fs, DNA-binding proteins that regulate cell cycle and apoptotic genes.2, 3, 4 E2f1, E2f2 and E2f3 are ‘activating E2fs' that drive quiescent cells into cycle and potently activate gene transcription. The E2f3 gene expresses two alternative proteins, E2f3a and E2f3b. In line with shared structure and function, E2f1 or E2f3b can substitute for E2f3a in vivo.5
Activating E2fs have long been linked to apoptosis but, unlike E2f1, whether E2f2/3 induce apoptosis directly is controversial. Initial work showed that E2f1 overexpression in vitro induces apoptosis,6, 7, 8 but E2f2 or E2f3 did not kill REF52 fibroblasts.9 Later work showed all three induce apoptosis of Rat1 fibroblasts,10 although E2f1 was the most potent effector.11 Transgenic E2f1 or E2f3a overexpression enhances skin apoptosis,12, 13 as does E2f1, E2f2 or E2f3a expression in lens,14, 15 but whereas E2f1 or E2f3 stimulate cardiomyocyte division and death, E2f2 induces division without death.16 Cross-talk between family members means E2f2/3-induced apoptosis may require E2f1 upregulation. Indeed, E2f3a-induced apoptosis, either in vitro or in the pituitary gland, requires E2f1 upregulation.17 In skin, E2f1 and E2f3a induce p53-dependent and p53-independent apoptosis, respectively, suggesting separate mechanisms,12, 13 but the E2f3a study noted that, like the pituitary, death requires E2f1.13 Potentially, therefore, E2f3a induces factors that cooperate with E2f1 to drive p53-independent death. These studies raise doubt as to whether E2f2 or E2f3 induce E2f1-independent death.
Overexpression may not mimic physiological E2f activation, thus others have assessed whether E2fs drive apoptosis in Rb−/− cells. Deleting E2f1, E2f2 or E2f3 reduces apoptosis in Rb−/− mouse embryo tissues,18, 19, 20 suggesting that exceeding a threshold of total E2f activity might trigger apoptosis.3 However, apoptosis in the Rb−/− embryo is mainly an indirect consequence of placental defects.21 Indeed, Rb−/− ES cells contribute as efficiently as wild-type (WT) cells to most tissues, except retina and lens.22 Rb deletion in retina causes rod, bipolar and ganglion neuron apoptosis,23, 24 which is E2f1-, but not E2f2- or E2f3-dependent.25 Deleting Rb in the cortex drives Cajul-Retzius neuron death, which is reversed by removing E2f1 or E2f3,26 but whether the latter is E2f1-dependent is unknown. Some E2f1-independent cell death has also been noted in Rb-deficient lens and muscle,27, 28 but whether this requires other E2fs or unrelated factors is unclear. A central role for E2f1 is extended by its requirement in negative selection of thymocytes29 and DNA damage-induced apoptosis.30 E2f3 can drive DNA damage-induced apoptosis, but dependency on E2f1 was, again, not tested.31 Unique apoptotic E2f1 domains, post-translational modifications and cofactors underscore its death function.30, 32, 33
Although there is no clear-cut support for E2f1-independent E2f-driven apoptosis in Rb- deficient cells, Myc-driven T-cell apoptosis requires E2f2, but not E2f1 or E2f3.34 The mechanism, however, is unclear. E2f1 can induce p53-dependent or independent apoptosis.2 E2f1 induces Arf, which suppresses Mdm2, thus reducing p53 ubiquitylation/degradation,2, 3, 4 and Atm and Chk2, which phosphorylate and activate p53.35 E2f1 also upregulates p53 cofactors, such as Aspp1/Aspp2, biasing p53 to activate death genes.36 It induces p53-independent apoptosis through pro-apoptotic genes such as Apaf1, Caspases, BH3-only proteins and the p53-relative p73, or by inhibiting NF-κB and Bcl-2 survival pathways.2 Whether other E2fs utilize these pathways for death is unclear, but at least in fibroblasts only E2f1, not E2f2, induces the Chk2-p53 axis.35
As noted above, neuronal death in Rb−/− retina requires E2f1 but not p53. Cones, required for color vision, survive Rb loss, but undergo apoptosis in the cancer-prone Rb/p107 double knockout (DKO) tissue.23 The mechanism is unknown. Here, compound mouse mutants, in vivo electroporation and extensive molecular analyses reveal that whereas E2f1 induces p53-independent death of three Rb/p107 null neuronal cell types, E2f2 drives p53-dependent apoptosis of Rb/p107-deficient cones.
Results
Cell type-specific E2f1-dependent neuronal cell death
To study which E2fs mediate Rb/p107 null retinal phenotypes, we generated mice carrying the α-Cre transgene, floxed Rb (Rbf), p107 null, E2f1 null, E2f2 null and/or E2f3f alleles. E2f1 and p107 are only 2.6 Mb apart, so we generated a recombinant carrying both null alleles.37 The α-Cre transgene is induced in the peripheral retina at E9.5 and we previously confirmed deletion of single and combined floxed Rb, E2f3, p53 and other alleles in this model.38 As E2f1 mediates ectopic division and death in RbKO retina,25 we asked whether E2f1 also drives these processes in Rb/p107 DKO retina. In Rb/p107/E2f1 triple null (triple knockout; TKO) retinas, division was assessed using Ki67 (all cycling cells), bromodeoxyuridine (BrdU) incorporation (S-phase) and phosphohistone H3 (PH3; mitosis), while apoptosis was detected using TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) or active caspase 3 (AC3) staining. At postnatal day 12 (P12), when normal progenitors have exited the cell cycle, deleting E2f1 suppressed most ectopic division (94.2% Ki67+, 95.6% BrdU+ and 90.1% PH3+ cells) and cell death (96.1% TUNEL+ cells) and there was a dramatic rescue of rod, bipolar and ganglion cells (Figures 1a–c, Supplementary Figures S1a–c). Unexpectedly, however, removing E2f1 did not rescue Rb/p107-deficient cones, marked either by S-antigen (rods and cones) or Cone-arrestin (cones only) (Figures 1a and c). In agreement, westerns indicated that both Rb/p107 and Rb/p107/E2f1-deficient retinas had more AC3 than control retina (Supplementary Figure S1d).
Figure 1.

Cell type-specific E2f-dependent neuronal cell death. (a) P12 horizontal retinal sections of the indicated genotypes were BrdU-labeled for 2 h, and stained for nuclei (DAPI, blue), S-phase (anti-BrdU, red), cell death (TUNEL, red), Rods and cones (S-antigen, green) and cones (Cone-arrestin, red). ONL: outer nuclear layer. INL: inner nuclear layer. GCL: ganglion cell layer. Scale bar is 50 μm. (b and c) Fold change in BrdU+ and TUNEL+ cells per 100 μm unit (b), and of S-antigen+ and Cone-arrestin+ cells (c) relative to control (Rb f/f;p107 −/−) retina. Error bars represent S.D. and asterisks indicate significant difference between Rb/p107 DKO and other retinas (*P<0.01. Analysis of variance (ANOVA) and Tukey honestly significant difference (HSD) test)
Newborn cones express thyroid hormone receptor β2 (Trβ2).39 There were many Trβ2+ cells in the postnatal day 0 (P0) Rb/p107 DKO and Rb/p107/E2f1 TKO retina, thus cones are specified (Figure 2a). As expected, many Rb/p107-deficient Trβ2+ cells were dividing aberrantly and undergoing apoptosis, but unlike other neuronal cells, cone division and death continued virtually unabated in the Rb/p107/E2f1 TKO retina (Figures 2a and b). Cone development was not simply delayed in the latter because akin to P12 (Figure 1a), almost no Cone-arrestin was detected at P21, P45 or as late as P60 (Figures 2a and c). Thus, as in the Rb−/− retina,25 E2f1 drives abnormal division of most Rb/p107 null neurons and apoptosis of rod, ganglion and bipolar cells, but is not required for division or death of Rb/p107 null cones.
Figure 2.

Rb/p107 null cones are specified but divide and die. (a) Horizontal retinal sections of the indicated genotypes and ages were stained for nuclei (DAPI, blue), division (Ki67, green), new born cones (Trβ2, red or green as indicated), cell death (TUNEL, red) and mature cones (Cone-arrestin, red). Arrows indicate dividing or dying cones. ONL: outer nuclear layer. INL: inner nuclear layer. GCL: ganglion cell layer. NBL: neuroblastic layer. Scale bar is 50 μm. (b) The ratio of Ki67+;Trβ2+ or TUNEL+;Trβ2+ cells over all Trβ2+ cells of the indicated genotypes at P0. (c) The average number of Cone-arrestin+cells per 100 μm of the indicated genotypes and ages. Error bars represent S.D. and asterisks indicate significant difference between control (Rbf/f;p107−/−) and other retinas (*P<0.05, **P<0.01. Analysis of variance (ANOVA) and Tukey honestly significant difference (HSD) test)
E2f2 mediates ectopic division and death of Rb/p107 or Rb/p107/E2f1 null cones
To investigate the mechanism underlying cone death in Rb/p107 and Rb/p107/E2f1 null retinas, we used RT-PCR to assess E2f1-3 gene expression at P0, when most newborn cones are differentiating (Figure 3a). Relative to control p107 null (Rbf/f; p107−/−, Slate Blue in Figure 3a) or control p107/E2f1 null (Rbf/f; p107−/−; E2f1−/−, Black in Figure 3a) retinas, E2f1, E2f2 and E2f3 genes were all upregulated in the Rb/p107 DKO retina, particularly E2f2 (brown in Figure 3a). Notably, however, only E2f2 remained upregulated significantly in the Rb/p107/E2f1 TKO retina (P<0.05; yellow in Figure 3a), indicating that unlike E2f3, E2f2 induction is E2f1-independent. Westerns confirmed E2f2 protein upregulation in Rb/p107 DKO and Rb/p107/E2f1 TKO retinas (Figure 3b). Immunostaining did not detect E2f2 in WT P0 retina even though E2f2 protein was observed in the western blot (Figure 3b), but in Rb/p107-deficient tissue it was seen in differentiating neurons including Trβ2+ cones, and staining was specific as it was absent in the Rb/p107/E2f2 TKO retina (Figure 3c). Increased E2f2 immunoreactivity could reflect either elevated protein, and/or epitope exposure due to loss of Rb/p107 and associated corepressors. E2f1 staining was not possible as numerous antibodies generated non-specific signals in the E2f1−/− retina, but we successfully detected a specific E2f3 signal before,25 and while this protein was seen in many cells, it was absent in Trβ2+ cones (Supplementary Figure S2a). In summary, E2f2 is upregulated in Rb/p107 null cones independent of E2f1.
Figure 3.

E2f2 induces cone death independent of E2f1 and E2f3. (a) E2f1-independent E2f2 induction in the Rb/p107 null retina. RT-qPCR was used to measure the relative levels of E2f1-3 mRNAs in P0 retinas of the indicated genotypes. (b) Elevated E2f2 protein in the Rb/p107 and Rb/p107/E2f1 null retina. Graph shows normalized E2f2 protein levels at P0 in the indicated genotypes, and a representative western is shown below (n=3). (c) E2f2 is expressed in emerging cones. Horizontal P0 retinal sections of the indicated genotypes were stained for nuclei (DAPI, blue), E2f2 (red), cone precursors (Trβ2, green). Arrows indicate examples of double-labeled E2f2+; Trβ2+ cells. Scale bar is 50 μm. (d) Ectopic E2f2-induces death in E2f3 or E2f1/3 null cones. P0 retinas of the indicated genotypes were electroporated with Mxie-GFP-Cre/CMV-E2f2 plasmids (1 : 4) or Mxie-GFP-Cre alone. At P3, horizontal retinal sections were stained for nuclei (DAPI, blue), differentiating cones (Trβ2, white), cell death (TUNEL, red), and transfected cells (GFP, green). (e) The ratio of TUNEL+;GFP+;Trβ2+ cells (dying transfected cones) to all GFP+;Trβ2+ cells (transfected cones) in (d). Error bars represent S.D. and asterisks indicate significant difference from control (Rbf/f, p107−/− in (a), WT in (b)) and E2f3f/f cells transfected with Mxie-GFP-Cre in (e) (*P<0.05, **P<0.01. Analysis of variance (ANOVA) and Tukey honestly significant difference (HSD) test)
Next, we investigated whether E2f2 and/or E2f3, or dual combinations of activating E2fs 1, 2 and 3 mediate Rb/p107 DKO cone defects. For this, TKO (Rb/p107 plus E2f2 or E2f3) or quadruple knockout (QKO; Rb/p107 plus E2f1/2, E2f2/3 or E2f1/3) retinas were generated. Strikingly, neither E2f1 nor E2f3 deletion rescued cones, but the Rb/p107/E2f2 null retina exhibited many Cone-arrestin+ cells at P12 (Figures 1a and c), P21 and P45 (Figures 2a and c). Moreover, we detected Ki67−, S- and M-opsin+ cells, indicating rescue of both cone subtypes (Supplementary Figure S1e). There was also marginal rescue of Cone-arrestin+ cells at P21-P45 in the Rb/p107/E2f1/3 QKO retina (Supplementary Figure S3), which was consistent with reduced E2f2 mRNA expression in this genotype (blue in Figure 3a). Although removing E2f2 did not affect the overall level of ectopic division/apoptosis at P0, reflecting E2f1-driven defects in other neurons, it reduced division and death dramatically in the small Trβ2+ cone population (Figures 2a and b). In contrast, removing E2f1 (Figures 2a and b), E2f3 alone or even both together to create a QKO retina (Supplementary Figures S3a and b) had no such effect.
Rescued Cone-arrestin+ cells in the P21 or P45 Rb/p107/E2f2 TKO retina were scattered, lacked inner and outer segments, and disappeared by P60 (c.f. rows 1 versus 4 in Figure 2a). Cones need rods to survive,40 thus we reasoned that removing E2f1, which rescues rods, may prevent secondary cone death. Indeed, normal cones were observed in the Rb/p107/E2f1/E2f2 QKO retina through P60 (Figure 2a, row 5). Cone numbers in the QKO retina matched that in the E2f1/E2f2 DKO retina, which was lower than WT because E2f1 removal mildly impairs progenitor division.38 Altogether, expression and genetic data indicate that although E2f1 kills most Rb/p107 null neurons, E2f2 drives ectopic division and death of newborn Rb/p107 null cones.
E2f2 gain-of-function drives cone death independent of E2f1 and E2f3
E2f2 is required to kill Rb/p107 DKO cones. To test whether E2f2 is also sufficient, we electroporated P0 E2f3f/f and E2f1−/−; E2f3f/f retinas in vivo with Mxie-GFP-Cre, expressing Cre-recombinase and GFP, alone or with an E2f2 expression vector (CMV-E2f2). Retinas were analyzed 2 days later for apoptosis in transfected (GFP+) Trβ2+ cones, which represented ∼0.5% of all GFP-Cre transfected cells. Recombination was effective, as cells with Mxie-GFP-Cre never co-stained for E2f3 protein, while some cells transfected with control Mxie-GFP vector were E2f3+ (Supplementary Figure S2b). Cre alone did not affect survival, but E2f2 expression killed cones in both E2f3 and E2f1/3 null retinas (Figures 3d and e). Thus, E2f2 is sufficient to drive cone apoptosis independent of E2f1 and E2f3.
Putative links between E2f2 and p53 activation
Next, we assessed the mechanism underlying cell type-specific death (Figures 4a–e). The rarity of cones complicates analysis of whole-tissue lysates. Moreover, because rods are abundant (∼80% of retina) and their death is E2f1-dependent, comparing Rb/p107 and Rb/p107/E2f2-deficient retina may mask E2f2-driven effects. Thus, as well as assessing Rb/p107-deficient retina (magenta bars, Figures 4a and b) and controls with WT cell death levels (light blue and gray bars, Figures 4a and b), the most critical comparison for whole-tissue analysis was between Rb/p107/E2f1 TKO (yellow bars, Figures 4a and b) and Rb/p107/E2f1/E2f2 QKO (pink bars, Figures 4a and b) samples, in which E2f1-driven molecular changes are nullified and the only phenotypic difference is cone survival (Figures 1, 2, and Supplementary Figure S1).
Figure 4.

Cone death is p53-dependent. (a) Relative expression of indicated genes in P0 retina of the indicated genotypes, measured by RT-qPCR. The dashed line indicates the level of controls. (b) Relative protein level of indicated proteins in the P0 retina of the indicated genotypes, normalized to actin. Representative westerns are shown in C (n=3). (c) Western of indicated proteins in indicated retinas. Control: Rbf/f;p107−/−. DKO: Rb/p107 DKO. TKO: Rb/p107/E2f1 TKO. QKO: Rb/p107/E2f1/E2f2 QKO. (d) P0 retinas of the indicated genotypes were stained for nuclei (DAPI, blue) and p53 (red). (e) The absolute numbers per 100 μm unit of p53+ cells in indicated P0 retinas. (f) Retinal sections of the indicated ages and genotypes were stained for nuclei (DAPI, blue), Cone-arrestin (red), Trβ2 (green) and cell death (TUNEL, red). (g) The absolute number of Cone-arrestin+ cells per 100 μm of the indicated genotypes in f. (h) The ratio of Trβ2+/TUNEL+ cells over all Trβ2+ cells in f. In d and f, NBL: neuroblast layer. ONL: outer nuclear layer. INL: inner nuclear layer. GCL: ganglion cell layer. Scale bar is 50 μm. In a, b, e, g and h, error bars represent S.D., and asterisks indicate significant difference between control (Rbf/f;p107−/−) and other genotypes (*P<0.05, **P<0.01. Analysis of variance (ANOVA) and Tukey honestly significant difference (HSD test). Red cross indicates significant difference between Rb/p107/E2f1 TKO (yellow bar) and Rb/p107/E2f1/E2f2 QKO (pink bar) (P<0.05, Student's t-test)
Analysis of E2f target levels revealed, as expected, that cell cycle genes (e.g., Mcm3, Tk1) were induced in the absence of Rb and p107 and, notably, were downregulated when E2f2 was removed, consistent with E2f2-dependent ectopic cone division (Figure 4a). The E2f-targets and p53 modifiers Aspp1 or Aspp2 were not induced, but p73 mRNA was markedly elevated in an E2f2-dependent manner (Figures 4a–c). We did not observe induction of Arf or p53 mRNA (Figure 4a), or the p53 targets Puma, Pidd and Gadd45 (Supplementary Figure S4a), but the p53 targets p21, Noxa and Siva were induced in P0 retinas where cones are undergoing apoptosis, all in an E2f2-dependent manner (Figure 4a). Consistent with these findings there was a striking nine-fold induction of p53 immunoreactivity in Rb/p107 null retinal sections compared with control, which was only slightly reduced in the Rb/p107/E2f1 null retina where cones still die, but was dramatically downregulated to WT levels in the Rb/p107/E2f2TKO and Rb/p107/E2f1/E2f2 QKO retinas where cones are rescued (Figures 4d and e).
Increased p53 immunoreactivity could reflect either elevated protein or epitope exposure.38 In strong support of the latter, westerns showed the same p53 levels in control and Rb/p107 null retinas (Figures 4b and c). Previously, p53 epitope exposure was linked to acetylation,38 but acetyl-p53 levels were unchanged in the Rb/p107 null retina, as were those of the p53 deacetylases Sirt1 and HDAC1, and the p53 acetyl-transferases Pcaf and Tip60 (Figure 4c, Supplementary Figure S4b). However, p53 is also activated by phosphorylation and this modification was elevated in the Rb/p107 null versus control retina (Figures 4b and c). Notably, high phospho-p53 levels were maintained in the Rb/p107/E2f1 TKO retina where cones are dying, but were curtailed significantly in the Rb/p107/E2f1/E2f2 QKO retina where cones are rescued (Figures 4b and c). A ∼3-fold drop in phospho-p53 was in part explained by a 1.5-fold drop in total p53 protein levels upon removal of E2f2 (Figures 4b and c), thus E2f2 maintains p53 levels and in addition, promotes its phosphorylation.
Next, we investigated how E2f2 might regulate p53 phosphorylation. The pattern of phospho-p53 matched that of γH2AX (Figure 4c) and both proteins are phosphorylated by the Chk or ATR/ATM kinases. We did not observe activation of Chk1, ATR or ATM and only observed weak induction of pChk2 (Supplementary Figures S4b and c); these negative/marginal results might reflect the rarity of cones and/or the phosphorylation of p53 and/or H2AX by other kinase(s).
We also examined the mechanism by which E2f2 maintains p53 protein levels. Arf protein was significantly reduced in the Rb/p107/E2f1/E2f2 QKO retina relative to the Rb/p107/E2f1 TKO retina, and in line with its known ability to reduce the p53 ubiquitin ligase Mdm2, we detected high levels of the latter in QKO retina (Figures 4b and c). E2f1 and E2f2 can induce Arf transcription9, 41 but surprisingly, Arf mRNA levels were similarly low in both the Rb/p107/E2f1 TKO and Rb/p107/E2f1/E2f2 QKO retina (Figure 4a). Nucleophosmin (Npm) and tumor necrosis factor receptor-1 associated death domain (Tradd) can prevent Trip12/Ulf (ubiquitin ligase for Arf)-mediated Arf degradation, independent of Arf mRNA level,42, 43 thus we wondered if E2f2 might affect these Arf regulators. Ulf protein levels were similar in all genotypes, but Tradd and Npm proteins were modestly reduced, specifically in the absence of E2f2 (Supplementary Figures S4b and c). Tradd antibodies did not work for immunostaining, thus we could not examine expression in cones. Nevertheless, the western data suggest a model in which E2f2 may induce Tradd, which is known to inhibit Ulf-mediated degradation of Arf. We did not detect changes in Tradd (or Npm) transcripts (Supplementary Figure S4a), suggesting another level of post-transcriptional control in this E2f2-dependent pathway. However, extensive additional work is required to determine whether this putative pathway is relevant specifically in Rb/p107 null cones.
Cone death requires p53 but not p73
These data implicate p53 and/or p73 as mediators of E2f2-induced cone death. Therefore, we utilized p73 null44 and p53 floxed alleles45 to address this issue. In the Rb−/− retina, apoptosis deletes Brn3b+ ganglion cells, Pkcα+ rod bipolar cells and∼half Rhodopsin+ rods.25 We confirmed prior data24 showing that their death is p53-independent (Supplementary Figure S5). New observations here include the findings that Rb−/− ganglion, rod or bipolar neuronal death is p73-independent, p53 and p73 are also dispensable for death of these three neuorns in Rb/p107 null retina, and Rb/p107 null cone apoptosis is also p73-independent (Supplementary Figure S5). In contrast, there were many Cone-arrestin+ cells in the Rb/p107/p53 TKO P8 retina (Figures 4f and g). Moreover, removing p53 dramatically reduced the TUNEL+ fraction of newborn Trβ2+ Rb/p107 null cones at P0 (Figures 4f and h), which paralleled reduced levels of the pro-apoptotic p53 targets Siva and Noxa (Green bar, Figure 4a). Rescued Rb/p107/p53 TKO cones had abnormal morphology, but this problem was corrected when rod apoptosis was blocked by removing E2f1 (Rb/p107/E2f1/p53 QKO retina, Figure 5), as before for rescued Rb/p107/E2f2 null cones (c.f. Rb/p107/E2f1/E2f2 QKO retina in Figure 2c). Thus, whereas some Rb and Rb/p107 null neurons undergo E2f1-dependent, p53-independent apoptosis, Rb/p107 cone death is instead driven by E2f2 and requires p53.
Figure 5.
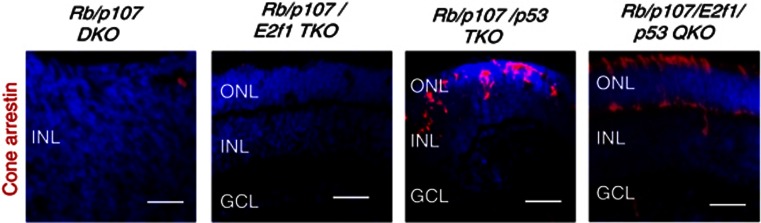
Rb/p107-deficient cones survive when p53 is removed, and have normal morphology when rods are also rescued by removing E2f1. P8 retinal sections of the indicated genotypes were stained for nuclei (DAPI, blue) and Cone-arrestin (red). ONL: outer nuclear layer. INL: inner nuclear layer. GCL: ganglion cell layer. Scale bar is 50 μm
Discussion
E2f2 induces apoptosis independent of other activating E2fs
Data here and prior work25 show that E2f1, but not E2f2 or E2f3, mediates rod, bipolar and ganglion cell death in the Rb and Rb/p107 null retina. Ectopic cone division did not require E2f1 or E2f3, but was instead E2f2-dependent. E2f2 mRNA and protein were upregulated in Rb/p107 DKO and Rb/p107/E2f1 TKO retinas, and E2f2 was expressed in newborn Rb/p107 DKO cones. Ectopic E2f2 expression was sufficient to kill cones in the absence of E2f1 and/or E2f3. These data provide unequivocal proof that E2f2 induces apoptosis independent of other activating E2fs, including E2f1.
Distinct mechanisms of E2f1- or E2f2-induced apoptosis
The only other example of E2f2-dependent death in E2f1/3 null cells is in Myc-induced lymphoma,34 but the mechanism is unknown. Prior work showed that E2f1-dependent death of Rb−/− retinal neurons is p53-independent.24, 25 We confirmed and extended that result to include p73. Similarly, p53 or p73 were dispensable for E2f1-induced death of Rb/p107-deficient retinal neurons. However, we detected E2f2-dependent p53 epitope-exposure and concomitant induction of the p53 targets Noxa and Siva. Deleting p53-blocked induction of these pro-apoptotic genes and cone death. Thus, in response to the identical oncogenic insult (Rb/p107 loss), E2f1 induces p53-independent apoptosis, whereas E2f2 stimulates p53-dependent death. Whether this reflects cell-specific expression of activating E2fs, or different pro-apoptotic properties of E2f1 and E2f2 will require additional analyses. E2f2 was easily detected in cones, and although it was not feasible to assess E2f1 by immunostaining, E2f3 was undetectable, suggesting that cell type-specific expression contributes to E2f2-mediated cone death. In contrast, E2f2 and E2f3 are expressed in cells other than cones that show E2f1-dependent death (this work and Chen et al.25), thus specificity may be mediated by post-translational modifications or cofactors that regulate E2f1 function (reviewed in Biswas and Johnson30).
Molecular analyses suggested two possible ways in which E2f2 facilitates p53-mediated apoptosis, although confirmation that these mechanisms are active in cones requires additional studies. First, E2f-dependent epitope exposure of p53 was linked to E2f2-dependent Ser15 phosphorylation. E2f1 overexpression or virally-mediated Rb family inactivation in fibroblasts activates Chk2-dependent p53 phosphorylation.35 E2f2 did not affect Chk2 in the latter context, but in Rb/p107 null retina E2f2 was required to maintain Chk2 protein levels and activation through T68 phosphorylation. E2f1 induces Chk2 mRNA in fibroblasts,35 but E2f2-sustained Chk2 protein post-transcriptionally in retina. E2f2 did not affect ATM or ATR levels or phosphorylation, so it remains to be seen whether they or other kinases target Chk2 in Rb/p107 null cones. Irrespective, these data suggest that both E2f1 and E2f2 may activate Chk2, a known p53 regulator, through distinct mechanisms. Phosphorylation of Chk2 was weak, perhaps reflecting the paucity of cones in the murine retina, and it will be important in the future to examine whether Chk2 is required for apoptosis of Rb/p107 null cones using, for example, in vivo electroporation of siRNAs and/or introduction of Chk2 inhibitors. As well as promoting phosphorylation, E2f2-sustained p53 protein levels. E2f2 maintained the levels of Tradd, which inhibits Trip12/Ulf-mediated Arf ubiquitylation and degradation.42, 43 Moreover, removing E2f2 reduced Arf and increased its target Mdm2. Future studies will be required to assess the implied functional relevance of each of these components in controlling p53 levels and apoptosis in Rb/p107 null neurons. We were unable to stain for these components with existing antibodies, thus it remains uncertain if this tentative pathway is active specifically in cones. It should be feasible in the future to assess whether knockdown of the putative pathway affects the survival of cones, or other cell types in which E2f2 is suspected of causing death. E2F1 can regulate the degradation of TRAF2, a downstream target of TRADD, and induce p53-independent cell death.46 Thus, E2fs may interact with multiple members of the TNF pathway to regulate cell death.
Implications for human disease
Activating E2fs inhibit tumorigenesis in some contexts, which may be linked to their pro-apoptotic activity.4 For example, removing one allele of E2f2 reduces apoptosis and promotes the formation of Myc-driven murine T-cell lymphomas.34 The origin of human Rb is unclear and, as with any cancer, is difficult to determine because, by definition, oncogenesis transforms cell properties.47 However, human Rb cells rely on key aspects of cone photoreceptor circuitry to survive and grow.48 As well as the inbuilt advantages, adopting this circuitry could have drawbacks, such as activation of the E2f2-Tradd-p53 pro-apoptotic pathway described herein. Notably, E2f2 maps to human chromosome 1p36 and Tradd maps to 16q22, both of which are commonly deleted in human Rb,49 and p53 inactivation by the Mdm family or Arf loss sustains this cancer.48, 50, 51, 52 Loss of E2f1, but not E2f2 or E2f3, blocks the initiation of mouse Rb,37 but whether E2f2 affects the growth rate of human Rb and/or p53 degradation remains to be seen.
Normal neurons are post-mitotic, but activation of cell cycle molecules, including E2fs, often precedes their degeneration.53 Inherited retinal degeneration has a prevalence of ∼1 in 3000, of which the most common subtype is retinitis pigmentosa (RP).40 RP results from a primary defect in rods, but leads to secondary cone loss. There are also defects that specifically target cones.40 Understanding how rods and cones die is important to design therapies. Our data indicate that E2f1-activation mediates rod death, whereas the E2f2-p53 axis promotes cone death. p53 is dispensable for rod and secondary cone death in the rd1 retina,54 but whether the E2f2-p53 pathway might contribute to cone death in other circumstances is unknown. Five retinal degeneration loci map to 1p36 (https://sph.uth.tmc.edu/retnet/disease.htm), the location of E2f2, thus future studies should consider whether this gene contributes to photoreceptor loss in humans.
Materials and Methods
Mouse strains and genotyping
Mice were treated according to local and national guidelines. α-Cre mice,55 Rb floxed,56 p107−/−,57 E2f1–/–,29 E2f2–/–,58 E2f3 floxed,59 p73−/− (ref. 44) and p53 floxed mice60 were maintained on a mixed (NMRI × C57/Bl × FVB/N × 129sv) background. Different genotypes were compared within the same litter and across at least three litters. We have not noted phenotypic differences in separate litters. Genotyping was performed as before.23, 25, 37
Histology, immunofluorescence and measurements
Eyeballs were fixed in 4% paraformaldehyde for 1 h at 4 °C, embedded in OCT (TissueTek 4583), frozen on dry ice and cut into 12–14 μm sections on superfrost slides (VWR). For S-phase analysis, BrdU (100 μg/g of body weight) was injected intraperitoneally 2 h before killing. BrdU+ cells were detected using a biotin-conjugated sheep polyclonal antibody (1 : 500, Maine Biotechnology Services, Portland, ME, USA). Other antibodies were Brn3 (Santa Cruz SC-6062, Dallas, TX, USA), Cone-arrestin (CM Craft and X Zhu, University South California), E2f2 (Santa Cruz SC-9967), E2f3 (Upstate Biotechnology 05–551, Lake Placid, NY, USA), GFP (Abcam, Toronto, ON, Canada, ab6662 and Molecular Probe, Life Technologies, CA, USA, A11122), Ki67 (BD science Pharmingen 550609, San Jose, CA, USA), M opsin (CM Craft and X Zhu, University South California), p53(NCL-p53-CM5p; Novocastra, Leica Microsystems Inc., Concord, ON, Canada), Phosphohistone H3 (Upstate Biotechnology 06–570), Protein kinase C α (Sigma, P5704, Oakville, ON, Canada), Rhodopsin (R. Molday, University British Columbia), S-antigen (PA Hargrave, University Florida), S opsin (CM Craft and X Zhu, University South California) and Trβ2 (D Forrest, NIH). Antigen retrieval was performed by boiling sections in citric acid solution for 15 min.25 TUNEL was performed as described.25 Briefly, sections were incubated for 1 h at 37 °C with 75 μl of mixture solution consisting of 0.5 μl of terminal deoxynucleotide transferase (TdT), 1 μl of biotin-16-dUTP, 7.5 μl of CoCl2, 15 μl of 5 × TdT buffer and 51 μl of distilled water. After three washes in 4 × SSC buffer, sections were incubated with Alexa-488 or Alexa-568−streptavidin (1 : 1000; Molecular Probes) for 1 h at room temperature. Primary antibodies or labeled cells were visualized using donkey anti-mouse, donkey anti-rabbit and donkey anti-goat antibodies conjugated with Alexa-488, Alexa-568 or Alexa-647, and Streptavidin Alexa-488, Alexa-568 or Alexa-647 (1 : 1000; Molecular Probes). Nuclei were counter-stained with 4′, 6-diamidino-2-phenyindole (DAPI; Sigma). Labeled cells were visualized using a Zeiss (Carl-Zeiss-Strasse, Oberkochen, Germany) Axioplan-2 microscope with Plan Neofluar objectives and images captured with a Zeiss AxionCam camera. For double-labeled samples, confocal images were obtained with a Zeiss LSM 5.0 Laser Scanning microscope. For quantification of stained sections, the retina was separated into bins. Measurements were performed with Axiovison software. Quantification used horizontal sections containing the optic nerve. At least three sections per eye and three eyes from different litters were counted.
RT–qPCR
Total RNA from dissected peripheral retina was prepared using RNeasy Mini kit (Qiagen, Toronto, ON, Canada), and treated with DNA-free (Ambion, Life Technologies, CA, USA) according to the manufacturer's instructions. First-strand cDNA was synthesized from 0.2–0.5 μg using SuperScript II (Invitrogen, Life Technologies); Primers are listed in Table 1. An Applied Biosystems (Life Technologies, CA, USA) PRISM 7900HT and SYBR Green PCR master mix was used for real-time PCR. Briefly, master stocks were prepared such that each 10 μl reaction contained 5 μl SYBR Green PCR master mix, 0.1 μl of each forward and reverse primer (stock 50 μM), 0.8 μl blue H2O (0.73% blue food color, McCormick, Canada) and 2 μl of diluted cDNA template and 2 μl of yellow H2O (0.73% yellow food coloring). PCR consisted of 40 cycles of denaturation at 95 °C for 15 s and annealing and extension at 55 °C for 30 s. An additional cycle (95 °C, 15 s, 60 °C) generated a dissociation curve to confirm a single product. The cycle quantity required to reach a threshold (Qt) in the linear range was determined and compared with a standard curve for each primer set generated by five three-fold dilutions of genomic DNA or cDNA samples of known concentration. Tests were run in duplicate on three biological samples. Values were normalized to β-actin.
Table 1. RT–qPCR primers.
| Genes | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| Actb | ACCACCACAGCTGAGAGGGA | GCCATCTCCTGCTCGAAGTC |
| Aspp1 | AAGACAAAAGCCCTGACGTG | GATGGTTTGAGGCTTTCCAA |
| Atm | GGTGGACAGGTGAACTTGCT | GTCACACCCAAGCTTTCCAT |
| Cdkn1a | GTGGCCTTGTCGCTGTCTT | GCGCTTGGAGTGATAGAAATCTG |
| Chk1 | GAAGCTCAGCGATGTTGTGA | TTCACCAGGAATGTGCAGAG |
| Chk2 | TGTGGCCCACTCCTTTAATC | CCTGGCTATCCTGGAACTCA |
| E2f1 | CTGCAGCAACTGCAGGAGAG | CTCCGAAAGCAGTTGCAGC |
| E2f2 | ACGGCGCAACCTACAAAGAG | GTCTGCGTGTAAAGCGAAGT |
| E2f3 | GGTCCTGGATCTGAACAAGGC | CCTTCCAGCACGTTGGTGAT |
| Gadd45 | GGAAGCTGCGAGAAAAGAGA | TGAAAGTAACCTGGCCATCC |
| Mcm3 | GCTGTGTCCTGCGTTTGTTA | CAACCTTGTCATCTGCCTGA |
| Mdm2 | CTGGCTTCCAGACGATAAGG | TGTCAGCTTTTTGCCATCAG |
| Noxa | TGCCAAAGGTTGTTGTCAAG | AAAGCAATCCCAAACGACTG |
| Npm | GGAACTCCACCCTTTACTTGG | CCATTTCCACATCTGACCACT |
| p19arf | CGCAGGTTCTTGGTCACTGT | TGTTCACGAAAGCCAGAGCG |
| Pidd | GGTACAGGCCTTGGAACAGA | TGCTGTCCTGGTACTTGTGG |
| Puma | GGGGGTCTGTGAAGAGCATA | CTGGGCACTGGGTTAAGAAG |
| Siva | CGACGATGGTGAGAAGACAC | TGAACATCTTGTGCTGTCTGTG |
| Tk1 | GATTGCCAAGATGCCTCAAT | ACCAGTGTCACAATGCTGGA |
| Tradd | ACTGTCGGGCACTGAGAGAT | TGGAAGGCCTGCTCGTATAG |
| Trp53 | TTGGACCCTGGCACCTACAAT | GGCTTTGCAGAATGGAAGGAA |
| Trp73 | GCCCATCAAAGAGGAGTTCA | TCCCACTTCCAAGAGCAGTT |
| Ulf | TTGTGCGGAAGACATTTGAA | TTTGTCCCGCATTATGTCAA |
Western blotting
For western blotting, peripheral mouse retinas were homogenized with a 30-gauge needle 5–10 times in 1 × cell lysis buffer (Cell Signaling, Danvers, MA, USA) 0.1 mM PMSF, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 2 mM NaN3, 1 μM Trichostatin A and 5 mM Niacinamide. Proteins were separated by 12% SDS-PAGE, transferred to nitrocellulose and analyzed by Li-Cor system (Lincoln, NE, USA) with antibodies against acetylated p53 (ab61241, Abcam), Active caspase 3 (9662; Cell Signaling), Aspp1 (ab71163; Abcam), Aspp2(ab36004; Abcam), ATM(2873S, Cell Signaling), p-ATM-S1981(5883p, Cell Signaling), ATR (2790S, Cell Signaling), p-ATR-S428(2853s, Cell Signaling), β-actin (A5441, Sigma), Chk1(ab47444, Abcam), p-Chk1-S317 (2344L, Cell Signalling), Chk2(3440S, Cell Signalling), p-Chk2-T68 (2197p, Cell Signalling), E2f2 (SC-633; Santa Cruz), γ-H2ax (05–636, Millipore, Billerica, MA, USA), HDAC1 (H-51; Santa Cruz), Mdm2 (K-20; Santa Cruz), Npm1/B23 (B0556; Sigma), p19Arf (NB200-106; Novus Biologicals, Oakville, ON, Canada), p53 (NCL-p53-CM5p; Novocastra), phosphorylated p53 (9286S, Cell Signalling), Pcaf (Sc 8999, Santa Cruz), Sirt1 (07–131, Upstate), Tip60 (ab23886, Abcam), Tradd (Sc7868; Santa Cruz) and Ulf/Trip12 (A301-814A; Bethyl laboratories, Montgomery, TX, USA).
In vivo electroporation
Cre-recombinase cDNA (L. van Parijs) was cloned into the BglII site of Mxie plasmid to obtain Mxie-GFP-Cre plasmids. CMV-E2f2 plasmids were from G Leone.59 P0 pups were anesthetized on ice. Two microliter of DNA (1–4 μg/μl) was injected into the subretinal space of P0 pups using a Hamilton syringe, and square electric pulses (80 V; five 50-ms with 950-ms intervals) applied with tweezer-type electrodes (CUY21 EDIT Electroporator).
Statistics
Multiple genotypes were assessed by analysis of variance then Tukey honestly significant difference test (http://www.xlstat.com). Paired genotypes were assessed by Student t-test.
Online supplementary material
Supplementary Figure S1 shows more information to support the idea that E2f1 drives most ectopic division and cell death, but E2f2 drives cone death. Supplementary Figure S2 shows E2f3 expression and Cre-mediated recombination. Supplementary Figure S3 shows Rb/p107 null cones die even when both E2f1 and E2f3 are removed. Supplementary Figure S4 shows RT-qPCR and western blot analysis of some genes including Puma, Pidd, Gadd45, Atm, Chk1, Chk2, Mdm2, Npm, Tradd and Ulf. Supplementary Figure S5 indicates that deleting p73 or p53 does not rescue Rb-null or Rb/p107 null ganglion, bipolar or rod neurons.
Acknowledgments
We thank G Leone and L van Parijs for plasmids; CM Craft, PA Hargrave, T Mak and R Molday for antibodies; G Leone and F McKeon for mice and P Monnier and R Slack for comments on the manuscript. This work was supported by grants from the Canadian Institutes for Health Research and the Foundation Fighting Blindness Canada to RB.
Author contributions
DC and RB designed the study and interpreted data. DC performed all the experiments. YC aided DC in Figure 4, Figure 5, Supplementary Figure S5. DF contributed reagents. DC and RB wrote the paper and all authors contributed to editing.
Glossary
- AC3
active caspase 3
- Brdu
bromodeoxyuridine
- DAPI
4′,6-diamidino-2-phenyindole
- DKO
double knockout
- E14
embryonic day 14
- E2f
adenovirus E2 promoter binding factor
- GCL
ganglion cell layer
- INL
inner nuclear layer
- IPL
inner plexiform layer
- KO
knockout
- ONL
outer nuclear layer
- OPL
outer plexiform layer
- P0
postnatal day 0
- PH3
phosphohistone H3
- PKCα
protein kinase Cα
- QKO
quadruple knockout
- Rb
retinoblastoma
- Tradd
tumor necrosis factor receptor-1 associated death domain
- Trβ2
thyroid hormone receptor β2
- TUNEL
terminal dUTP nick-end labeling
- TKO
triple knockout
- Ulf
ubiquitin ligase for Arf
- WT
wild-type
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Edited by BD Dynlacht
Supplementary Material
References
- Pacal M, Bremner R. Insights from animal models on the origins and progression of retinoblastoma. Curr Mol Med. 2006;6:759–781. doi: 10.2174/1566524010606070759. [DOI] [PubMed] [Google Scholar]
- DeGregori J, Johnson DG. Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr Mol Med. 2006;6:739–748. doi: 10.2174/1566524010606070739. [DOI] [PubMed] [Google Scholar]
- Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9:785–797. doi: 10.1038/nrc2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SY, Opavsky R, Sharma N, Wu L, Naidu S, Nolan E, et al. Mouse development with a single E2F activator. Nature. 2008;454:1137–1141. doi: 10.1038/nature07066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Levine AJ. p53 and E2F-1 cooperate to mediate apoptosis. Proc Natl Acad Sci USA. 1994;91:3602–3606. doi: 10.1073/pnas.91.9.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalik TF, DeGregori J, Schwarz JK, Nevins JR. E2F1 overexpression in quiescent fibroblasts leads to induction of cellular DNA synthesis and apoptosis. J Virol. 1995;69:2491–2500. doi: 10.1128/jvi.69.4.2491-2500.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin XQ, Livingston DM, Kaelin WG, Jr., Adams PD. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci USA. 1994;91:10918–10922. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci USA. 1997;94:7245–7250. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigo E, Muller H, Prosperini E, Hateboer G, Cartwright P, Moroni MC, et al. CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol Cell Biol. 1999;19:6379–6395. doi: 10.1128/mcb.19.9.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni MC, Hickman ES, Denchi EL, Caprara G, Colli E, Cecconi F, et al. Apaf-1 is a transcriptional target for E2F and p53. Nat Cell Biol. 2001;3:552–558. doi: 10.1038/35078527. [DOI] [PubMed] [Google Scholar]
- Pierce AM, Gimenez-Conti IB, Schneider-Broussard R, Martinez LA, Conti CJ, Johnson DG. Increased E2F1 activity induces skin tumors in mice heterozygous and nullizygous for p53. Proc Natl Acad Sci USA. 1998;95:8858–8863. doi: 10.1073/pnas.95.15.8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson QX, McArthur MJ, Johnson DG. E2F3a stimulates proliferation, p53-independent apoptosis and carcinogenesis in a transgenic mouse model. Cell Cycle. 2006;5:184–190. doi: 10.4161/cc.5.2.2307. [DOI] [PubMed] [Google Scholar]
- Chen Q, Hung FC, Fromm L, Overbeek PA. Induction of cell cycle entry and cell death in postmitotic lens fiber cells by overexpression of E2F1 or E2F2. Invest Ophthalmol Vis Sci. 2000;41:4223–4231. [PubMed] [Google Scholar]
- Chen Q, Liang D, Yang T, Leone G, Overbeek PA. Distinct capacities of individual E2Fs to induce cell cycle re-entry in postmitotic lens fiber cells of transgenic mice. Dev Neurosci. 2004;26:435–445. doi: 10.1159/000082285. [DOI] [PubMed] [Google Scholar]
- Ebelt H, Hufnagel N, Neuhaus P, Neuhaus H, Gajawada P, Simm A, et al. Divergent siblings: E2F2 and E2F4 but not E2F1 and E2F3 induce DNA synthesis in cardiomyocytes without activation of apoptosis. Circ Res. 2005;96:509–517. doi: 10.1161/01.RES.0000159705.17322.57. [DOI] [PubMed] [Google Scholar]
- Denchi EL, Helin K. E2F1 is crucial for E2F-dependent apoptosis. EMBO Rep. 2005;6:661–668. doi: 10.1038/sj.embor.7400452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai KY, Hu Y, Macleod KF, Crowley D, Yamasaki L, Jacks T. Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol Cell. 1998;2:293–304. doi: 10.1016/s1097-2765(00)80274-9. [DOI] [PubMed] [Google Scholar]
- Ziebold U, Reza T, Caron A, Lees JA. E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes Dev. 2001;15:386–391. doi: 10.1101/gad.858801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra HI, Wu L, de Bruin A, Timmers C, Rosol TJ, Weinstein M, et al. Specificity of E2F1, E2F2, and E2F3 in mediating phenotypes induced by loss of Rb. Cell Growth Differ. 2002;13:215–225. [PubMed] [Google Scholar]
- Wu L, De Bruin A, Saavedra HI, Starovic M, Trimboli A, Yang Y, et al. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature. 2003;421:942–947. doi: 10.1038/nature01417. [DOI] [PubMed] [Google Scholar]
- Robanus-Maandag EC, Van der Valk M, Vlaar M, Feltkamp C, O'Brien J, Van Roon M, et al. Developmental rescue of an embryonic-lethal mutation in the retinoblastoma gene in chimeric mice. EMBO J. 1994;13:4260–4268. doi: 10.1002/j.1460-2075.1994.tb06746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Livne-Bar I, Vanderluit JL, Slack RS, Agochiya M, Bremner R. Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death-resistant cell-of-origin in retinoblastoma. Cancer Cell. 2004;5:539–551. doi: 10.1016/j.ccr.2004.05.025. [DOI] [PubMed] [Google Scholar]
- MacPherson D, Sage J, Kim T, Ho D, McLaughlin ME, Jacks T. Cell type-specific effects of Rb deletion in the murine retina. Genes Dev. 2004;18:1681–1694. doi: 10.1101/gad.1203304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Opavsky R, Pacal M, Tanimoto N, Wenzel P, Seeliger MW, et al. Rb-mediated neuronal differentiation through cell-cycle-independent regulation of E2f3a. PLoS Biol. 2007;5:e179. doi: 10.1371/journal.pbio.0050179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan KA, Ruzhynsky VA, Douda DN, Vanderluit JL, Ferguson KL, Chen D, et al. Unique requirement for Rb/E2F3 in neuronal migration: evidence for cell cycle-independent functions. Mol Cell Biol. 2007;27:4825–4843. doi: 10.1128/MCB.02100-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Liang P, Leng R, Guo Z, Liu Y, Liu X, et al. E2F1 and p53 are dispensable, whereas p21(Waf1/Cip1) cooperates with Rb to restrict endoreduplication and apoptosis during skeletal myogenesis. Dev Biol. 2000;227:28–41. doi: 10.1006/dbio.2000.9892. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zacksenhaus E. E2F1 mediates ectopic proliferation and stage-specific p53-dependent apoptosis but not aberrant differentiation in the ocular lens of Rb deficient fetuses. Oncogene. 2000;19:6065–6073. doi: 10.1038/sj.onc.1203996. [DOI] [PubMed] [Google Scholar]
- Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG, Jr, Livingston DM, et al. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell. 1996;85:549–561. doi: 10.1016/s0092-8674(00)81255-6. [DOI] [PubMed] [Google Scholar]
- Biswas AK, Johnson DG. Transcriptional and nontranscriptional functions of E2F1 in response to DNA damage. Cancer Res. 2012;72:13–17. doi: 10.1158/0008-5472.CAN-11-2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez LA, Goluszko E, Chen HZ, Leone G, Post S, Lozano G, et al. E2F3 is a mediator of DNA damage-induced apoptosis. Mol Cell Biol. 2010;30:524–536. doi: 10.1128/MCB.00938-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallstrom TC, Nevins JR. Jab1 is a specificity factor for E2F1-induced apoptosis. Genes Dev. 2006;20:613–623. doi: 10.1101/gad.1345006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick FA, Dyson N. pRB contains an E2F1-specific binding domain that allows E2F1-induced apoptosis to be regulated separately from other E2F activities. Mol Cell. 2003;12:639–649. doi: 10.1016/s1097-2765(03)00344-7. [DOI] [PubMed] [Google Scholar]
- Opavsky R, Tsai SY, Guimond M, Arora A, Opavska J, Becknell B, et al. Specific tumor suppressor function for E2F2 in Myc-induced T-cell lymphomagenesis. Proc Natl Acad Sci USA. 2007;104:15400–15405. doi: 10.1073/pnas.0706307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogoff HA, Pickering MT, Frame FM, Debatis ME, Sanchez Y, Jones S, et al. Apoptosis associated with deregulated E2F activity is dependent on E2F1 and Atm/Nbs1/Chk2. Mol Cell Biol. 2004;24:2968–2977. doi: 10.1128/MCB.24.7.2968-2977.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogal V, Kartasheva NN, Trigiante G, Llanos S, Yap D, Vousden KH, et al. ASPP1 and ASPP2 are new transcriptional targets of E2F. Cell Death Differ. 2005;12:369–376. doi: 10.1038/sj.cdd.4401562. [DOI] [PubMed] [Google Scholar]
- Sangwan M, McCurdy SR, Livne-Bar I, Ahmad M, Wrana JL, Chen D, et al. Established and new mouse models reveal E2f1 and Cdk2 dependency of retinoblastoma, and expose effective strategies to block tumor initiation. Oncogene. 2012;31:5019–5028. doi: 10.1038/onc.2011.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Pacal M, Wenzel PL, Knoepfler PS, Leone G, Bremner R. Division and apoptosis of E2f-deficient retinal progenitors. Nature. 2009;462:925. doi: 10.1038/nature08544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng L, Hurley JB, Dierks B, Srinivas M, Salto C, Vennstrom B, et al. A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat Genet. 2001;27:94–98. doi: 10.1038/83829. [DOI] [PubMed] [Google Scholar]
- Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet. 2010;11:273–284. doi: 10.1038/nrg2717. [DOI] [PubMed] [Google Scholar]
- Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, et al. p14ARF links the tumour suppressors RB and p53. Nature. 1998;395:124–125. doi: 10.1038/25867. [DOI] [PubMed] [Google Scholar]
- Chen D, Shan J, Zhu WG, Qin J, Gu W. Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nature. 2010;464:624–627. doi: 10.1038/nature08820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio II, Sasaki M, Ghazarian D, Moreno J, Done S, Ueda T, et al. TRADD contributes to tumour suppression by regulating ULF-dependent p19Arf ubiquitylation. Nat Cell Biol. 2012;14:625–633. doi: 10.1038/ncb2496. [DOI] [PubMed] [Google Scholar]
- Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404:99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- Phillips AC, Ernst MK, Bates S, Rice NR, Vousden KH. E2F-1 potentiates cell death by blocking antiapoptotic signaling pathways. Mol Cell. 1999;4:771–781. doi: 10.1016/s1097-2765(00)80387-1. [DOI] [PubMed] [Google Scholar]
- Bremner R. Retinoblastoma, an inside job. Cell. 2009;137:992–994. doi: 10.1016/j.cell.2009.05.034. [DOI] [PubMed] [Google Scholar]
- Xu XL, Fang Y, Lee TC, Forrest D, Gregory-Evans C, Almeida D, et al. Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell. 2009;137:1018–1031. doi: 10.1016/j.cell.2009.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corson TW, Gallie BL. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosomes Cancer. 2007;46:617–634. doi: 10.1002/gcc.20457. [DOI] [PubMed] [Google Scholar]
- Elison JR, Cobrinik D, Claros N, Abramson DH, Lee TC. Small molecule inhibition of HDM2 leads to p53-mediated cell death in retinoblastoma cells. Arch Ophthalmol. 2006;124:1269–1275. doi: 10.1001/archopht.124.9.1269. [DOI] [PubMed] [Google Scholar]
- Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–66. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- Conkrite K, Sundby M, Mu D, Mukai S, MacPherson D. Cooperation between Rb and Arf in suppressing mouse retinoblastoma. J Clin Invest. 2012;122:1726–1733. doi: 10.1172/JCI61403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K, Yang Y. Cell cycle regulation in the postmitotic neuron: oxymoron or new biology. Nat Rev Neurosci. 2007;8:368–378. doi: 10.1038/nrn2124. [DOI] [PubMed] [Google Scholar]
- Wu J, Trogadis J, Bremner R. Rod and cone degeneration in the rd mouse is p53 independent. Mol Vis. 2001;7:101–106. [PubMed] [Google Scholar]
- Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell. 2001;105:43–55. doi: 10.1016/s0092-8674(01)00295-1. [DOI] [PubMed] [Google Scholar]
- Vooijs M, Berns A. Developmental defects and tumor predisposition in Rb mutant mice. Oncogene. 1999;18:5293–5303. doi: 10.1038/sj.onc.1202999. [DOI] [PubMed] [Google Scholar]
- LeCouter JE, Kablar B, Hardy WR, Ying C, Megeney LA, May LL, et al. Strain-dependent myeloid hyperplasia, growth deficiency, and accelerated cell cycle in mice lacking the Rb-related p107 gene. Mol Cell Biol. 1998;18:7455–7465. doi: 10.1128/mcb.18.12.7455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone G, Sears R, Huang E, Rempel R, Nuckolls F, Park CH, et al. Myc requires distinct E2F activities to induce S phase and apoptosis. Mol Cell. 2001;8:105–113. doi: 10.1016/s1097-2765(01)00275-1. [DOI] [PubMed] [Google Scholar]
- Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, et al. The E2F1-3 transcription factors are essential for cellular proliferation. Nature. 2001;414:457–462. doi: 10.1038/35106593. [DOI] [PubMed] [Google Scholar]
- Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.