

Summary
We had previously reported that RBEL1A, a novel Ras-like GTPase, was overexpressed in multiple human malignancies and that its depletion suppressed cell growth. However, the underlying molecular mechanism remained to be elucidated. Here we report that depletion of endogenous RBEL1A results in p53 accumulation due to increased p53 half-life whereas increased expression of RBEL1A reduces p53 levels under unstressed and genotoxic stress conditions. RBEL1A directly interacts with p53 and MDM2, and strongly enhances MDM2-dependent p53 ubiquitylation and degradation. We also found that RBEL1A modulation of p53 ubiquitylation by MDM2 does not depend on its GTPase activity. We have also defined the p53 oligomeric domain and RBEL1A GTPase domain to be the crucial regions for p53–RBEL1A interactions. Importantly, we have found that RBEL1A strongly interferes with p53 transactivation function; thus our results indicate that RBEL1A appears to function as a novel p53 negative regulator that facilitates MDM2-dependent p53 ubiquitylation and degradation.
Key words: GTPase; MDM2; p53 tumor suppressor; Ubiquitylation, Ras superfamily; GTPase
Introduction
p53 is a key tumor suppressor that is frequently inactivated by mutations, deletions or by other mechanisms such as protein degradation in a variety of human malignancies. p53 has proven itself to be one of the most important molecules implicated in various key cellular processes including cell cycle, apoptosis, senescence and DNA repair (Zilfou and Lowe, 2009). It predominantly functions as a transcription factor containing a N-terminal transactivation domain, a central DNA-binding domain and a C-terminal oligomerization domain. In response to cellular stresses, p53 binds to its regulatory elements in homotetrameric configuration and regulates the expression of its downstream target genes whose products are important in controlling cell cycle and apoptosis (Zilfou and Lowe, 2009).
p53 is primarily regulated at the protein level through the ubiquitin–proteasome pathway. In unstressed cells, p53 protein is maintained at low levels via rapid protein degradation by ubiquitin ligases. A number of p53 E3 ligases have been identified including MDM2, COP1 and Pirh2 among others (Corcoran et al., 2004). MDM2 is one of the most important regulators of p53. MDM2 is a RING domain E3 ligase that ubiquitylates and degrades p53. Under genotoxic conditions, p53 is quickly stabilized and transcriptionally regulates a number of target genes, such as p21 and Puma that are involved in cell cycle regulation and apoptosis. DNA damage is thought to promote p53 phosphorylation through various kinases and p53 phosphorylation disrupts its interaction with MDM2 thereby preventing its degradation (Jiang et al., 2010; Meek and Anderson, 2009).
We have recently reported the identification and characterization of a novel Rab-like GTP-binding protein named RBEL1A (Montalbano et al., 2007; Montalbano et al., 2009). RBEL1A protein harbors a N-terminal Ras/Rab-like GTPase domain followed by a GTP-binding regulatory domain, a proline-rich region and a C-terminal nuclear localization signal (Montalbano et al., 2009). RBEL1A protein is predominantly GTP bound and functions as a GTPase. RBEL1A was found to be overexpressed in big portion of primary breast (67%) and colon (47%) cancer samples when compared to matching normal tissues (Montalbano et al., 2007). Furthermore, RBEL1A knockdown triggered severe growth suppression, cell death and activation of caspase 3 in various cancer cell lines suggesting that the expression of RBEL1A appeared to be important for maintaining cell growth and survival (Montalbano et al., 2009). RBEL1A is novel protein and the molecular mechanisms via which it regulates cell growth and survival remain to be fully elucidated.
In the present study, we demonstrate that RBEL1A plays an important role in facilitating MDM2-mediated p53 ubiquitylation. RBEL1A directly interacts with both MDM2 and p53 proteins and enhances MDM2-dependent p53 ubiquitylation and degradation. RNAi-mediated knockdown of RBEL1A prolongs p53 half-life which partly accounts for increased p53 protein levels. RBEL1A also inhibits transactivation potential of p53 and suppresses p53 activation in response to DNA damage induced by UV radiation. Together, our results provide mechanistic insights into the function of RBEL1A and demonstrate that RBEL1A is an important novel negative regulator of p53.
Results
RBEL1A negatively regulates p53 protein levels and depletion of RBEL1A prolongs p53 protein half-life
We have previously shown that RBEL1A is overexpressed in human primary breast (67%) and colon cancers (47%) and that RBEL1A suppression via shRNA knockdown triggers cell death and growth suppression in human cancer cells (Montalbano et al., 2007; Montalbano et al., 2009). To further study the function of RBEL1A and to investigate how elevated levels of RBEL1A would affect cellular functions, we examined the effect of exogenous RBEL1A on a number of tumor-related proteins; results presented in Fig. 1 indicate that increased expression of RBEL1A significantly reduced the protein expression levels of p53, p21, PUMA (left panel) but had no effect on p85 (PI3-K subunit), ERK1/2 and cyclin D3 (right panel). Because p21, PUMA and MDM2 are downstream targets of p53, we suspected that RBEL1A may negatively affect p53 and its function. We therefore examined the effect of RBEL1A knockdown (KD) on p53 protein expression. Fig. 1B left middle panel shows that RBEL1A protein expression was sufficiently depleted by the expression of lentiviral-mediated RBEL1A shRNA that was used in our previous studies (Montalbano et al., 2009). And it is of note that our previous studies have shown that RBEL1A is a glycosylated protein (Montalbano et al., 2007; Montalbano et al., 2009) and it exhibits multiple band patterns ∼100–110 kDa and 125–130 kDa on the western blotting depending on the degree of glycosylation of the protein. Importantly, we found that knockdown of RBEL1A in these cells led to increased p53 protein expression. We further examined whether RBEL1A KD alters p53 protein half-life and the results presented in Fig. 1C,D indicate that depletion of RBEL1A significantly increased p53 protein half-life from ∼15 to ∼73 minutes; and that was concurrent with increased protein expression of p53 downstream target genes p21 and PUMA (Fig. 1B). Conversely, overexpression of RBEL1A resulted in decreased p21 and PUMA mRNA levels (Fig. 1E). To study the mechanism via which RBEL1A affected p53 protein stability, MCF-7 cells, stably expressing exogenous RBEL1A or vector-only, were treated with MG132 (proteasome inhibitor) and Nutlin-3 (MDM2-specific inhibitor) independently and p53 expression was then analyzed. Fig. 1F shows that RBEL1A-mediated decline in p53 protein levels was clearly abrogated in cells treated with MG132 or Nutlin-3 (compare lane 2 with lanes 4 and 6). It is also noted that RBEL1A overexpression had minimal effect on p53 transcript levels (Fig. 1E, panel 1). These results suggest that RBEL1A-mediated downregulation of p53 appears to involve MDM2 and proteasome-dependent mechanism.
Fig. 1.

RBEL1A negatively regulates expression of p53 and its target genes p21 and PUMA. (A) Increased RBEL1A expression is associated with reduced p53 protein levels in RBEL1A-overexpressing cells. Western blot analyses showing the expressions of p53, Puma, p21, MDM2, Erk1/2, PI3K, cyclin D3, HA–RBEL1A in cells stably overexpressing RBEL1A or empty vector. β-actin served as loading control. All results shown in the left panel and those in the top right panel were from the same membrane; images in the right bottom panel were from the duplicate membrane using the same lysates. (B) RBEL1A knockdown increases p53 levels and expression of its target genes. MCF-7 cells were infected (MOI = 0.3) with lentivirus carrying either scrambled shRNA or RBEL1A-specific shRNA. Ten days post-infection, cells were harvested and western blot analyses were performed using the indicated antibodies. β-actin was also detected on the same membrane as a loading control. (C) RBEL1A knockdown increases p53 protein half-life. MCF-7 cells infected with lentivirus-mediated RBEL1A shRNA or with the scramble shRNA (10 days) were treated with cycloheximide (50 µg/ml) and harvested at various times as indicated. The RBEL1A, p53 and β-actin protein signals were detected by western blotting on the same membrane. (D) The values of relative intensity were obtained based on the densitometry measurements of p53 and β-actin levels on the western blotting shown in C. (E) Results of real-time qPCR of p53, p21 and Puma transcripts in RBEL1A-overexpressing and control cells. Real-time qPCR assays were performed as described in the Materials and Methods. The data are from three independent experiments in which each sample was tested in triplicate. **P<0.01 as determined by a t-test. (F) RBEL1A-mediated p53 downregulation is blocked by the proteasome inhibitors MG132 and Nutlin-3. Cells stably overexpressing RBEL1A or empty vector (control) were either not treated or treated with proteasome inhibitor MG132 (10 µM) or Nutlin-3 (10 µM) for 5 hours. The expression levels of endogenous p53 and β-actin were detected on the same membrane. The numbers below the p53 panel indicate the relative intensity of p53 expression.
RBEL1A directly interacts with both p53 and MDM2
Next, we sought to investigate the mechanism via which RBEL1A modulates p53 protein and to that end, we first determined whether RBEL1A interacts with p53. Results shown in Fig. 2A indicate that exogenously expressed RBEL1A and p53 mutually interact with each other; and the endogenous RBEL1A also interacted with the endogenous p53 (Fig. 2B). To determine whether RBEL1A directly interacts with p53, we utilized the bacterial purified recombinant p53 and purified S-tagged RBEL1A to perform protein pull-down assays. Results presented in Fig. 2C indicate that the purified RBEL1A and p53 proteins directly interacted with each other and thus, RBEL1A and p53 interactions are direct in nature.
Fig. 2.
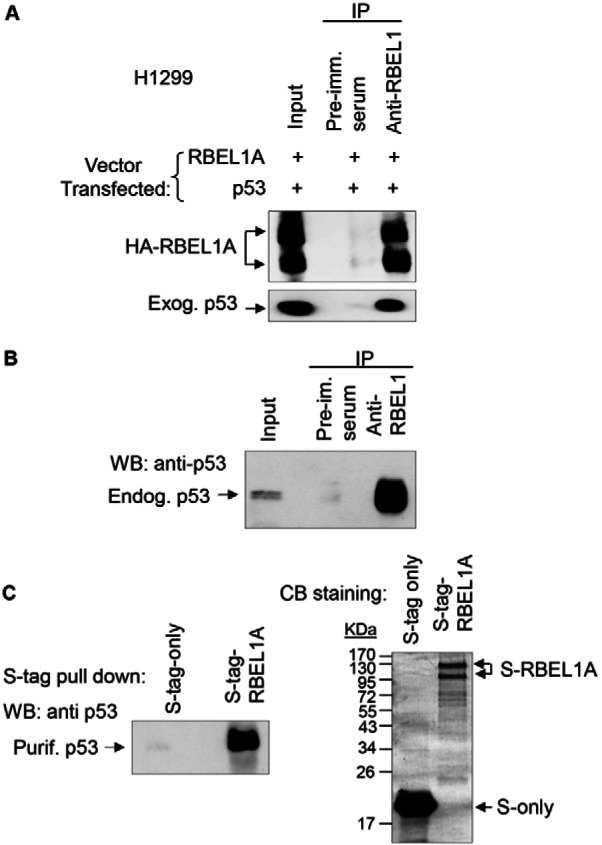
RBEL1A interacts with p53. (A) H1299 cells were transiently transfected with RBEL1A and p53 expression constructs. Twenty-four hours post-transfections, cells were lysed and immunoprecipitations were performed using anti-RBEL1 antibody or pre-immune serum as negative control. The protein precipitants were analyzed by immunoblotting using anti-p53 antibodies. (B) Endogenous RBEL1A co-immunoprecipitates with endogenous p53. Endogenous RBEL1A was immunoprecipitated using anti-RBEL1 antibodies; pre-immune serum was used as a negative control. RKO cell lysates were used in these experiments. The cell lysate input (5%) and the interaction between RBEL1A and p53 were detected by western blotting using anti-p53 antibodies. (C) Recombinant purified p53 directly interacts with recombinant purified S-tagged RBEL1A protein. Recombinant bacterial purified p53 protein was incubated with either the recombinant bacterial purified S-protein or the bacterial purified S-tag-RBEL1A protein. Approximately 6 hours after incubation, S-tag pull-down assays were performed and the interactions between p53 and RBEL1A were detected by western blotting using anti-p53 antibodies. Right panel: the Coomassie Blue (CB) staining shows the inputs of S-tagged RBEL1A and S-tag alone proteins used for the pull-down assay.
Aforementioned results indicate that RBEL1A negatively regulates p53 in a proteasome-dependent manner. However, RBEL1A sequence lacks structural motif(s) of E3 ligase suggesting that it is less likely to be an ubiquitin ligase. Therefore, we postulated that RBEL1A might be engaging an E3 ligase to regulate p53 levels. MDM2 is a major E3 ligase for p53 and thus we investigated whether RBEL1A engages MDM2 to regulate p53. Next, we examined whether RBEL1A interacts with MDM2. We found that the bacterial purified GST–MDM2, but not GST alone, pulled down endogenous RBEL1A and endogenous p53 (Fig. 3A). Furthermore, endogenously expressed RBEL1A also co-immunoprecipitated with endogenous MDM2 and p53 (Fig. 3B). To determine whether RBEL1A directly interacts with MDM2, protein pull-down assays were also performed using the purified recombinant S-tagged RBEL1A and GST–MDM2. Results shown in Fig. 3C indicate that the recombinant MDM2 protein was pulled down by the recombinant S-tagged RBEL1A, but not by the S-tag only, confirming the direct interactions between these two proteins. These results strongly indicate that RBEL1A forms a complex with p53 and MDM2.
Fig. 3.
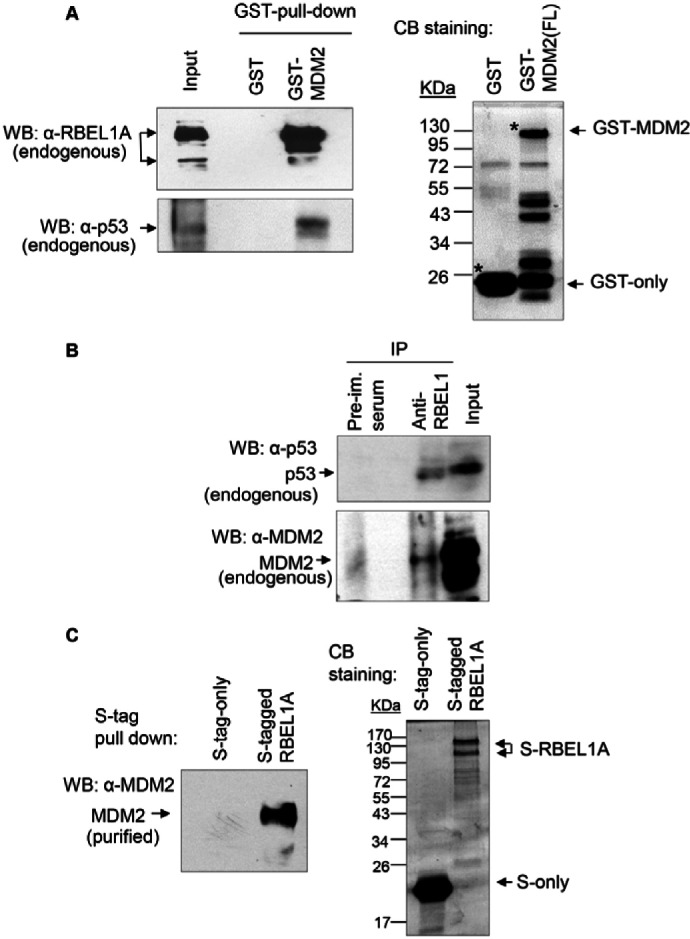
RBEL1A interacts with MDM2. (A) Purified recombinant MDM2 pulls down endogenous RBEL1A and p53. About 25 µg of GST–MDM2 (full-length) or GST control were used to perform GST pull down using MCF7 cell lysates, and the precipitated proteins were analyzed by immunoblottings using RBEL1A- and p53-specific antibodies. Coomasssie Blue staining of GST–MDM2 and GST alone is shown in the right panel. Asterisks indicate the correctly sized purified protein products. (B) Endogenous RBEL1A co-immunoprecipitates with endogenous MDM2 and p53. Endogenous RBEL1A and p53 from RKO cell lysates were immunoprecipitated using anti-RBEL1 antibodies; pre-immune serum was used as a negative control. Interactions of RBEL1A with MDM2 and p53 were detected by western blotting using anti-MDM2 and anti-p53 antibodies. (C) Recombinant purified MDM2 directly interacts with recombinant purified S-tagged RBEL1A protein. The MDM2 protein was incubated with either the S-protein or the S-tagged RBEL1A protein. Approximately 6 hours after incubation, S-tag pull-down assays were performed and the interactions between MDM2 and RBEL1A were detected by immunoblotting using anti-MDM2 antibodies. Right panel: Coomassie Blue staining shows the input of S-tagged RBEL1A and S-tag alone used for the pull-down assay.
RBEL1A-mediated p53 downregulation requires MDM2
To determine the role of MDM2 in RBEL1A-mediated p53 downregulation, we utilized the p53/MDM2 double knockout mouse embryonic fibroblasts (hereafter referred to as MEF2KO). To that end, we used the exogenously expressed wild-type and mutant p53 (L22Q/W23S) that was shown to be defective in interaction with MDM2 (Dobbelstein et al., 1999; Kussie et al., 1996; Lin et al., 1994). As shown in Fig. 4A, p53 was not detected in the MEF2KO cells (lane 1) but detected only when exogenous wild-type p53 was reintroduced (lanes 2–7). Interestingly, expression of exogenous RBEL1A minimally altered wild-type (wt) p53 levels in MDM2-deficient background (compare lanes 2 and 3). Expression of exogenous MDM2 alone led to a modest reduction in the wild-type p53 levels (compare lanes 2 and 4); however, p53 levels were further reduced when RBEL1A and MDM2 were co-expressed (Fig. 4A, compare lane 5 with lane 4). Importantly, our results also show that L22Q/W23S p53 mutant was resistant to MDM2-mediated downregulation even in the presence of exogenous RBEL1A (Fig. 4A, compare lane 6 with 4 and lane 7 with 5, respectively) which suggests that the effect of RBEL1A on p53 depends on MDM2–p53 interaction. Further, that MEF2KO cells exhibit easily detectable levels of endogenous RBEL1A (data not shown) would suggest that exogenous MDM2-only-mediated downregulation of p53 levels (Fig. 4A, lane 4) could involve some contribution from the endogenous RBEL1A.
Fig. 4.

RBEL1A-mediated p53 downregulation requires MDM2. (A) p53−/−/MDM2−/− double knockout MEFs were transfected with the indicated vectors (+). Twenty-four hours post-transfection, cells were harvested and western blot analyses were performed using the indicated antibodies. It is of note that the mutant p53 (L22Q/W23S) is known to be defective in interaction with MDM2 (Kussie et al., 1996). (B,C) RBEL1A downregulates p53 in MCF12A normal human breast cells in an MDM2-dependent manner. MCF12A cells were transiently transfected with the indicated vectors in various combinations. Transfectants were treated with or without Nutlin-3 (10 µM) for 6 hours and cell lysates were analyzed by western blotting using the indicated antibodies. The numbers shown under the p53 western blots indicate the intensity of the p53 signal measured by densitometry and normalized with respect to the intensity of β-actin for each lane.
We further investigated whether RBEL1A modulates p53 in a MDM2-dependent manner in a human mammary cell system. MCF12A normal breast cells were transfected with the exogenous wild-type p53 (GFP-p53), MDM2-resistant mutant p53 (L22Q/W23S), HA-RBEL1A and HA-only control vectors in different combinations. Transfected cells were treated with or without MDM2 inhibitor Nultin-3 and cell lysates were then analyzed. Fig. 4B,C show that overexpressed RBEL1A in MCF12A cells also downregulated exogenous p53 (Fig. 4B) and endogenous p53 (Fig. 4C) and such effect was blocked by Nutlin-3 (Fig. 4B,C). In addition, the p53 mutant (L22Q/W23S) resisted MDM2-mediated downregulation with or without exogenous RBEL1A (Fig. 4B, lanes 5 and 6). Together these results indicate that RBEL1A-mediated p53 downregulation is MDM2 dependent and that RBEL1A functions to facilitate MDM2-mediated modulation of p53.
RBEL1A enhances MDM2-mediated p53 ubiquitylation
Next, we investigated the effect of RBEL1A on MDM2-mediated ubiquitylation of p53 using purified recombinant RBEL1A, p53 and MDM2 proteins employing an in vitro ubiquitylation assay. As shown in Fig. 5A, p53 ubiquitylation was not detected without MDM2, which served as a negative control for this assay (lanes 1–3, both upper and lower panels). p53 was modestly ubiquitylated in the presence of MDM2 without RBEL1A, as noted by (i) the appearance of a light smear on the anti-p53 western blot membrane (lanes 4 and 6, upper panel) and (ii) the anti-ubiquitin-specific signals on the duplicated western blot membrane (lanes 4 and 6, lower panel). Interestingly, RBEL1A alone without adding MDM2 had no effect on p53 ubiquitylation (lane 5). However, p53 ubiquitylation was substantially enhanced when both MDM2 and RBEL1A were present (lane 7). These findings corroborate the aforementioned results indicating that MDM2 by itself is capable of ubiquitylating p53; however, its effect on p53 is considerably enhanced by RBEL1A. Additionally, the effect of RBEL1A on in-cell p53 ubiquitylation (Fig. 5B) is consistent with its effect in in vitro assays (Fig. 5A) further substantiating that increased expression of RBEL1A does indeed enhance intracellular p53 ubiquitylation.
Fig. 5.

RBEL1A enhances MDM2-mediated p53 ubiquitylation. (A) In vitro ubiquitylation of p53. In vitro ubiquitylation assays were performed as described in the Materials and Methods. Purified recombinant p53, GST-tagged MDM2 and S-tagged RBEL1A were incubated with E1/E2/Ubiquitin (Ub) mixture in the indicated combinations (+). The reaction products were analyzed by western blotting using anti-p53 (upper panel) and anti-ubiquitin antibodies (lower panel). (B) RBEL1A increases in-cell p53 ubiquitylation. An in-cell ubiquitylation assay was performed as described in Materials and Methods. RKO cells were transfected with His-tagged ubiquitin vector together with HA-tagged RBEL1A or empty vectors. Twenty-four hours later, cells were treated with MG132 (10 µM) for 6 hours, then proteins were extracted and a His-tag protein pull-down (Ni2+ pull-down) assay was performed to precipitate the ubiquitylated proteins. The precipitants were analyzed by western blotting using p53-specific antibodies to detect the extent of p53 ubiquitylation. The smearing pattern indicates the intensity of poly-ubiquitylation of p53 (upper panel). Western blotting analyses showing inputs of p53 and β-actin from the whole cell lysates are included (middle and lower panels) to show that equal amount of proteins were used in the pull-down assays. (C) Nutlin-3 blocks RBEL1A′s effect on p53 ubiquitylation. MCF-7 cells stably expressing HA-RBEL1A or HA-only vector were treated with MG132 only or MG132 plus Nutlin-3 (both used 10 µM) for 6 hours prior to protein extraction. Immunoprecipitations were then performed using ubiquitin-specific antibodies and the immunoprecipitants were analyzed by western blotting using p53-specific antibodies to detect the extent of p53 ubiquitylation. p53 and β-actin levels from the whole cell lysates were analyzed by western blotting on another membrane to indicate the inputs. The expression of HA–RBEL1A is also shown. (D) RBEL1A knockdown decreases p53 ubiquitylation. MCF-7 cells infected with lentivirus carrying either scrambled shRNA or RBEL1A-specific shRNA were treated with MG132 (10 µM) for 6 hours prior to protein extraction. Immunoprecipitations were then performed using p53-specific antibodies. The immunoprecipitants were analyzed by western blotting using ubiquitin-specific antibodies to detect the extent of p53 ubiquitylation. A shorter exposure of the p53 spots on the same membrane is shown on the left.
We also used MDM2 inhibitor Nutlin-3 to investigate the effect of RBEL1A on p53 ubiquitylation inside the cells. Fig. 5C shows that p53 ubiquitylation was enhanced in the presence of exogenous RBEL1A (compare lane 2 with lane 1) and the effect of RBEL1A on p53 ubiquitylation was strongly inhibited in the presence of Nutlin-3 (compare lane 4 with lane 2). We also analyzed the effect of RBEL1A knockdown on p53 ubiquitylation and our results indicated that depletion of endogenous RBEL1A reduced p53 ubiquitylation inside the cells (Fig. 5D). Collectively, these results demonstrate that RBEL1A enhances p53 ubiquitylation via MDM2-dependent manner.
Mapping of interaction regions on p53 and RBEL1A
Next, we sought to map the interacting regions of p53 and RBEL1A. Fig. 6A shows the schematic illustration of the GST-tagged p53 (full-length or deletion variants). Fig. 6B left panel shows the expression of recombinant p53 proteins (correct size marked by asterisks). Some degradation of the purified p53 proteins is observed as has also been seen in other studies (Buchhop et al., 1997; Hofmann et al., 2002; Sui et al., 2004), but it did not affect their interactions with RBEL1A. As also seen in Fig. 6B (right panel), as expected, the full-length p53 interacted with the purified RBEL1A (lane 3). However, of the deletion variants of p53, only one containing residues 301–393 interacted with RBEL1A protein (Fig. 6B, lane 7) while the other variants devoid of this region did not. These results indicate that the carboxyl terminus of p53 containing residues 301–393 appears to be important for its interaction with RBEL1A.
Fig. 6.
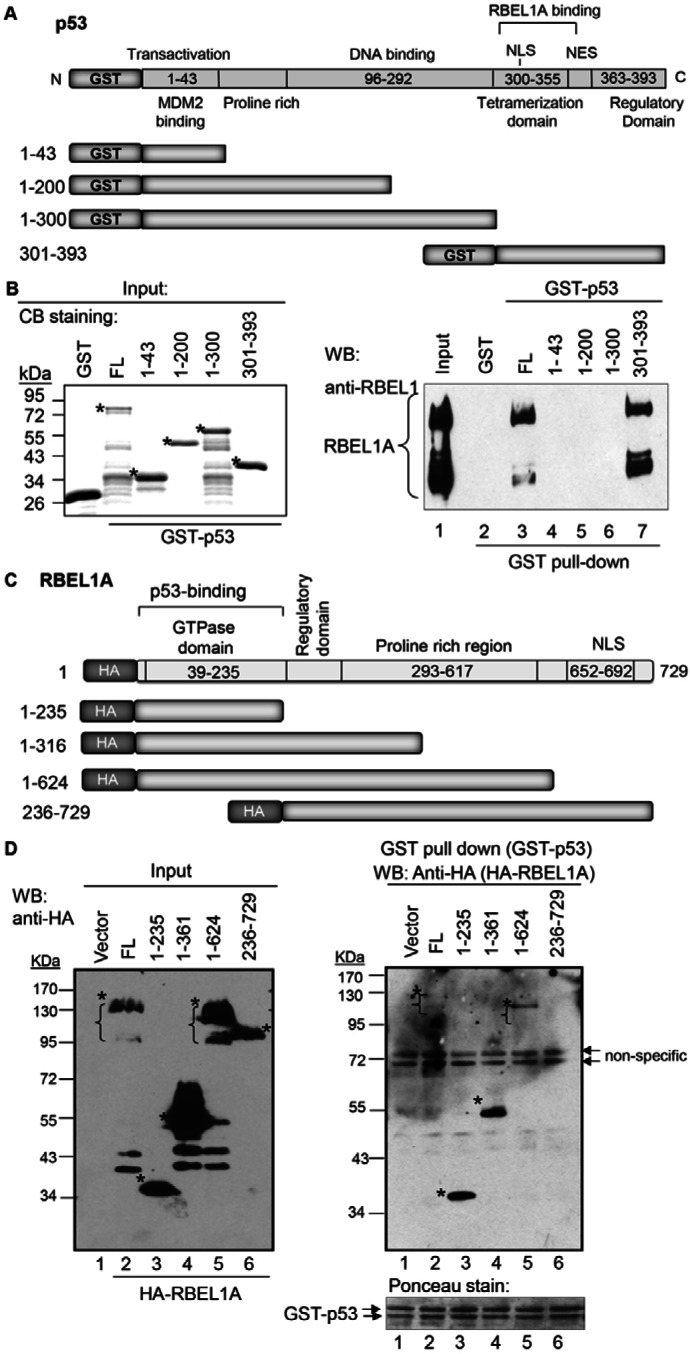
Mapping p53 and RBEL1A interaction domains. (A) A schematic illustration of the full-length p53 protein and various deletion variants. The RBEL1A-binding region is also indicated based on the results shown in B. (B) Left panel: protein expression of recombinant GST-tagged full-length and deletion variants of p53 detected by Coomassie Blue (CB) staining. Asterisks indicate the correctly sized purified protein products. Right panel: mapping of the RBEL1A-interaction region of p53. Purified GST-tagged p53 (full-length and deletion variants) were incubated with RBEL1A recombinant protein lysates and GST pull-down assays were performed as described in Materials and Methods. The pull-down protein products were analyzed by western blotting using anti-RBEL1 antibodies. Approximately 1% input of whole cell lysates was loaded in lane 1. (C) A schematic illustration of the full-length and various deletion variants of RBEL1A protein. The p53 binding region is also indicated based on the results shown in D. (D) Mapping of the p53-interaction region of RBEL1A. Left panel: expression of HA-tagged RBEL1A full-length and deletion variants in HEK293 cells, detected by western blotting using anti-HA antibodies. Asterisks indicate the correctly sized RBEL1A protein products. Right upper panel: purified GST-tagged p53 (wild type) was incubated with lysates from cells expressing the HA-tagged RBEL1A (full-length or deletion variant) vectors. GST pull-down assays were performed as described in Materials and Methods and the pull-down protein products were analyzed by western blotting using anti-HA antibodies. Right lower panel: Ponceau staining of the GST–p53 input in each sample.
Next, we sought to determine the p53-interacting region on RBEL1A. A schematic illustration of a set of deletion variants and full-length (FL) HA-tagged RBEL1A proteins is shown in Fig. 6C. Expression of RBEL1A variants in HEK293T cells was confirmed by WB (Fig. 6D, left panel; correct size indicated by the asterisks). GST pull-down assays were then performed using the purified GST-tagged p53 incubated with HEK293T cell lysates expressing the FL-RBEL1A or its deletion variants. Fig. 6D (right panel) shows that the GST-tagged p53 pulled down the FL-RBEL1A as well as all other RBEL1A deletion variants except the HA–236–729 variant lacking the N-terminal 1–235 residues (right panel, lane 6). These findings indicate that the residues 1–235 are crucial for RBEL1A interaction with p53.
GTP binding is not essential for RBEL1A modulation of p53
Our previous studies demonstrated that RBEL1A is a GTPase (Montalbano et al., 2007; Montalbano et al., 2009), we next investigated whether GTP-binding status of RBEL1A is important for its modulation of p53. For this purpose, we used the wild-type RBEL1A, the mutant RBEL1A T57N and a RBEL1A protein fragment comprising residues 1–235. We have previously demonstrated that (i) wild-type RBEL1A predominantly binds to GTP, (ii) RBEL1A T57N is unable to bind to both GTP or GDP (Montalbano et al., 2007) and (iii) the RBEL1A fragment 1–235 predominantly binds to GDP (Montalbano et al., 2009). HEK293T cells were transfected with above mentioned constructs and cell lysates were utilized for GST protein pull-down assays using the purified GST–p53. Consistent with our previous findings (Montalbano et al., 2007), the mutant RBEL1A T57N was unstable and expressed at much lower levels (Fig. 7A, left panel, lane 3); that is possibly due to its inability to bind nucleotides, which affects the protein stabilization (Montalbano et al., 2007). As shown in Fig. 7A right panel, GST–p53 was able to pull down the RBEL1A T57N and 1–235 mutant proteins. Interestingly, we also consistently noted that even with less input of the mutant T57N protein (because it is less stable), GST–p53 pulled down more RBEL1A T57N mutant protein (Fig. 7A, right panel). These results would suggest that loss of GTP-binding ability of RBEL1A did not inhibit but rather enhanced interactions between RBEL1A and p53. We also determined whether RBEL1A mutants were capable of binding to MDM2. Results presented in supplementary material Fig. S1A,B indicate that GST–MDM2, but not GST protein alone, pulled down both RBEL1A T57N and 1–235 mutant proteins and thus indicated that these RBEL1A variants were capable of interacting with MDM2 protein. Importantly, we also found that both RBEL1A mutants were capable of enhancing p53 ubiquitylation that was blocked by Nutlin-3 (Fig. 7B), suggesting that such effect was MDM2 dependent. In addition, overexpression of RBEL1A mutant variants also mediated reduction in p53 levels (Fig. 7C). Collectively, these findings indicate that GTP binding does not appear to be essential for RBEL1A modulation of p53 ubiquitylation.
Fig. 7.

GTP-binding does not appear to be essential for RBEL1A modulation of p53. (A) RBEL1A GDP-bound (1-235) and nucleotide-binding deficient (T57N) variants are able to interact with p53. Left panel: protein expression of HA-tagged wild-type (Wt), T57N and 1–235 variant RBEL1A in HEK293T cells. Cells were transfected with the indicated vectors and their expressions were detected by western blotting using anti-HA antibodies. Right upper panel: purified GST-tagged p53 (wild type) was incubated with lysates from cells expressing the HA-tagged RBEL1A (full-length and T57N and 1–235 variants) vectors or the control empty vector. GST pull-down assays were performed and the protein pull-down products were analyzed by western blotting using anti-HA antibodies. Right lower panel: inputs of GST–p53 in each sample were detected by Ponceau staining. (B) RBEL1A mutants are able to modulate p53 ubiquitylation in an MDM2-dependent manner. MCF-7 cells stably expressing HA–RBEL1A (WT, T57N, 1–235) or HA-only vector were treated with MG132 with or without Nutlin-3 (10 µM) for 6 hours prior to protein extraction. Immunoprecipitations were then performed using anti-ubiquitin antibodies followed by p53 western blotting (left upper panel). Left middle and lower panels show input of p53 protein and β-actin on a separate membrane. Input of HA-RBEL1A (WT, T57N, and 1-235) are shown in the right panel. (C) Western blot analysis shows endogenous p53 levels in MCF-7 cells stably overexpressing HA–empty-vector and WT, T57N and 1–235 HA–RBEL1A.
RBEL1A negatively regulates p53 transcriptional function and DNA-damage-induced activation of p53
Next, we sought to investigate whether RBEL1A affects p53 transcriptional function inside the cells. H1299 cells (no endogenous p53) were transiently transfected with PUMA (p53 target gene) promoter–luciferase construct (Luo et al., 2003) together with p53 and RBEL1A expression vectors in various combinations; and the luciferase activity that reflects p53 regulation on PUMA promoter was determined. As shown in Fig. 8A, exogenous p53 clearly increased PUMA promoter activity (compare lanes 1 and 2), however, in the presence of increased levels of RBEL1A (0.5 and 1 µg), p53-mediated PUMA promoter activity was strongly suppressed (lanes 3 and 4). These results thus demonstrate that RBEL1A interferes with the transactivation function of p53.
Fig. 8.
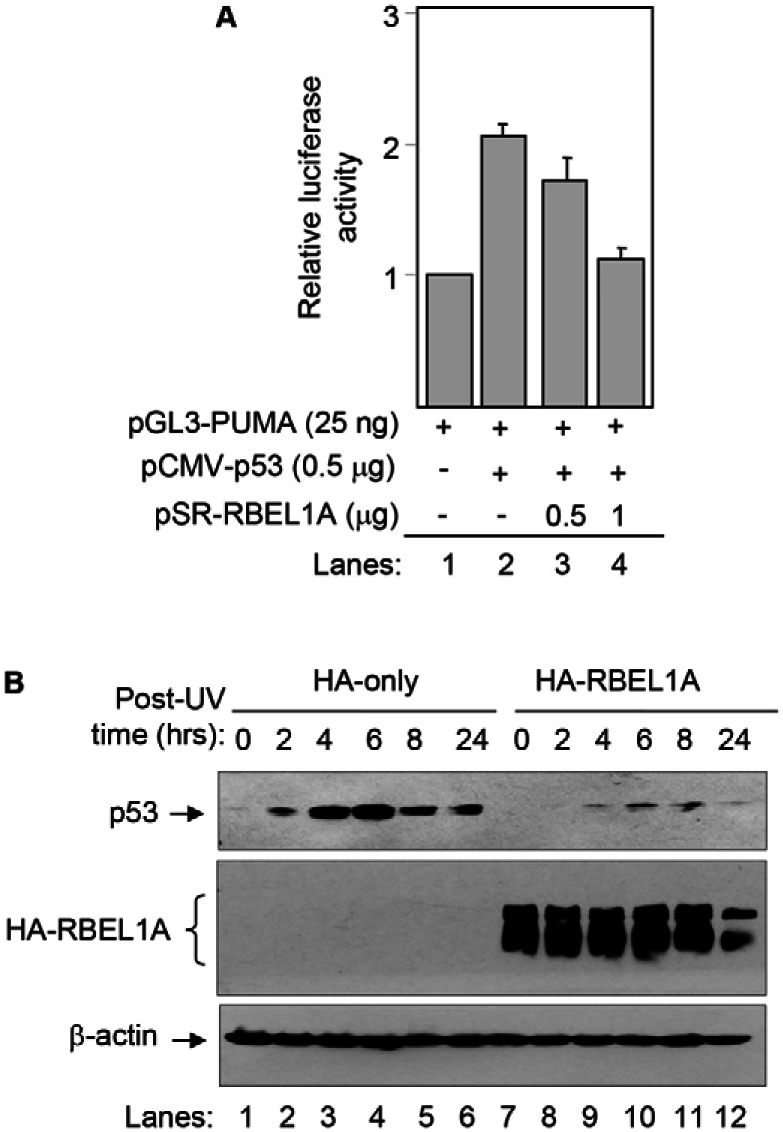
RBEL1A negatively regulates p53 transcriptional function and DNA-damage-induced activation. (A) RBEL1A suppresses p53 transactivation of the PUMA promoter. The p53-null H1299 cells were transfected with different amounts of the indicated vectors and p53 promoter luciferase activity was assayed as described in the Materials and Methods. The plotted results are mean (±s.e.m.) of triplicates in one experiment; similar results were obtained from several additional independent experiments. (B) RBEL1A suppresses p53 induction following UV-induced DNA damage. MCF-7 cells stably expressing the vector alone or the RBEL1A expression construct were exposed to UV (20 J/m2, twice instantly). Cells were harvested at the indicated times and western blotting were performed using the indicated antibodies on the same membrane.
It is well established that p53 protein induction and activation following DNA damage is important for its various physiological functions. We therefore, sought to investigate whether elevated expression of RBEL1A [as seen in human cancer (Montalbano et al., 2007)] would affect p53 induction following DNA damage. MCF-7 cells, stably expressing HA vector only or HA-tagged RBEL1A, were exposed to UV radiation and the effect of RBEL1A on p53 expression was determined. As seen in Fig. 8B, UV exposure triggered a strong p53 induction that peaked at 6 hours and remained until 24 hours after UV exposure in the control cells (lanes 1–6); however, p53 induction was remarkably suppressed in RBEL1A-expressing cells (lanes 7–12). Taken together our results indicate that RBEL1A negatively regulates p53 both in unstressed cells (Fig. 1) and in cells that are exposed to DNA damage (Fig. 8B) and the functions of p53 is modulated by increased RBEL1A expression.
Discussion
In the present study, we have demonstrated that RBEL1A is an important negative regulator of p53 tumor suppressor. Our results indicate that RBEL1A negatively regulates p53 by enhancing p53 protein ubiquitylation and degradation mediated by MDM2. To this end, our results demonstrate that RBEL1A directly interacts with both p53 and MDM2 (Figs 2, 3). Our results also indicate that increased expression of RBEL1A causes reduction in p53 protein levels whereas depletion of RBEL1A leads to increase in p53 protein level due to enhanced p53 protein half-life (Fig. 1C,D). The RBEL1A-mediated effect on p53 ubiquitylation and degradation appears to be MDM2 dependent as our results demonstrate that (i) RBEL1A strongly enhances MDM2-dependent ubiquitylation and degradation of p53 (Figs 4, 5, 7); (ii) RBEL1A has no effect on the mutant form of p53 that cannot interact with MDM2 (Fig. 4B); (iii) RBEL1A has minimal effect on the wild-type p53 that is expressed in a MDM2-deficient background (Fig. 4A); and (iv) MDM2 inhibitor blocks the negative effect of RBEL1A on p53 (Figs 1, 4, 5). A possibility remains that RBEL1A may also negatively modulate p53 function in a MDM2-independent manner. For example, our results also demonstrate that RBEL1A interacts with p53 via the tetramerization domain of p53 (Fig. 6A,B). It is known that p53 functions as a tetramer to bind within the promoter regions of its target genes. It is therefore possible that, by interacting with the p53 oligomerization domain, RBEL1A may also affect the oligomerization and the transactivational activity of p53 in a MDM2-independent fashion.
It is established that p53 is a transcription factor that regulates its downstream target genes, such as p21 and PUMA which are important for cell cycle control and apoptosis (Riley et al., 2008). Our results demonstrate that RBEL1A suppression of p53 is functionally relevant as RBEL1A inhibits the ability of p53 to transactivate its target genes such as p21 and PUMA and that RBEL1A knockdown leads to an increased expression of p21 and PUMA. Our results demonstrate that intracellular (endogenous) RBEL1A plays an important role in regulation of p53 protein as our results indicate that RBEL1A silencing (i) significantly enhances p53 protein stability (Fig. 1C,D) and (ii) also decreases p53 ubiquitylation (Fig. 5D). Furthermore, proteaseome and MDM2-specific inhibitors abrogate RBEL1A-mediated suppression of p53 levels (Fig. 1F; Fig. 4B,C).
Induction and activation of p53 following stress mediated by DNA damage is a physiological cellular response. p53 is a potent tumor suppressor and its induction and activation in response to DNA damage leads to expression of genes needed for cell cycle arrest, DNA repair and/or cell death. The role of p53 in regulation of these cellular processes is important to inhibit transmission of genetic mutations to next generation of cells as defects in p53 activation are linked to genomic instability (Jiang et al., 2010). In this context, our findings that RBEL1A negatively regulates p53, in the unstressed cells (Fig. 1) as well as in cells stressed via DNA damage (Fig. 8B), are potentially important and highlight the pathophysiological significance of negative regulation of p53 by RBEL1A. It is conceivable that under normal conditions in the normal cells, the functions of both RBEL1A and p53 are balanced as both proteins are expressed at the low levels. However, in pathological conditions, such as in tumors, RBEL1A levels are elevated at moderate to very high levels (Montalbano et al., 2007). It is possible that such increased levels of RBEL1A would suppress p53 function, thereby conferring a growth advantage to RBEL1A-overexpressing tumors. Therefore, our results demonstrate that RBEL1A-mediated negative regulation of p53 is relevant in context to pathophysiology of human malignancy.
RBEL1A is a Ras superfamily GTPase and to the best of our knowledge, this is the first report demonstrating that Ras-like GTPase is directly involved in the regulation of p53 ubiquitylation and degradation. RBEL1A harbors an N-terminal GTPase domain that is highly conserved among the Ras superfamily proteins, although it is more homologous to the Ran/Rab subfamily GTPases (Montalbano et al., 2007; Montalbano et al., 2009). Here we show that the minimal p53-interaction region of RBEL1A is at the core GTP-binding region (i.e. residues 1–235). RBEL1A deletion variant (RBEL1A 236–729) lacking the N-terminal region (residues 1–235) is not able to interact with p53 (Fig. 6D). Interestingly, our results also indicate that GTP binding does not appear to be essential for RBEL1A interaction with and regulation of p53 because (i) the GDP-bound variant RBEL1A 1–235, (ii) the GTP-bound variants (RBEL1A wt, RBEL1A 1–361, RBEL1A 1–624) (Montalbano et al., 2009) and (iii) the nucleotide-binding-deficient variant (RBEL1A-T57N) are all able to interact with p53 and enhance p53 ubiquitylation (Fig. 7A). We also consistently note that RBEL1A T57N variant interacts with p53 better than the wild-type counterpart (Fig. 7A). Such results suggest that the inability of GTP/nucleotide binding may enhance RBEL1A association with p53 and GTP binding may alter the ability of RBEL1A to interact with p53. Further studies would be needed to fully dissect this issue in the future.
A protein named YY1 has also been reported to function as a p53 negative regulator that promotes MDM2-mediated p53 ubiquitylation and degradation (Sui et al., 2004). It appears that RBEL1A has certain functional characteristics resembling those of YY1. For example, RBEL1A also directly interacts with p53 as well as MDM2; RBEL1A interacts with p53 at its C-terminal domain and depletion of RBEL1A also induces growth arrest and apoptosis. It is thought that for YY1 to negatively regulate p53, its interactions with MDM2 and formation of the YY1–p53–MDM2 complex are crucial. Whether RBEL1A functions in a similar fashion to facilitate p53–MDM2 association is an issue that remains to be further investigated. RBEL1A is a large and glycosylated protein with a molecular mass within 80–130 kDa range (Montalbano et al., 2007). Based on our data and those presented by Sui et al., (Sui et al., 2004), both RBEL1A and YY1 directly interact with p53 at its C-terminal region; however, it is possible that RBEL1A and YY1 may exist in different protein complexes because it would seem less likely that both of these proteins simultaneously interact with one molecule (i.e. p53) within rather small region. It is also possible that RBEL1A may modulate p53 ubiquitylation and degradation by other mechanisms. For example, RBEL1A may influence MDM2 ligase activity by protein interaction. In addition, RBEL1A may play a role in facilitating p53 protein transport from the nucleus to the cytosol for protein degradation. In our recently published results (Montalbano et al., 2009) we have shown that RBEL1A is a Rab and Ran-like protein; the Rab and Ran-like proteins are known to play important roles in protein transport and distribution. Future studies are needed to explore these possible mechanisms. Nevertheless, our experimental evidence highlights RBEL1A to be the first GTPase in the Ras superfamily to directly and negatively regulate p53.
p53 mutations and other alterations have been found in about half of human tumors, however, the other half of tumors harbor wild-type p53. Expression and function of p53 is believed to be antagonized by negative regulators, such as MDM2. Small synthetic peptide inhibitors, which block the p53–MDM2 interactions and reactivate the expression and function of wild-type p53 have shown promise as potential therapeutics for the treatment of cancers harboring wild-type endogenous p53 (Lane et al., 2010; Shangary and Wang, 2009; Vassilev, 2007). Various MDM2 inhibitors such as MI-219 and Nutlin-3 have advanced to preclinical and early phase clinical trials (Shangary and Wang, 2009). Likewise, as shown in our present study, RBEL1A also plays an important role in negative regulation of p53 by strongly enhancing MDM2-mediated p53 ubiquitylation and also by inhibiting transactivation of p53. RBEL1A is found to be overexpressed in multiple human cancers (Montalbano et al., 2007) and our current findings also show that MDM2 inhibitor (Nutlin-3) inhibits the effect of RBEL1A on p53. It is possible that inhibitors of MDM2 and RBEL1A that block the mutual interactions among RBEL1A, p53 and MDM2 may have therapeutic potential in reactivating the wild-type p53 and hence, our novel findings may be important for the future development of novel therapeutics to restore the function of p53.
Materials and Methods
Antibodies and reagents
HA-tag (HA.11) antibody was from Convance (Berkeley, CA, USA). Antibodies specific to ubiquitin, p21, MDM2 (SMP14), p53 (DO-1, FL393) and GFP, were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies for Puma, Erk1/2, PI3K (p85) and cyclin D3 were from Cell Signaling Inc. Anti-RBEL1 (pan-RBEL1) antibodies were generated in our laboratory as previously described (Montalbano et al., 2007). Proteasome inhibitor MG132 and Nutlin-3 were from Sigma (St. Louis, MO, USA). Human recombinant ubiquitins (Ubs), E1 and E2 mix (UbcH5a, b, c) were from Boston Biochem (Cambridge, MA, USA).
Expression constructs
Construction of HA-S-tagged full-length RBEL1A, RBEL1A T57N mutant, and its deletion variants 1–235, 1–361, and 1–624 have been reported previously (Montalbano et al., 2009). Deletion variant 236–729 was PCR amplified and subcloned into pSRα vector. Expression vectors carrying p53 (pCMV-p53) and MDM2 (HA-MDM2) were kindly provided by Dr Bert Vogelstein (Jones Hopkins University, USA) and Dr Stanley Cohen (Stanford University) respectively. pGL3-PUMA promoter luciferase reporter vector was kindly provided by Dr Thomas Chittenden (ImmunoGen Inc., Cambridge, MA, USA). pGFP-p53 and pGFP-MDM2 were generated by inserting the PCR-amplified cDNAs of p53 and MDM2 into the pGFP-c1 mammalian expression vector (BD Bioscience Clontech, San Jose, CA, USA). pGFP-p53 (L22Q, W23S) mutant was generated using QuikChange Site-directed Mutagenesis kit (Stratagene, La Jolla, CA, USA) using pGFP-wild-type p53 as a template. pCEP4-HA-S-RBEL1A was constructed by excision of HAS-RBEL1A DNA fragment from pSR-HA-S-RBEL1A (WT, T57N, 1-235) using BamHI and HindIII restriction enzymes and subsequently subcloned it into pCEP4 vector. GST-tagged wild-type p53 and MDM2 and p53 deletion mutants were generated by inserting the PCR-amplified full-length p53 or MDM2 cDNAs into pGEX6P-2 expression vector (GE healthcare, Pittsburgh, PA, USA). Bacterial expression vector of S-tagged RBEL1A was generated by inserting the PCR-amplified full-length RBEL1A into pET32b expression vector. All expression vectors were sequenced to validate their authenticity.
Cell culture, transfection and RBEL1A knockdown
Cell lines MCF-7 (breast cancer), RKO (colon cancer), H1299 (lung cancer), HEK293T (human embryonic kidney cells) and p53−/−/MDM2−/− mouse embryonic fibroblasts (kindly provided by Dr Guillermina Lozano, U. T. M. D. Anderson Cancer Center, USA) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Gemini Bio-Products Inc., West Sacramento, CA, USA). MCF12A human normal breast cell line (kindly provided by Dr G. Wayne Zhou, Marine Biological Lab, USA) was maintained in DMEM supplemented with 10% FBS and 10 µg/ml insulin, 20 ng/ml hEGF, 100 ng/ml cholera toxin, 500 ng/ml hydrocortisone, 10 IU/ml penicillin–streptomycin (Mediatech, Inc., Manassas, VA, USA) and 200 µM L-glutamine (Mediatech, Inc.). pCEP4-RBEL1A (WT, T57N, 1–235)-expressing cells (pooled clones) or vector-alone-expressing cells (pooled clones) were generated by stable transfection. All transfections were done using either Lipofectamine 2000 (Invitrogen/Life Technologies, Grand Island, NY, USA) or TransIT-LT1 (Mirus, Madison, WI, USA). RBEL1A shRNA knockdown was performed as we previously described (Montalbano et al., 2009).
Recombinant protein purification
Wild-type and deletion variants of GST-tagged p53 and MDM2 as well as S-tagged RBEL1A were expressed in E. coli strain BL21 derivative, Rosetta (Novagen, EMD, Bioscience, Madison, WI, USA) and induced by using 1 mM IPTG (isopropyl-1-thio-β-D-galactopyranoside) for 5 hours at 30°C. Bacteria were lysed by using lysis buffer containing 28 mM Tris, 135 mM NaCl, 1 mM dithiothreitol (DTT), 1% Triton X-100 (w/v), protease inhibitor cocktail and 400 µg/ml lysosome at room temperature for 30 minutes, followed by 26,000 g centrifugation for 30 minutes (Sorvall RC5C Plus). GST-tagged recombinant proteins were purified by using glutathione–Sepahrose™ 4B beads (GE healthcare) according to manufacturer's protocol. S-tagged RBEL1A as well as GST-tagged p53 (both were used in in vitro ubiquitylation and S-tag pull down) were gel-purified as previously described (Corcoran et al., 2009).
Luciferase assays
Luciferase assays were performed as we previously reported (Luo et al., 2005). Briefly, H1299 lung cancer cells were transiently transfected with the indicated vectors for 24 hours by using TransIT-LT1 transfection reagent (Mirus, Madison, WI) according to manufacturer's protocol. Cells were harvested, washed and lysed, followed by centrifugation at 16,000 g for 30 minutes. The luciferase activity was measured using equal amounts of total protein corresponding to each sample by a luminometer (LUMAT LB9507, Berthold Technologies, Germany).
Immunoblotting and immunoprecipitation analyses
Immunoblotting and immunoprecipitation were performed as we have previously described (Montalbano et al., 2009; Rong et al., 2007).
Quantitative real-time PCR
Total RNAs from RBEL1A-overexpressing or vector-only-expressing cells were extracted using the Trizol Reagent (Invitrogen) as previously described (Lui et al., 2006). About 10 µg of the total RNAs were reverse-transcribed to first strand cDNAs by using iScript™ Select cDNA Synthesis Kit (Bio-Rad Life Science Research, Hercules, CA, USA). The expression levels of p53, MDM2, p21, Puma were assayed by iQ™ SYBR green PCR reagent (Bio-Rad Life Science Research) using iCycler iQ5™ real-time PCR (Bio-Rad Life Science Research). The primers used as follows: p21 forward primer: 5′-CAGACCAGCATGACAGATTTC-3′ and p21 reverse primer 5′-TTAGGGCTTCCTCTTGGAGA-3′. Puma forward primer: 5′-AGAGGGAGGAGTCTGGGAGTG-3′ and Puma reverse primer: 5′-GCAGCGCATATACAGTATCTTACAGG-3′; MDM2 forward primer: 5′-CCCTTAATGCCATTGAACCT-3′ and MDM2 reverse primer: 5′-CATACTGGGCAGGGCTTATT-3′; p53 forward primer: 5′-GCCCCCAGGGAGCACTA-3′ and p53 reverse primer: 5′-GGGAGAGGAGCTGGTGTTG-3′; β-actin forward primer: 5′-GCTCGTCGTCGACAACGGCTC-3′ and β-actin reverse primer: 5′-CAAACATGATCTGGGTCATCTTCTC-3′ (Baumbusch et al., 2006; Li et al., 2010). Each sample was analyzed in triplicate and repeated at least by three independent experiments. β-actin was included for normalization. For data analysis, comparative ΔΔCt method was applied (Livak and Schmittgen, 2001). Briefly, the raw Ct (threshold cycle) values were averaged and normalized to averaged β-actin Ct to obtain ΔCt, then the normalized ΔCt was compared with the control samples ΔCt to obtain ΔΔCt.
In-cell ubiquitylation assay
Experiments were performed using three different methodologies. Method 1: cells were transfected with expression vectors carrying His-tagged ubiquitin plus either HA-tagged RBEL1A or HA empty vector. Twenty-four hours after transfection, cells were treated with 10 µM MG132 for 6 hours prior to harvesting. Cell lysates (1 mg) from the transfectants were used for His-tagged protein pull down and the precipitated ubiquitylated proteins were analyzed by p53-specific antibodies (Fig. 5B). Method 2: cells, stably overexpressing RBEL1A or control vector, were treated with MG132 (10 µM) or MG132 (10 µM) plus Nultin-3 (10 µM) for 6 hours. Cells were harvested and lysed prior to pull-down assays using anti-ubiquitin antibody, and the precipitates were analyzed by western blot using anti-p53-specific antibodies (Fig. 5C). Method 3: cells infected with lentivirus carrying either scrambled shRNA or RBEL1A-specific shRNA were treated with MG132 (10 µM) for 6 hours. Cells were harvested and lysed prior to protein pull-down assays using anti-p53 antibodies, and the precipitated p53 proteins were analyzed by western blot using anti-ubiquitin-specific antibodies (Fig. 5D).
In vitro ubiquitylation assay
Purified recombinant p53 (700 ng), RBEL1A (2.5 µg), MDM2 (1.5 µg) proteins in various combinations were incubated with a cocktail containing E1 (150 ng), E2 mix (200 ng) and Ubs (2 µg) in the reaction buffer [Tris-HCl (50 mM, pH 7.4), MgCl2 (5 mM 2 mM), ATP, and DTT (2 mM)] at 30°C for 90 minutes. About 1/5 of the reaction mix was separated by 8–15% gradient SDS-PAGE gel followed by immunoblotting with p53 and ubiquitin-specific antibodies.
Supplementary Material
Acknowledgments
We thank Dr Guillermina Lozano (The University of Texas MD Anderson Cancer Center, TX), Thomas Chittenden (ImmunoGen, Inc., Cambridge, MA), Bert Vogelstein (Johns Hopkins Kimmel Cancer Center, MD), and Stanley Cohen (Stanford University School of Medicine, CA) for providing the p53−/−/MDM2−/− mouse embryonic fibroblasts, pGL3-PUMA-luciferase, pCMV-p53 and HA-MDM2 constructs, respectively. We also thank Dr Rosemary Rochford (SUNY Upstate Medical University) for facilitating the real-time PCR experiments.
Footnotes
Author contributions
K.L. and Y.H. conceived and designed the study. K.L. performed most of the experiments. J.A., J.M., J.S., C.C., Q.H., and H.S. also performed experiments and analyzed the data. K.L., M.S.S. and Y.H. intellectually contributed to the study, analyzed the results and wrote the manuscript. All authors read and commented on the manuscript.
Funding
This work was supported by the National Institutes of Health [grant number CA113868 to Y.H.]; a predoctoral fellowship from the US Department of Defense [grant number BC083017 to K.L.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.118117/-/DC1
References
- Baumbusch L. O., Myhre S., Langerød A., Bergamaschi A., Geisler S. B., Lønning P. E., Deppert W., Dornreiter I., Børresen-Dale A. L. (2006). Expression of full-length p53 and its isoform Deltap53 in breast carcinomas in relation to mutation status and clinical parameters. Mol. Cancer 5, 47 10.1186/1476-4598-5-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchhop S., Gibson M. K., Wang X. W., Wagner P., Stürzbecher H. W., Harris C. C. (1997). Interaction of p53 with the human Rad51 protein. Nucleic Acids Res. 25, 3868–3874 10.1093/nar/25.19.3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran C. A., Huang Y., Sheikh M. S. (2004). The p53 paddy wagon: COP1, Pirh2 and MDM2 are found resisting apoptosis and growth arrest. Cancer Biol. Ther. 3, 721–725 10.4161/cbt.3.8.1068 [DOI] [PubMed] [Google Scholar]
- Corcoran C. A., Montalbano J., Sun H., He Q., Huang Y., Sheikh M. S. (2009). Identification and characterization of two novel isoforms of Pirh2 ubiquitin ligase that negatively regulate p53 independent of RING finger domains. J. Biol. Chem. 284, 21955–21970 10.1074/jbc.M109.024232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbelstein M., Wienzek S., König C., Roth J. (1999). Inactivation of the p53-homologue p73 by the mdm2-oncoprotein. Oncogene 18, 2101–2106 10.1038/sj.onc.1202512 [DOI] [PubMed] [Google Scholar]
- Hofmann T. G., Möller A., Sirma H., Zentgraf H., Taya Y., Dröge W., Will H., Schmitz M. L. (2002). Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 4, 1–10 10.1038/ncb715 [DOI] [PubMed] [Google Scholar]
- Jiang L., Sheikh M. S., Huang Y. (2010). Decision making by p53: Life versus death. Mol. Cell Pharmacol. 2, 69–77 [PMC free article] [PubMed] [Google Scholar]
- Kussie P. H., Gorina S., Marechal V., Elenbaas B., Moreau J., Levine A. J., Pavletich N. P. (1996). Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274, 948–953 10.1126/science.274.5289.948 [DOI] [PubMed] [Google Scholar]
- Lane D. P., Cheok C. F., Lain S. (2010). p53-based cancer therapy. Cold Spring Harb. Perspect. Biol. 2, a001222 10.1101/cshperspect.a001222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Cheng Q., Li Z., Chen J. (2010). p53 inactivation by MDM2 and MDMX negative feedback loops in testicular germ cell tumors. Cell Cycle 9, 1411–1420 10.4161/cc.9.7.11255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J., Chen J., Elenbaas B., Levine A. J. (1994). Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev. 8, 1235–1246 10.1101/gad.8.10.1235 [DOI] [PubMed] [Google Scholar]
- Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Lui K., Huang Y., Choi H. L., Yu S., Wong K. B., Chen S., Chan F. L. (2006). Molecular cloning and functional study of rat estrogen receptor-related receptor gamma in rat prostatic cells. Prostate 66, 1600–1619 10.1002/pros.20429 [DOI] [PubMed] [Google Scholar]
- Luo X., Huang Y., Sheikh M. S. (2003). Cloning and characterization of a novel gene PDRG that is differentially regulated by p53 and ultraviolet radiation. Oncogene 22, 7247–7257 10.1038/sj.onc.1207010 [DOI] [PubMed] [Google Scholar]
- Luo X., He Q., Huang Y., Sheikh M. S. (2005). Cloning and characterization of a p53 and DNA damage down-regulated gene PIQ that codes for a novel calmodulin-binding IQ motif protein and is up-regulated in gastrointestinal cancers. Cancer Res. 65, 10725–10733 10.1158/0008-5472.CAN-05-1132 [DOI] [PubMed] [Google Scholar]
- Meek D. W., Anderson C. W. (2009). Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 1, a000950 10.1101/cshperspect.a000950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalbano J., Jin W., Sheikh M. S., Huang Y. (2007). RBEL1 is a novel gene that encodes a nucleocytoplasmic Ras superfamily GTP-binding protein and is overexpressed in breast cancer. J. Biol. Chem. 282, 37640–37649 10.1074/jbc.M704760200 [DOI] [PubMed] [Google Scholar]
- Montalbano J., Lui K., Sheikh M. S., Huang Y. (2009). Identification and characterization of RBEL1 subfamily of GTPases in the Ras superfamily involved in cell growth regulation. J. Biol. Chem. 284, 18129–18142 10.1074/jbc.M109.009597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley T., Sontag E., Chen P., Levine A. (2008). Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 9, 402–412 10.1038/nrm2395 [DOI] [PubMed] [Google Scholar]
- Rong R., Jiang L. Y., Sheikh M. S., Huang Y. (2007). Mitotic kinase Aurora-A phosphorylates RASSF1A and modulates RASSF1A-mediated microtubule interaction and M-phase cell cycle regulation. Oncogene 26, 7700–7708 10.1038/sj.onc.1210575 [DOI] [PubMed] [Google Scholar]
- Shangary S., Wang S. (2009). Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 49, 223–241 10.1146/annurev.pharmtox.48.113006.094723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui G., Affar B., Shi Y., Brignone C., Wall N. R., Yin P., Donohoe M., Luke M. P., Calvo D., Grossman S. R. et al. (2004). Yin Yang 1 is a negative regulator of p53. Cell 117, 859–872 10.1016/j.cell.2004.06.004 [DOI] [PubMed] [Google Scholar]
- Vassilev L. T. (2007). MDM2 inhibitors for cancer therapy. Trends Mol. Med. 13, 23–31 10.1016/j.molmed.2006.11.002 [DOI] [PubMed] [Google Scholar]
- Zilfou J. T., Lowe S. W. (2009). Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Biol. 1, a001883 10.1101/cshperspect.a001883 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.