

Summary
In this report, we have shown that miR146b promotes the maintenance of pregnancy-derived mammary luminal alveolar progenitors. MiR146b expression was significantly higher in the mammary glands of pregnant and lactating mice than in virgin mice. Furthermore, miR146b levels were significantly higher in mouse mammary glands exposed to the sex hormones, estrogen and progesterone, compared with those of untreated control animals. Pregnancy-derived primary mouse mammary epithelial cells in which miR146b was knocked down showed a significant reduction in the number of hollow acinar organoid structures formed on three-dimensional Matrigel and in β-casein expression. This demonstrates that miR146b promotes the maintenance of pregnancy-derived mammary luminal alveolar progenitors. It has been shown that mouse mammary luminal progenitors give rise to hollow organoid structures, whereas solid organoid structures are derived from stem cells. Among several miR146b targets, miR146b knockdown resulted in preferential STAT3β overexpression. In the primary mouse mammary epithelial cells, overexpression of STAT3β isoform caused mammary epithelial cell death and a significant reduction in β-casein mRNA expression. Therefore, we conclude that during pregnancy miR146b is involved in luminal alveolar progenitor cell maintenance, at least partially, by regulating STAT3β.
Key words: MicroRNA-146b, Alveolar Progenitors, STAT3
Introduction
Development of the mammary gland is a highly dynamic process and involves distinct embryonic, pubertal, pregnancy, lactation and involution stages. Hormones and growth factors undoubtedly play key roles in all stages of mammary development. The essential role of micro-RNAs (miRNAs) in the various stages of mammary development is being recognized. Not surprisingly, a disruption in miRNAs, has been implicated in human breast cancers (Watson and Khaled, 2008).
MiRNAs have been linked to both normal physiology and pathology, including regulation of developmental processes, proliferation, differentiation and apoptosis. Loss of mature miRNAs resulted in a failure of the cells to differentiate (Andl et al., 2006; Bernstein et al., 2003; Kanellopoulou et al., 2005; Muljo et al., 2005), providing evidence that miRNAs are involved in stem cell maintenance, self-renewal, and/or differentiation (Greene et al., 2010). MiRNAs have been found in co-expressed clusters during mammary gland development, suggesting co-regulation of miRNA groups (Avril-Sassen et al., 2009; Piao and Ma, 2012). Furthermore, it has been shown that mRNAs encoding receptors of endocrine hormones are direct targets of specific miRNAs, which also indicates the role of miRNAs in mammary gland development (Cui et al., 2011).
Mouse microRNA-146b-5p (miR146b) is located on chromosome 19 at position 19qC3, whereas miR146a is located on chromosome 11 at position 11qA5 (Fujita et al., 2011). Structurally, the mature miR146a and miR146b differ by only two nucleotides at the 3′ end, a region that is believed to play a minor role in target recognition, and in many instances miR146a and miR146b have demonstrated redundant activities (Lewis et al., 2003). The role of miR146b in cancer progression is currently controversial. MiR146b overexpression has been shown to be associated with aggressive behavior in papillary thyroid carcinomas (PTC) (He et al., 2005) and triple negative breast cancers (Garcia et al., 2011). Conversely, miR146b has also been shown to inhibit carcinogenesis. Welch and colleagues recently classified miR146a/b as a ‘metastasis-suppressing metastamir’ (Hurst et al., 2009). The term ‘metastamir’ refers to metastasis regulatory miRNAs that have an impact on critical steps in the metastatic cascade, such as epithelial–mesenchymal transition (EMT), apoptosis and angiogenesis (Hurst et al., 2009). For example, miR146b was shown to inhibit glioblastoma cell migration (Xia et al., 2009) and to suppress breast cancer metastasis (Bhaumik et al., 2008; Hurst et al., 2009). These findings suggest that miR146b may play a dual role in cancer and that much remains to be learned about the mechanism by which this miRNA may participate during carcinogenesis.
Recently, our group isolated distinct populations of ductal-limited, alveolar-limited and multipotent mammary progenitors by single-cell cloning of the COMMA-D (CD) cell line (Kittrell et al., 2011). Transplantation of the alveolar-limited clone, referred to herein as an ‘alveolar progenitor cells/clone’, generated functional alveolar structures in vivo. Transplantation of ductal-limited clone, referred to hereinafter as a ‘ductal progenitor clone’, generated only ductal structures devoid of functional alveolar differentiation potential. Transplantation of two of the clones, referred to hereinafter as ‘multipotent progenitor clones’, generated ducts and alveolar structures (Kittrell et al., 2011). Using this model system, we identified a hormonally regulated miRNA, miR146b, that plays a pivotal role in alveolar progenitor cell maintenance.
As mentioned above, altered expression of miR146b has been linked to an invasive and metastatic capacity in diverse cancers. Finding a hormonally regulated miRNA that regulates both the processes of alveolar cell maintenance and breast cancer may provide one missing link in the molecular pathways implicated in hormonal regulation of human breast cancer.
Results
MiR146b is highly expressed in the alveolar progenitor cells
The distinct mammary alveolar, ductal and multipotent progenitor clones were used to screen for changes in the expression of 84 unique miRNAs (Fig. 1A) by an RT2 miRNA PCR array. This screening showed a set of 74 upregulated miRNAs in the clones with in vivo morphogenic potential (including the alveolar progenitor, ductal progenitor and multipotent progenitors) when compared with clones that exhibited no in vivo morphogenic potential. Twenty miRNAs showed a 10 to 20-fold increase, and 15 miRNAs showed >20-fold increase (Fig. 1A). Among those upregulated, miR146b showed >72-fold increase, and miR-203 showed >77-fold increase. To examine the expression of upregulated miRNAs in the specific progenitor clones, RT-qPCR was used on RNA derived from progenitor clones grown on Matrigel in three dimensions (3D). MiR146b was the only miRNA that showed differential expression as it was highly expressed by the alveolar progenitor clone with a 10-fold increase compared with the level in the ductal and the multipotent progenitor clones (10±1.8 versus 1.29±0.7 and 1±0.23, respectively; Fig. 1B). Others such as miR203 expression showed no difference between the distinct progenitor clones (data not shown).
Fig. 1.

MiR146b expression is upregulated in the CD-derived alveolar progenitor cells and in mouse mammary glands during pregnancy and lactation. (A) An RT2 miRNA PCR array was used to screen for miRNAs differentially expressed in CD clones with in vivo growth potential normalized to CD clones lacking in vivo growth potential. (B) Quantitative PCR of miR146b expression in three CD-derived clones grown for 7 days on Matrigel. Ductal and alveolar progenitor clones were normalized to the multipotent clone (n = 4; *P<0.05). (C) Quantitative PCR of miR146b and miR146a expression in PMECs from virgin, pregnant, lactating or 10 days post-weaning mice (n = 3; different letters indicate a statistical difference; P<0.05). (D) Representative histogram of flow cytometry analysis of the three mammary cell subpopulations, lin−CD24+CD29hi (stem cells/basal cells), lin−CD24+CD29loCD61+ (luminal progenitor cells) and lin−CD24+CD29loCD61− (differentiated luminal cells), from virgin and pregnant mice. The subpopulations were sorted by FACS. (E) miR146b expression in the subpopulations analyzed by qPCR (n = 3, *P<0.05 compared with the stem cells). Data are means ± s.e.m.; different letters indicate a statistical difference, P<0.05.
To confirm these findings, miR146b expression levels were examined in the primary mammary epithelial cells (PMECs) derived from virgin, pregnant, lactating and 10 days post-weaning (involuting) female BALB/c mice (Fig. 1C). These studies showed a 6.3-fold increase in miR146b expression levels in the PMECs derived from pregnant mice compared with those from virgin and post-weaning mice (6.3±0.83 versus 1.0±0.44 and 1.3±1.17, respectively, P<0.05) and a 12-fold increase in PMEC derived from lactating compared with virgin and post-weaning mice (12.03±1.45 versus 1.0±0.44 and 1.3±1.17, respectively, P<0.05). MiR146b expression levels were normalized to keratin-18, in order to exclude the contribution of stromal cells. Because the pattern of expression of miR146a and miR146b are similar and the isoforms are reported to be co-regulated (Garcia et al., 2011), miR146a levels were similarly examined by RT-qPCR. The results showed that miR146a levels increased similarly during pregnancy and lactation. However, the increase in miR146a expression levels, were not as significant as miR146b (Fig. 1C).
Recently, the progenitor and differentiated cells in the mouse mammary glands and human breast were phenotypically and functionally characterized by fluorescence-activated cell sorting (FACS) followed by transplantation. These studies defined the subpopulations enriched in stem cells by the expression of unique surface markers, lineage-negative (lin−) CD24+CD29hi and lin−CD24+CD49fhi, luminal progenitors by lin−CD29loCD24+CD61+, differentiated estrogen receptor-positive (ER+) luminal cells by CD24+CD133+ and myoepithelial progenitors by CD29loCD24lo (Asselin-Labat et al., 2007; Shackleton et al., 2006; Stingl et al., 2006).
To find the mammary epithelial sub-population with the highest level of miR146b expression, the luminal progenitors (lin−CD29loCD24+CD61+), basal/stem cells (lin−CD29hiCD24+) and differentiated luminal cells (lin−CD29loCD24+CD61−) were FACS sorted from the mammary glands of virgin and pregnant BALB/c mouse, followed by RNA extraction and RT-qPCR. The results revealed that during pregnancy miR146b expression levels were ∼2-fold higher in the luminal progenitors when compared with the basal/stem cells (10.6±2.17 versus 4.6±0.19; P<0.05) and 1.6-fold higher when compared with the differentiated luminal cells (10.6±2.17 versus 6.3±0.68; P<0.05; Fig. 1D,E). However, there were no differences in miR146b expression levels between these epithelial subpopulations derived from virgin mouse mammary glands (Fig. 1E). Interestingly, miR146b was 10.5-fold higher in the luminal progenitors (alveolar) of pregnant compared with virgin (ductal) mice (10.6±2.17 versus 1.01±0.38; P<0.05). Since expression was significantly higher in the luminal alveolar progenitors than in stem cells and differentiated luminal cells, we focused our studies on the functional role of miR146b in the luminal alveolar progenitor cells.
Sex hormones and prolactin result in the upregulation of miR146b levels in primary mammary epithelial cells
Since miR146b expression levels were significantly higher in all epithelial subpopulations from pregnant compared with virgin mice, we hypothesized that pregnancy hormones may participate in its upregulation. To investigate the influence of hormones on miR146b, we exposed glands from virgins to estrogen (E) and progesterone (P) in vivo. E plus P hormonal pellets were placed subcutaneously in virgin BALB/c mice for 3 weeks. Following treatment, the mammary glands were removed followed by PMEC isolation. RT-qPCR revealed that E plus P treatment resulted in a 2.7-fold increase in miR146b expression levels, normalized to keratin-18, compared with the level in the non-treated controls (2.7±0.08 versus 1±0.54; P<0.05; Fig. 2A). To rule out the contribution of stroma, PMECs derived from virgin mouse mammary glands were treated with E plus P, prolactin alone, or all three hormones for 10 days in vitro. A similar rise in miR146b levels was found upon exposure to E plus P, prolactin alone, or all three hormones compared with the level in the untreated cells (1.46±0.09 and 1.38±0.09, respectively, versus 1±0.26; P<0.05; Fig. 2C). These results suggested that sex hormones and or prolactin, directly or indirectly, mediate the upregulation in miR146b levels.
Fig. 2.
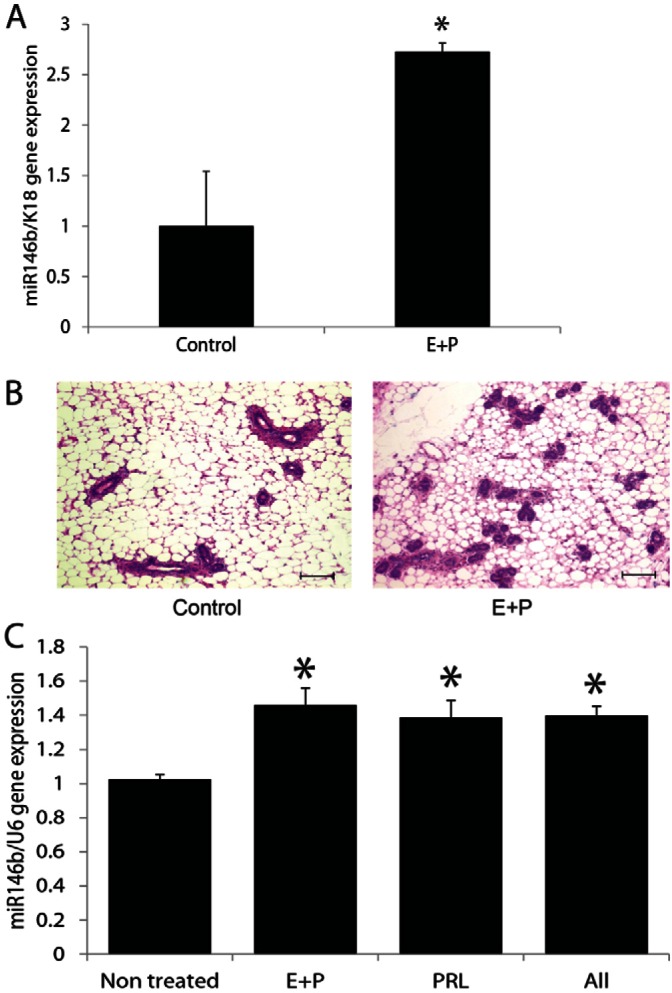
In vivo administration of estrogen and progesterone resulted in upregulation of miR-146b expression in the mammary epithelial cells. (A) Quantitative PCR of miR-146b in PMECs from virgin mice treated with estrogen and progesterone for 3 weeks compared with non-treated virgin mice. Data are means ± s.e.m. (n = 4, *P<0.05). (B) Representative Hematoxylin and Eosin-stained sections from the hormone-treated and control mice. Scale bars: 100 µm. (C) Quantitative PCR of miR-146b in PMECs from virgin mice treated with estrogen plus progesterone (E+P), with prolactin (PRL) or both for 10 days in vitro compared with non-treated PMECs. Data are means ± s.e.m. (n = 3, *P<0.05 compared with the control).
MiR146b mediates the maintenance of alveolar luminal progenitor cells
To assess the functional role of miR146b, ‘GFP’-labeled Locked Nucleic Acid oligonucleotides (LNA™, Exiqon) complementary to miR146b were used to knockdown miR146b expression. For these studies, PMECs derived from BALB/c mouse mammary glands and/or CD-derived alveolar progenitor cells were used. In order to examine the specificity of the miR146b LNA, RT-qPCR was used to examine miR146a levels following miR146b knockdown. The data showed a non-significant reduction in miR146a, confirming that the knockdown was selective against miR146b (Fig. 3A). Furthermore, time-course experiments using the CD-derived alveolar progenitor cells followed by RT-qPCR showed stable miR146b knockdown up to 72 hours post-transfection (supplementary material Fig. S1A). To assess effects on alveolar progenitor cell survival, the CD-derived alveolar progenitor cells were collected at 48, 60 and 72 hours post-transfection followed by staining with the LIVE/DEAD® Fixable Dead Cell Staining Kit. The fluorescent dye can permeate the compromised membranes of dead cells and react with free amines both in the interior and on the cell surface. A significant reduction in the alveolar progenitor cells was seen at 72 hours post-transfection (Fig. 3C,D; supplementary material Fig. S1B). Effects on cell death was confirmed by a western blot analysis, which showed an increase in cleaved caspase 3 in the alveolar progenitor cells knocked down of miR146b compared with the control groups (Fig. 3B). Our data showed selective effect on cell survival in the alveolar progenitor cells when compared with the CD-derived ductal limited and multipotent progenitor cells (Fig. 3C,D; supplementary material Fig. S1B). As seen in supplementary material Fig. S1C, >80% of the total population of CD cells were transfected with the miR146b LNA. Even though ∼60% of the PMECs were transfected with the miR146b LNA, there was not a significant effect on survival of the total population of PMECs derived from pregnant mouse mammary glands (data not shown). However, as seen in Fig. 3E,F, there was a significant reduction in the percentage of alveolar luminal progenitor cells knocked down of miR146b (lin−CD29loCD24hiCD61+) compared with the control (0.55±0.097% versus 1.0±0.18%; means ± s.e.m., P<0.05).
Fig. 3.

Knockdown of miR-146b leads to a reduction in the luminal progenitor subpopulation during pregnancy. (A) Quantitative RT-PCR of miR146a and miR146b in PMECs at 72 hours post-transfection with a miR146b LNA inhibitor (miR146b KD) and with non-silencing controls (NS; n = 3, P<0.05). (B) Representative western analysis of cleaved caspase 3, and β-actin in the alveolar progenitor cell line 48 hours post-transfection in non-transfected (NT), NS and miR146b KD cells. (C) Representative histograms of flow cytometry analysis for cells stained with the LIVE/DEAD® Fixable Dead Cell Staining Kit. (D) Quantification of the percentage of live cells from the live–dead analysis of the alveolar progenitor cell line at 72 hours post-transfection (n = 3, P<0.05). (E) Representative flow cytometry dot plots of PMECs at 72 hours post-transfection analyzed for the Lin−CD24+CD29loCD61+ population. (F) The reduction in the CD24+CD61+ progenitor population from the lin−CD29lo population of PMECs following knockdown of miR-146b (n = 3, *P<0.05). Data are means ± s.e.m.
To further examine the functional role of miR146b in the luminal alveolar progenitors, an assay involving three-dimensional (3D) hollow or solid acinar organoid formation efficiency on Matrigel was performed. Based on previously published studies, the mammary stem cells give rise to solid acinar organoids, whereas luminal progenitors form hollow acinar organoid structures when grown in 3D Matrigel cultures (Guo et al., 2012; Lim et al., 2009; Shackleton et al., 2006; Stingl et al., 2006). For these studies, PMECs were isolated from pregnant mouse mammary glands followed by transfection with miR146b LNA. At 24 hours post-transfection, the cells were transferred from tissue culture plates to Matrigel and treated with prolactin for 7 days. The epithelial organoids were counted and classified into solid or hollow acinar organoid structures. In addition, some cells were recovered from each group to test for knockdown efficiency using RT-qPCR. As seen in Fig. 4A, the PMECs were efficiently knocked down of miR146b up to 7 days post-LNA transfection. Furthermore, there was a 5.3-fold reduction in the number of hollow acinar organoid structures upon miR146b knockdown compared with the control groups (2±0 versus 10.6±2.4; means ± s.e.m., P<0.05), reflecting a reduction either in the number of luminal alveolar progenitors or their ability to give rise to the differentiated luminal structures on Matrigel (Fig. 4B,C). However, the number of the solid structures remained constant in all of the treatment groups. The number of hollow acinar structures were higher with prolactin treatment (16±4 versus 7±1.15; means ± s.e.m., P<0.05). However, the effect of miR146b knockdown was similar with or without treatment with prolactin.
Fig. 4.

MiR146b knockdown reduced the alveolar luminal progenitor cell number when grown in Matrigel®. (A) Quantitative PCR of the PMECs at 24 hours post-transfection (before growth on Matrigel®) and after 7 days on Matrigel. (B) Immunofluorescent images of the solid and hollow acinar structures. The nucleus is counterstained with DAPI (blue). (C) The solid organoid- and hollow acinus-forming efficiencies of PMECs transfected with the miR146b-LNA inhibitor (miR146b KD) compared with non-silencing controls (NS) and non-transfected cells (NT). The cells were cultured on Matrigel for 7 days, either with or without prolactin treatment (n = 3, P<0.05). (D,E) RT-qPCR of the CD-derived alveolar progenitor cells (D), and primary mammary epithelial cells (E) transfected with the miR146b-LNA inhibitor (miR146b KD) compared with non-silencing controls (NS) and non-transfected cells (NT). The cells were cultured on Matrigel for 72 hours, with prolactin treatment. Cells were recovered and analyzed for β-casein expression by qPCR (n = 3, *P<0.05 compared with other cells). Data are means ± s.e.m.
To further investigate the effect of miR146b knockdown on the alveolar progenitor cell function we tested the ability of the CD-derived alveolar progenitor cell line to express β-casein milk protein following miR146b knockdown. β-casein is endogenously expressed in the CD-cell line and can be rapidly induced by prolactin in vitro (Ball et al., 1988). The CD-derived alveolar progenitor cells and primary mouse epithelial cells were transfected with miR146b LNA inhibitor in 2D culture. At 24 hours post-transfection the cells were transferred to Matrigel and treated with prolactin (PRL) for another 72 hours. The cells were then recovered from the Matrigel and an RT-qPCR for β-casein mRNA levels was performed. MiR146b knockdown produced a significant reduction in β-casein expression compared with the level in the control CD-derived alveolar progenitor cells treated with PRL (2.3E4±0.1E4 versus 3.7E4±0.2E4; mean normalized expression ± s.e.m., P<0.05; Fig. 4D), as well as in the primary mouse mammary epithelial cells (0.1±0.03 versus 1±0.04; mean normalized expression ± s.e.m., P<0.05; Fig. 4E). These data show that miR146b regulated β-casein expression, a marker of alveolar cell viability and function.
MiR146b promotes alveolar progenitor cell maintenance, at least partially, through downregulation of STAT3β
We hypothesized that miR146b may promote alveolar progenitor cell survival and function by suppressing transcription factors with key roles during mammary gland development during pregnancy and or involution. Previous studies have shown miR146b targets to include interleukin (IL)-1 receptor associated kinase (IRAK1), TNF receptor-associated factor 6 (TRAF6), nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB) and SMAD family member 4 (SMAD4) (Bhaumik et al., 2008; Geraldo et al., 2012). Since some miRNAs might also upregulate expression (Ghosh et al., 2008; Vasudevan et al., 2007), we also examined signal transducer and activator of transcription 5a (STAT5a) and Elf5, two transcription factors that promote alveologenesis (Bouras et al., 2008; Siegel and Muller, 2010). For these experiments, the CD-derived alveolar progenitor cells were knocked down of miR146b to screen for targets. As shown in Fig. 5A, knockdown of miR146b by LNA inhibitors decreased the expression of SMAD4, IRAK1 and NFkB, but had no effects on STAT5a, TRAF6 and Elf5. These data suggest that the previously identified targets of miR146b may not be directly regulated in the mammary glands.
Fig. 5.

Effect of knockdown on miR146b known targets as well as on STAT3α/β. (A) Western analysis of Smad4, IRAK1, STAT5a, NFκB, TRAF6, ELF5 and β-actin in the alveolar progenitor cell line 48 hours post-transfection in non-transfected (NT), NS and miR146b KD cells. (B,C) qPCR showing successful knockdown (top panels, and representative western analysis (bottom panels) of total-STAT3 (top row), STAT3β-specific antibody (middle row in B) and β-actin (lower row) in the alveolar progenitor cell line and in non-transfected (NT), NS and miR146b KD cells (n = 4) 48 hours post-transfection.
STAT3 is a transcription factor that has been implicated previously in promoting mammary epithelial cell apoptosis at the onset of involution (Chapman et al., 1999). This finding prompted us to examine whether miR146b might target STAT3. The 3′UTR of the STAT3 gene was analyzed with Ensembl 2012 (Flicek et al., 2012) and RNA22 target prediction program (Miranda et al., 2006), and two putative miR146b binding sites were identified in the STAT3α 3′UTR (1.9 kb), while three miR146b binding sites were identified in STAT3β 3′UTR (3.6 kb; Fig. 6A). The two binding sites in STAT3α 3′UTR are similar to STAT3β 3′UTR.
Fig. 6.

MiR-146b targets the 3′UTR of STAT3α/β. (A) A diagram of the 3′UTRs of STAT3α and β computationally predicted binding sites for miR-146b. (B) Vector backbone of STAT3α/β 3′UTR reporter construct, STAT3α and β 3′UTR sequences were inserted downstream of the firefly luciferase reporter gene, driven by the SV40 promoter. Renilla luciferase was used as an internal control. (C) Luciferase reporter activity of the STAT3α and β 3′UTR in 293T cells expressing miR146b, the non-silencing negative control and the luciferase positive control. Reporter activity decreases by 30% when cells are overexpressing miR146b. Each sample was normalized to Renilla luciferase activity (n = 6,*P<0.0001). (D) Luciferase reporter activity of the STAT3β 3′UTR in PMECs from pregnant and virgin mice without treatment, and in PMECs treated with the luciferase positive control (n = 6,*P<0.0001). (E) RT-qPCR of PMECs transfected with the miR146b-LNA inhibitor (miR146b KD) compared with non-silencing controls (NS). At 72 hours post-transfection the cells were analyzed for STAT3α and STAT3β expression by qPCR (n = 3, P>0.05). Data are means ± s.e.m.
Knockdown of miR146b in the CD-derived alveolar progenitor cells, resulted in a significant increase in STAT3β expression compared with STAT3α (Fig. 5B; supplementary material Fig. S1E). A western blot using STAT3β-specific antibody (generated in the laboratory of D. J. T.) showed that the upregulated lower band in total STAT3 western blot was indeed STAT3β and not a product of STAT3α proteolytic degradation (Fig. 5B; middle panel). The STAT3β-specific antibody was raised against the unique C-terminal 7 residues in STAT3β protein. The increase in STAT3β expression upon miR146b knockdown in PMECs derived from pregnant mouse mammary glands was up to 2-fold compared with the controls (Fig. 5C, and densitometry data not shown). These data suggest that miR146b preferentially regulates STAT3β protein expression.
MiR146b binds to the 3′UTR of STAT3α and STAT3β
To determine if the putative miR146b binding sites resided within the 3′UTR of STAT3, we utilized luciferase constructs in which the 3′UTRs of STAT3α or STAT3β were inserted downstream of the firefly luciferase reporter gene (driven by SV40 promoter). A chimeric mRNA is transcribed consisting of the firefly luciferase and the STAT3α or β 3′UTR sequence. The vector also contains a Renilla luciferase as an internal control downstream of the CMV promoter (Fig. 6B). To examine whether miR146b directly regulates STAT3α and β, 293T cells were transfected either with the STAT3α or β 3′UTR reporter construct along with the miR146b precursor and the Pre-miR™ negative control. Firefly luciferase activity was then measured and normalized to Renilla luciferase activities in the same wells. The results showed a statistically significant reduction in luciferase reporter activity of STAT3α (70.2±1.7% versus 100±2.4%; mean normalized luciferase units ± s.e.m., P<0.0001) and in luciferase reporter activity of STAT3β (72.9±3.2% versus 100±3.1%; mean normalized luciferase units ± s.e.m., P<0.0001) when cells overexpressed miR146b compared with cells that expressed the Pre-miR™ negative control (Fig. 6C). Luciferase reporter activity of the STAT3α (data not shown) and STAT3β3′UTR was also significantly lower in PMECs from pregnant mice, which had significantly higher miR146b expression, compared with luciferase activity in PMECs from virgin mice (71±9.8% versus 100±5.6%; mean normalized luciferase units ± s.e.m., P<0.0001; Fig. 6D). These data suggest that miR146b directly binds the 3′UTR of STAT3α and STAT3β. In order to examine effects on STAT3 splicing, real time RT-PCR using specific primers to STAT3α and STAT3β was performed. The results showed that, upon miR146b knockdown, STAT3α and STAT3β mRNA levels were unchanged (Fig. 6E). Therefore, most likely, miR146b does not affect STAT3 splicing.
STAT3β may play a functional role during mammary gland involution
The contribution of specific STAT3 isoforms during mammary epithelial cell apoptosis at the onset of involution is not known at this time. To correlate miR146b expression with STAT3α/β levels during mammary development, we performed total STAT3 western blots using CD-derived progenitor clones as well as BALB/c-derived mammary glands from virgin, pregnant, lactating, and 1, 3 and 6 days post-weaning (involution) mice. Interestingly, STAT3β levels were inversely correlated with miR146b expression in the CD-derived progenitor clones (compare Fig. 7A and Fig. 1B). Furthermore, STAT3β levels were inversely correlated with miR146b levels at each physiological stage, while STAT3α was equally expressed throughout the distinct mammary developmental stages (compare Fig. 7B and Fig. 1C). As shown previously (Chapman et al., 1999) and in this study (Fig. 7C,D), there is an increase in STAT3α and STAT3β tyrosine phosphorylation (phosphor-Y705-STAT3, present in both STAT3α and STAT3β) prior to a significant rise in STAT3α serine phosphorylation (phosphor-S727-STAT3 present only in STAT3α) beginning at day 2 post-weaning. These findings suggests that STAT3β, either as homodimers or heterodimer with STAT3α or other transcription factors, may contribute to the onset of mammary involution. The specific STAT3 isoform knockout mice have been generated, but the contribution of each isoform to mammary involution has not been reported (Maritano et al., 2004). In order to relate the role of miR146b to STAT3β, we studied the effect of STAT3β overexpression on alveolar luminal cell viability and function. A cDNA overexpressing STAT3β was transfected into the CD-derived alveolar progenitor cells. The cells were collected at 24 hours post-transfection and stained with the LIVE/DEAD® Fixable Dead Cell Staining Kit. As expected, similar to the effects seen with miR146b knockdown, STAT3β overexpression reduced cell viability in the CD-derived alveolar progenitors by 20% compared with the controls (76.6±1.04% versus 95.7±1.5%; means ± s.e.m.; Fig. 7E,F). Furthermore, we examined whether overexpression of STAT3 isoforms resulted in a similar reduction in β-casein mRNA levels as seen with miR146b knockdown. For these experiments, virgin-derived mouse mammary epithelial cells were transfected with the STAT3β- and STAT3α-overexpressing cDNA clones, transferred onto Matrigel, and treated with PRL for 72 hours. The cells were then recovered for RT-qPCR. Fig. 7G, shows that similar to miR146b knockdown, overexpression of STAT3α and to a higher extent STAT3β resulted in a significant reduction in β-casein mRNA levels compared with the control (0.2±0.01 and 0.06±0.02 versus 1±0.13; mean normalized expression ± s.e.m., P<0.05). These data suggests that miR146b may regulate alveolar progenitor cells, at least partially, through direct regulation of STAT3β. Since we did not find a significant regulation of STAT3α protein expression by miR146b, we believe that miR146b effects are at least partially, through regulation of STAT3β.
Fig. 7.

STAT3β is activated in the mammary epithelial cells during involution. (A) Western blot analysis of total STAT3α/β in the CDβ cell line and the CD-derived clones. (B–D) Western blot analysis of total STAT3α/β (B); phosphorylated tyrosine 705 (phospho-Tyr-STAT3; C); phosphorylated serine 727 (phosphor-ser-STAT3; D) in total glands from virgin (V), pregnant (P) and lactating (L) mice and from mice at different physiological stages of involution: days 1, 3 and 6 (B–D) and 10 (C) days post-weaning (D-PW). (E) Representative histogram of flow cytometry analysis of the alveolar progenitor cell line stained with the LIVE/DEAD® Fixable Dead Cell Staining Kit at 24 hours post-transfection with STAT3β overexpression clone. (F) Quantification of the percentage of live cells (n = 3, P<0.05). (G) RT-qPCR of mouse mammary epithelial cells transfected with STAT3α(STAT3α-OE)- or STAT3β (STAT3β-OE)-overexpression clone compared with cells transfected with the empty vector (control). The cells were cultured on Matrigel for 72 hours, with prolactin treatment. Cells were recovered and analyzed for β-casein expression by qPCR (n = 3, *P<0.05 compared with other cells). Data are means ± s.e.m.
Discussion
In this study, it is demonstrated that miR146b expression is upregulated in the luminal alveolar progenitor cells during pregnancy and lactation and upon hormonal stimulation with (E plus P) and prolactin. Furthermore, transient knockdown of miR146b reduced survival of luminal alveolar progenitor cells. These data suggest that miR146b participates in alveologenesis under the influence of sex hormones and prolactin. The effects of miR146b may, at least partially, be through suppression of key transcription factors involved in alveolar cell death during involution, such as STAT3β.
Although many studies report the role of miR146b in cancer, its role in mammary gland development is still unknown. Bockmeyer et al. studied the miRNA profiles of human basal and luminal epithelial cells as well as basal and luminal breast cancers (Bockmeyer et al., 2011). These studies showed miR146b to be a basal-specific miRNA and to be upregulated in basal-like breast cancers when compared with luminal A and B breast cancer subtypes (Bockmeyer et al., 2011). Other than in this report, expression of miR146b and its role in mammary epithelial cell development have not been described.
We reasoned that miR146b might play a role in alveolar cell maintenance by regulating a known target such as IRAK1/TRAF6, NF-κB, or SMAD4 or by regulating transcription factors with known roles during alveologenesis such as STAT5a and E74-like factor 5 (ELF5). A review of the literature showed miR146b to regulate TGF-β signaling by repressing SMAD4 in thyroid cancers (Geraldo et al., 2012). TGF-β has been shown to play an important role during mammary involution by inducing cellular apoptosis and alveolar collapse in the early stages of involution (Flanders and Wakefield, 2009). Furthermore, miR146b has been shown to inhibit breast cancer cell invasion and migration by targeting IRAK1/TRAF6 and by downregulating NF-kB signaling (Bhaumik et al., 2008). NF-kB has also been shown to destabilize STAT5a, a key transcription factor that promotes alveologenesis (Floyd et al., 2007). Interestingly, miR146b knockdown did not result in the upregulation of any of the known suspected targets. Therefore, the previously identified miR146b targets may not be similarly regulated in normal mammary glands.
Since STAT3 transcription factors were reported to play key roles in mammary epithelial cell apoptosis at the onset of involution, we explored the role of miR146b in regulating STAT3 isoforms. STAT3 has two main isoforms: the full-length STAT3α and the truncated STAT3β, by alternative mRNA splicing of exon 23 (Huang et al., 2007; Zammarchi et al., 2011). In STAT3β, the 55-amino acid C-terminal acidic transactivation domain of STAT3α is replaced by seven unique amino acid residues (Zammarchi et al., 2011). An important factor in STAT3 functional heterogeneity may be the existence of these two alternatively spliced isoforms. As suspected, miR146b knockdown resulted in upregulation of STAT3 expression in both PMECs derived from pregnant mammary glands and in the alveolar progenitor cells. However, the upregulation of the STAT3β isoform was higher than that of STAT3α. There are several possibilities for preferential regulation of STAT3β protein expression by miR146b. One is the existence of an additional miR146b binding site on the predicted STAT3β 3′UTR, resulting in a different secondary structure and higher efficiency of mir146b binding (Flicek et al., 2012). Another mechanism, may be the regulation of alternative splicing events. The interplay between the miRNA and splicing regulation has been demonstrated previously. These studies showed that miR124 and miR133 regulate the RNA-binding protein, polypyrimidine tract-binding protein 1 (PTBP1). PTBP1 is a global repressor of alternative pre-mRNA splicing in non neuronal cells (Boutz et al., 2007; Makeyev et al., 2007). Additionally, ESEfinder and PESX algorithms predicted a putative exonic splicing enhancer (ESE), which overlaps with one of the predicted miR146b binding sites. MiR146b binding to this overlapped region may regulate ESE. MiR146b could also indirectly regulate STAT3 alternative splicing by targeting key splicing factors. Real-time RT-PCR results showed that STAT3α and STAT3β mRNA levels remain constant in miR146b knockdown cells despite striking changes in protein expression, suggesting that miR146b most likely affects STAT3 protein translation.
STAT3 is characterized by its capacity to activate different sets of genes in different cell types (Levy and Lee, 2002), and the different STAT3 isoforms seem to regulate cell survival and maintenance in a cell-type- and context-specific manner by suppressing or activating a variety of target genes (Pensa et al., 2009). STAT3β has been considered a dominant-negative factor by inhibiting STAT3α gene transactivation (Caldenhoven et al., 1996; Schaefer et al., 1995). More recent studies have demonstrated that STAT3β may activate a specific set of genes and possess non-redundant functions to STAT3α. Zammarchi et al. used morpholino oligomers to redirect endogenous STAT3 alternative splicing from STAT3α to STAT3β (Zammarchi et al., 2011). This study showed that inducing the STAT3β isoform in MDA-MB-435 cells caused cell death and tumor regression in the xenograft models. The same study showed that STAT3 canonical target genes were not affected by the switch from STAT3α to STAT3β. Rather, a unique expression signature seems to be associated with the physiological α-to-β splicing shift (Zammarchi et al., 2011). In mammary gland development, it has become clear that STAT3 regulates a complex series of processes involving alveolar epithelial cell death during involution by maintaining the balance of inflammatory and anti-inflammatory signaling (Pensa et al., 2009). The contribution of specific STAT3 isoforms to the mammary involution process is still not known. Knockout mice of specific STAT3 isoforms have been generated (Maritano et al., 2004). It is reported that STAT3β knockout mice are viable, fertile and capable of nursing their pups (Maritano et al., 2004). However, a detailed characterization of the mammary glands during involution has not been reported in these mice. Our study clearly demonstrates that both isoforms of STAT3 are upregulated during involution, and overexpression of STAT3β in the alveolar luminal progenitor cells reduced their survival. It would be interesting to decipher the contribution of specific STAT3 isoforms to the mammary involution process in specific STAT3-isoform knockout mice.
In conclusion, we have demonstrated that estrogen plus progesterone and prolactin result in the upregulation miR146b. Furthermore, miR146b levels rise in the mouse mammary glands during lactation and pregnancy. MiR146b upregulation during pregnancy and lactation may mediate the maintenance of alveolar luminal progenitor cells by selectively suppressing STAT3β and to a lesser extent STAT3α. Following weaning and during the process of involution, miR146b levels drop to basal levels, which result in the upregulation of STAT3 isoforms and induction of alveolar cell death (Fig. 8). The LNA inhibitors induce a transient knockdown in miR146b; thus they are not suitable for long term in vivo transplantation studies. It will be essential to confirm our results by more stable miR146b knockdown and overexpression in mammary epithelial cells followed by in vivo transplantation studies.
Fig. 8.

A proposed model for the role of miR-146b in alveolar progenitor cell maintenance. Mammary epithelial cell hierarchy begins by asymmetric self-renewal in the stem cells, which generates multipotent and bipotent ductal and alveolar progenitor cells. Ductal and alveolar progenitor cells give rise to luminal- and myoepithelium-restricted progenitors. During pregnancy and lactation, miR146b is upregulated under the influence of estrogen, progesterone and prolactin. Upregulation of miR146b results in survival of luminal alveolar progenitor cells, at least partially, through suppression of STAT3β/α. During involution, miR-146b is downregulated in the alveolar progenitors to de-repress STAT3β/α, followed by death of the luminal alveolar cells resulting in the onset of involution.
Material and Methods
Cell lines
CommaDβ-derived clones (SP-1, SP-3, SP-4, NSP-1, NSP-2, NSP-3, NSP-4 and NSP-5) were used. The alveolar progenitor (SP-3), ductal progenitor (NSP-2) and multipotent progenitor clones (NSP-5) have previously been described and characterized (Kittrell et al., 2011). 293T cells (Clontech, cat. no. 632180) were used for luciferase reporter assays.
Animals
We used female BALB/c mice which were either bred or purchased from Harlan Laboratories, Inc., IN, USA. We used mice in late pregnancy, as well as mice 24 hours, 3 days and 6 days post-weaning (involuting mammary glands), and virgin mice in the metestrous phase of the estrous cycle as confirmed by vaginal cytology (Caligioni, 2009). Animal experiments were conducted following protocols approved by the University of Kansas Medical Center Institutional Animal Care and Use Committee.
Mouse surgeries
Slow-releasing pellets containing 50 µg of estradiol and 20 mg of progesterone for hormonal treatment were placed subcutaneously on the lateral side of the neck between the ear and shoulder. After three weeks, mice were sacrificed, and the mammary glands were excised. One mammary gland was processed for embedding, and the other mammary gland was used to recover the epithelial cells according to established protocol in our laboratory (Valdez et al., 2011).
Cell culture: 2D/3D (Matrigel™)
Primary mouse cells were grown and maintained in F12 (Invitrogen, cat. no. 11765) supplemented with 5% fetal bovine serum (Invitrogen, cat. no. 10438026), 1% antibiotic/antimycotic reagents (AA; Invitrogen, cat. no. 15240), 50 µg/ml gentamicin (Sigma-Aldrich cat. no. G1272), 1 mg/ml hydrocortisone (Stem Cell Technologies, cat. no. 07904), and 5 ng/ml epidermal growth factor. The CD-derived clones (i.e. SP-3, NSP-2 and NSP-5) were grown in DMEM/F12 (Invitrogen, cat. no. 11320) containing 2% fetal bovine serum, 5 ng/ml epidermal growth factor, 10 mM HEPES (Invitrogen, cat. no.15630), 5 µg/ml insulin, 50 µg/ml gentamicin and 1% antibiotic/antimycotic.
293T cells were grown in DMEM (Sigma-Aldrich, cat. no. D5796) containing 10% fetal bovine serum, and 1% antibiotic/antimycotic. Hormonal treatment include: 100 nM progesterone (Sigma, cat. no. P0130-25G), 100 µM β-estradiol (Sigma, cat. no. E8875-1G) and 3 µg/ml ovine prolactin (Harbor-UCLA Research and education institute). The cells were grown in humidified incubator at 37°C and 5% CO2. For 3D culture, cells were plated in media containing 2% BD Basement Membrane Matrix Matrigel™ (BD Biosciences, cat. no. 356234) on a thin film of Matrigel™. Cell recovery was achieved using BD Cell Recovery Solution (BD Biosciences, cat. no. 354253).
Hematoxylin and Eosin staining
Excised mammary glands were fixed in 4% paraformaldehyde for 2 hours on ice, then in 70% ethanol at 4°C until processing, and then embedded in paraffin according to established protocols. The slides containing 5-µm sections were deparaffinized in xylene (X3P-1GAL, Fisher scientific) and hydrated through graded ethanol series (100%, 95%, 80% and 70%), followed by hematoxylin staining (s212A-32OZ Harris hematoxylin with glacial acetic acid; Poly Scientific) for 30 seconds and destaining in acid ethanol (0.3% of concentrated HCl in 70% ethanol), followed by eosin staining (S176 Eosin Phloxine stain; Poly Scientific) for 30 seconds and dehydration in graded ethanol (95% and 100%) and xylene three times for 15 minutes each, followed by placement of a coverslip onto the slide with xylene-based permount (SP15-100 histological mounting medium, Fisher Scientific) (Valdez et al., 2011). Light microscopy was performed by using an Axio Imager.M2 microscope (Carl Zeiss MicroImaging, Inc., Thornwood, NY, USA). Pictures were taken at 10× magnification (objective N-Achroplan; 10×/0.25 ∞/0.17) with an AxioCam MRc5 High Resolution Camera (Carl Zeiss MicroImaging, Inc., Thornwood, NY, USA), and the acquisition software used was Ziess Axiovision GmbH (Carl Zeiss MicroImaging, Inc.).
RNA Isolation and Quantitative PCR (qPCR)
Total RNA was isolated with miRNeasy Mini Kit (Qiagen cat. no. 217004) using the manufacturer's protocol, and cDNA was synthesized from 250 ng of total RNA with miScript Reverse Transcription Kit (Qiagen, cat. no. 218061). MiRNA and mRNA were measured using miScript SYBR Green PCR Kit (Qiagen, cat. no. 218073), power SYBR® green PCR Master Mix (Applied Biosystems, cat. no. 4367659), TaqMan® Gene Expression Master Mix (Applied Biosystems, cat. no. 4369016) and primers specific for hsa-miR-146b-5p (IDT, Ref. no. 52053397), hsa-miR-146a (IDT, Ref. no. 51289005), STAT3a/b forward (IDT, Ref. no. 86187028), STAT3a reverse (IDT, Ref. no. 86187029), STAT3b reverse (IDT, Ref. no. 86187030), β-casein (Applied Biosystems, cat. no. Mm04207880-m1). Reactions were performed in the StepOnePlus™ Real-Time PCR System and software (Applied Biosystems) in 96-well plates. Target gene expression was normalized to snU6: snU6 forward (IDT, Ref. no. 50315659) and snU6 reverse (IDT, Ref. no. 50315660), actin forward (IDT, Ref. no. 86146439), actin reverse (IDT, Ref. no. 86146440) keratin 18 forward (IDT, Ref. no. 89270345), actin (Applied Biosystems, cat. no. Mm01204962-gH), keratin 18 reverse (IDT, Ref. no. 89270346). The standard curve method was used for quantification.
Statistical analysis
Data are presented as mean normalized expression ± s.e.m. One-way analysis of variance (ANOVA) was used for statistical comparisons. A value of P≤0.05 was considered significant.
Microarray studies
RNA from CDβ clones with growth potential (SP-3, NSP-1, NSP-2, NSP-5) and those without growth potential (NSP-3, NSP-4, SP-1, SP-4) were combined. RNA quantity and quality (RIN >8) was checked using the Agilent Bioanalyzer. Reverse transcription was done using SA Biosciences RT2 miRNA First Strand Kit, RT2 miRNA PCR Array System (human cancer array, SABiosciences MAH-102E). PCR array was loaded according to SA Biosciences protocol for loading map. The data were analyzed using the ΔΔCT method. (www.sabiosciences.com/pcrarraydataanalysis.php).
FACS analysis and sorting
Primary antibodies used were anti-mouse CD29 (APC/Cy7)-conjugated antibody (Biolegend, cat. no. 102226), anti-mouse CD24 (PE)-conjugated antibody (BD Biosciences, cat. no. 553262), anti-mouse CD61 (Alexa Fluor 647)-conjugated (Biolegend, cat. no. 104314), biotin-conjugated anti-mouse CD31 (Biolegend, cat. no. 102404), biotin-conjugated anti-mouse CD140a (Biolegend, cat. no. 135910), biotin-conjugated Rat anti-mouse CD45 (BD Pharmingen, cat. no. 553077), biotin-conjugated rat anti-mouse Ter-119 (BD Pharmingen, cat. no. 559971). The biotin-conjugated antibodies were labeled using streptavidin-V450 (BD Biosciences, cat. no. 560797). Isotype control staining was performed using PE-conjugated anti-rat immunoglobulin G2bk (IgG2bk) antibody (Biolegend, cat. no. 400635), APC/Cy7-conjugated mouse IgG2bk antibody (BD Pharmingen, cat. no. 558061) and biotin-conjugated rat IgG2ak (IgG2ak) antibody (BD Pharmingen, cat. no. 553928). The cells were stained at a final concentration of 1∶200 for 30 minutes on ice followed by washes in Hanks' balanced salt solution (Invitrogen, Carlsbad, CA, USA) containing 2% fetal bovine serum. For the live–dead assay, LIVE/DEAD® Fixable Dead Cell Stain Kit (Invitrogen, cat. no. L34955) was used according to the manufacturer's protocol. FACS and data analysis were performed using the BD LSR II flow cytometer and FlowJo software (Tree Star, Inc., Ashland, OR, USA). Cell sorting was performed using the FACS Aria (BD Biosciences, San Jose, California, USA).
MiR146b knockdown and overexpression in CDβ clones and in PMECs
To overexpress miR-146b, 40 nM of hsa-miR-146b-5p synthetic pre-miR precursors (Ambion, RefNO. AM17100) were transfected into PMECs, along with the Pre-miR™miRNA Precursor Negative Control #1 (Ambion RefNO. AM17110). To knockdown miR-146b, 40 nM of miRCURY LNA microRNA inhibitor (Exiqon, cat. no. 410066-04) for hsa-miR-146b-5p was transfected into the cell lines and PMECs.
Because the human and murine miR-146b-5p have identical sequences, the use of pre-miRs and LNA inhibitor products designed for human are a perfect match for the mouse. MiRCURY LNA microRNA inhibitor control (Exiqon, cat. no. 199004-04) was used as a negative control. In both cases, cells were seeded in a six-well plate and transfected 24 hours later with Lipofectamine™ 2000 Transfection Reagent (Invitrogen, cat. no. 11668-019) in the media described above, without serum or antibiotic present. Twenty-four hours after transfection, the cells were nourished with 1% serum. Seventy-two hours post-transfection, the cells were washed with PBS and recovered for analysis as described above.
STAT3a and b overexpression in 293T cells
293T cells were transiently transfected with cDNA encoding mouse STAT3β (OriGene Technologies, cat. no. MC221089), mouse STAT3α (OriGene Technologies, cat. no. MC221487) and control (OriGene Technologies, cat. no. PS100001), according to the manufacturer's protocol.
Western blot
Cells were lysed on ice using RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EGTA, 0.25% deoxycholate and 1% Triton X-100) containing protease inhibitors (Calbiochem, cat. no. 535140) and phosphatase inhibitors (Calbiochem, cat. no.524627). The protein extracts were prepared by collecting supernatants after centrifugation at 12,000 g at 4°C for 30 minutes. The protein concentrations were measured using the BCA protein assay kit according to the manufacturer's protocol (Thermo Scientific, cat. no. 23250). Ten µg of protein was separated on an 8–10% SDS-PAGE gel, transferred onto PVDF membrane (Millipore, 0.45 mm) in Tris-glycine (25 mM Tris and 192 mM glycine, pH 8.3; BioRad, cat. no. 161-0771) containing 20% v/v methanol at 100 V for 2 hours. The PVDF membrane was blocked with 5% non-fat dry milk in TBS containing 0.05% Tween 20 (TBST) at room temperature for 1 hour. The blots were washed three times for 10 minutes with TBST before incubating with primary antibody in TBST for 1 hour at room temperature or overnight at 4°C. The primary antibodies STAT5a (Invitrogen, cat. no. 13-3600), STAT3 (K-15; Santa Cruz, cat. no. sc-483), p(Ser)-STAT3 (23G5; Santa Cruz, cat. no. sc-56747), p(Tyr)-STAT3 (Cell signaling, cat. no. 9131S) TRAF6 (H-274; Santa Cruz, cat. no. sc-7221), IRAK1 (H-273; Santa Cruz, cat. no. sc-7883), STAT5a (ST5a-2H2; Invitrogen, cat. no. 13-3600), NFkB p65 (C22B4; Cell Signaling, cat. no. 4764), ELF5 (N-20; Santa Cruz, cat. no. sc-9645) and Smad4 (B-8; Santa Cruz, cat. no. sc-7966) were used at a dilution of 1∶1000, mouse monoclonal antibody clone specific for the C-terminal 7 residues of STAT3β was generated in D.J.T.'s laboratory, and was used at a dilution of 1∶100 overnight. Following primary antibody incubation, the membranes were washed thoroughly with TBST (3×, 10 minutes each) and incubated with horseradish-peroxidase (HRP)-conjugated secondary antibodies donkey anti-rabbit-HRP (Jackson, cat. no. 711-035-152) at 1∶10,000 or goat anti-mouse-HRP (Santa Cruz, cat. no. sc2005) at 1∶10,000 in TBST containing 1% milk for 1 hour at room temperature. After washing with TBST (3×, 10 minutes each), the bands were visualized with enhanced chemiluminescence (ECL), SuperSignal West Femto Kit (Thermo Scientific, cat. no. 34095) and the blots were digitalized using Auto Chemi systems (UVP Inc., Upland, CA). The membranes were stripped by incubating with stripping buffer (50 mM Tris-HCl, pH 6.8, 2% SDS and 100 mM β-marcaptoethanol) at 50°C for 30 minutes followed by washing with TBST and re-probed with β-actin antibody (Santa Cruz, cat. no. sc1616, 1∶1500 dilution) followed by anti-goat IgG HRP (Santa Cruz, cat. no. sc2350, 1∶10,000 dilution) for loading control. Densitometry analysis was performed using LabWorks software (UVP Inc., Upland, CA).
Luciferase reporter constructs
293T cells (Clontech, cat. no. 632180) were plated in 6-well plates. After 24 hours, the cells were transfected with 1.0 µg of STAT3α 3′UTR (GeneCopoiea, cat. no. MmiT043695) or with STAT3β 3′UTR (GeneCopoiea, cat. no. CS-MmiT102J-MT01) miRNA target sequence expression clone in pEZX-MT01 vector with fLuc along with 40 nM of miR-146b precursor miRNA (Ambion, RefNO. AM17100) and miRNA control (Ambion RefNO. AM17110). The cells were transferred to a 96-well plate 18 hours after transfection and cultured for another 24 hours. Both firefly luciferase and Renilla luciferase activities were measured using GeneCopoeia Luc-Pair™ miR Luciferase Assay kit (GeneCopoiea, cat. no. LPFR-M030) according to the manufacturer's protocol, and data were recorded on a BioTek Microplate Reader using Gen5™software. Firefly luciferase activity was normalized with Renilla luciferase activities in the same well.
Supplementary Material
Acknowledgments
We thank Drs Timothy Fields, Fang Fan, Nikki Cheng and Roy Jensen, University of Kansas Medical Center for their advice and guidance throughout this project; Richard Hastings and Alicia Zeiger, University of Kansas Medical Center for flow cytometry technical assistance and guidance.
Footnotes
Author contributions
H.E. participated in the design of the study, carried out the transfections, animal surgeries, FACS sorting and analysis, western blots, imaging, RT-PCR and drafted the manuscript; Y.H. participated in animal hormonal treatment and surgeries, western blots and H&E staining; K.V. participated in imaging and manuscript figures; M.C. performed the miRNA microarrays; U.B. generated and validated the STAT3β-specific antibody. S.S., M.M., D.J.T., P.P., D.T. and L.K.C. participated in the conception and design of the study; F.B. conceived the study, and participated in its design and coordination. All authors read and approved the final manuscript.
Funding
This work was supported by the National Institutes of Health [grant number 5 R00 CA127462 to F.B.]; a Center of Excellence Fellowship (to K.V.); a Center of Biomedical Research Excellence Pilot award [grant number P20 RR024214 to F.B.]; the Libyan North American Scholarship Program-Canadian Bureau for International Education (to H.E.); and the Department of Defense [grant number 5W81XWH-11-2-0018 P0004 to D.J.T.]. We acknowledge the Flow Cytometry Core Laboratory, which is sponsored, in part, by the National Institutes of Health/National Institute of General Medical Sciences Centers of Biomedical Research Excellence [grant number P30 GM103326]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.119214/-/DC1
References
- Andl T., Murchison E. P., Liu F., Zhang Y., Yunta-Gonzalez M., Tobias J. W., Andl C. D., Seykora J. T., Hannon G. J., Millar S. E. (2006). The miRNA-processing enzyme dicer is essential for the morphogenesis and maintenance of hair follicles. Curr. Biol. 16, 1041–1049 10.1016/j.cub.2006.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselin-Labat M. L., Sutherland K. D., Barker H., Thomas R., Shackleton M., Forrest N. C., Hartley L., Robb L., Grosveld F. G., van der Wees J. et al. (2007). Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat. Cell Biol. 9, 201–209 10.1038/ncb1530 [DOI] [PubMed] [Google Scholar]
- Avril-Sassen S., Goldstein L. D., Stingl J., Blenkiron C., Le Quesne J., Spiteri I., Karagavriilidou K., Watson C. J., Tavaré S., Miska E. A. et al. (2009). Characterisation of microRNA expression in post-natal mouse mammary gland development. BMC Genomics 10, 548 10.1186/1471-2164-10-548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball R. K., Friis R. R., Schoenenberger C. A., Doppler W., Groner B. (1988). Prolactin regulation of beta-casein gene expression and of a cytosolic 120-kd protein in a cloned mouse mammary epithelial cell line. EMBO J. 7, 2089–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E., Kim S. Y., Carmell M. A., Murchison E. P., Alcorn H., Li M. Z., Mills A. A., Elledge S. J., Anderson K. V., Hannon G. J. (2003). Dicer is essential for mouse development. Nat. Genet. 35, 215–217 10.1038/ng1253 [DOI] [PubMed] [Google Scholar]
- Bhaumik D., Scott G. K., Schokrpur S., Patil C. K., Campisi J., Benz C. C. (2008). Expression of microRNA-146 suppresses NF-kappaB activity with reduction of metastatic potential in breast cancer cells. Oncogene 27, 5643–5647 10.1038/onc.2008.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockmeyer C. L., Christgen M., Müller M., Fischer S., Ahrens P., Länger F., Kreipe H., Lehmann U. (2011). MicroRNA profiles of healthy basal and luminal mammary epithelial cells are distinct and reflected in different breast cancer subtypes. Breast Cancer Res. Treat. 130, 735–745 10.1007/s10549-010-1303-3 [DOI] [PubMed] [Google Scholar]
- Bouras T., Pal B., Vaillant F., Harburg G., Asselin-Labat M-L., Oakes S. R., Lindeman G. J., Visvader J. E. (2008). Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell 3, 429–441 10.1016/j.stem.2008.08.001 [DOI] [PubMed] [Google Scholar]
- Boutz P. L., Chawla G., Stoilov P., Black D. L. (2007). MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev. 21, 71–84 10.1101/gad.1500707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldenhoven E., van Dijk T. B., Solari R., Armstrong J., Raaijmakers J. A., Lammers J. W., Koenderman L., de Groot R. P. (1996). STAT3beta, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J. Biol. Chem. 271, 13221–13227 [DOI] [PubMed] [Google Scholar]
- Caligioni C. S. (2009). Assessing reproductive status/stages in mice. Curr Protoc Neurosci. 48, A.4I.1–A.4I.8 10.1002/0471142301.nsa04is48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman R. S., Lourenco P. C., Tonner E., Flint D. J., Selbert S., Takeda K., Akira S., Clarke A. R., Watson C. J. (1999). Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of Stat3. Genes Dev. 13, 2604–2616 10.1101/gad.13.19.2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W., Li Q., Feng L., Ding W. (2011). MiR-126-3p regulates progesterone receptors and involves development and lactation of mouse mammary gland. Mol. Cell. Biochem. 355, 17–25 10.1007/s11010-011-0834-1 [DOI] [PubMed] [Google Scholar]
- Flanders K. C., Wakefield L. M. (2009). Transforming growth factor-(beta)s and mammary gland involution; functional roles and implications for cancer progression. J. Mammary Gland Biol. Neoplasia 14, 131–144 10.1007/s10911-009-9122-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P., Amode M. R., Barrell D., Beal K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fairley S., Fitzgerald S. et al. (2012). Ensembl 2012. Nucleic Acids Res. 40, D84–D90 10.1093/nar/gkr991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd Z. E., Segura B. M., He F., Stephens J. M. (2007). Degradation of STAT5 proteins in 3T3-L1 adipocytes is induced by TNF-alpha and cycloheximide in a manner independent of STAT5A activation. Am. J. Physiol. Endocrinol. Metab. 292, E461–E468 10.1152/ajpendo.00334.2006 [DOI] [PubMed] [Google Scholar]
- Fujita P. A., Rhead B., Zweig A. S., Hinrichs A. S., Karolchik D., Cline M. S., Goldman M., Barber G. P., Clawson H., Coelho A. et al. (2011). The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 39, D876–D882 10.1093/nar/gkq963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia A. I., Buisson M., Bertrand P., Rimokh R., Rouleau E., Lopez B. S., Lidereau R., Mikaélian I., Mazoyer S. (2011). Down-regulation of BRCA1 expression by miR-146a and miR-146b-5p in triple negative sporadic breast cancers. EMBO Mol. Med 3, 279–290 10.1002/emmm.201100136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraldo M. V., Yamashita A. S., Kimura E. T. (2012). MicroRNA miR-146b-5p regulates signal transduction of TGF-β by repressing SMAD4 in thyroid cancer. Oncogene 31, 1910–1922 10.1038/onc.2011.381 [DOI] [PubMed] [Google Scholar]
- Ghosh T., Soni K., Scaria V., Halimani M., Bhattacharjee C., Pillai B. (2008). MicroRNA-mediated up-regulation of an alternatively polyadenylated variant of the mouse cytoplasmic beta-actin gene. Nucleic Acids Res. 36, 6318–6332 10.1093/nar/gkn624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene S. B., Herschkowitz J. I., Rosen J. M. (2010). Small players with big roles: microRNAs as targets to inhibit breast cancer progression. Curr. Drug Targets 11, 1059–1073 10.2174/138945010792006762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Keckesova Z., Donaher J. L., Shibue T., Tischler V., Reinhardt F., Itzkovitz S., Noske A., Zürrer-Härdi U., Bell G. et al. (2012). Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148, 1015–1028 10.1016/j.cell.2012.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H., Jazdzewski K., Li W., Liyanarachchi S., Nagy R., Volinia S., Calin G. A., Liu C. G., Franssila K., Suster S. et al. (2005). The role of microRNA genes in papillary thyroid carcinoma. Proc. Natl. Acad. Sci. USA 102, 19075–19080 10.1073/pnas.0509603102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Qiu J., Dong S., Redell M. S., Poli V., Mancini M. A., Tweardy D. J. (2007). Stat3 isoforms, alpha and beta, demonstrate distinct intracellular dynamics with prolonged nuclear retention of Stat3beta mapping to its unique C-terminal end. J. Biol. Chem. 282, 34958–34967 10.1074/jbc.M704548200 [DOI] [PubMed] [Google Scholar]
- Hurst D. R., Edmonds M. D., Welch D. R. (2009). Metastamir: the field of metastasis-regulatory microRNA is spreading. Cancer Res. 69, 7495–7498 10.1158/0008-5472.CAN-09-2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellopoulou C., Muljo S. A., Kung A. L., Ganesan S., Drapkin R., Jenuwein T., Livingston D. M., Rajewsky K. (2005). Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 19, 489–501 10.1101/gad.1248505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittrell F. S., Carletti M. Z., Kerbawy S., Heestand J., Xian W., Zhang M., Lamarca H. L., Sonnenberg A., Rosen J. M., Medina D. et al. (2011). Prospective isolation and characterization of committed and multipotent progenitors from immortalized mouse mammary epithelial cells with morphogenic potential. Breast Cancer Res. 13, R41 10.1186/bcr2863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D. E., Lee C. K. (2002). What does Stat3 do? J. Clin. Invest. 109, 1143–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis B. P., Shih I. H., Jones-Rhoades M. W., Bartel D. P., Burge C. B. (2003). Prediction of mammalian microRNA targets. Cell 115, 787–798 10.1016/S0092-8674(03)01018-3 [DOI] [PubMed] [Google Scholar]
- Lim E., Vaillant F., Wu D., Forrest N. C., Pal B., Hart A. H., Asselin-Labat M. L., Gyorki D. E., Ward T., Partanen A. et al. (2009). Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 15, 907–913 10.1038/nm.2000 [DOI] [PubMed] [Google Scholar]
- Makeyev E. V., Zhang J., Carrasco M. A., Maniatis T. (2007). The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 27, 435–448 10.1016/j.molcel.2007.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maritano D., Sugrue M. L., Tininini S., Dewilde S., Strobl B., Fu X., Murray-Tait V., Chiarle R., Poli V. (2004). The STAT3 isoforms alpha and beta have unique and specific functions. Nat. Immunol. 5, 401–409 10.1038/ni1052 [DOI] [PubMed] [Google Scholar]
- Miranda K. C., Huynh T., Tay Y., Ang Y. S., Tam W. L., Thomson A. M., Lim B., Rigoutsos I. (2006). A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 126, 1203–1217 10.1016/j.cell.2006.07.031 [DOI] [PubMed] [Google Scholar]
- Muljo S. A., Ansel K. M., Kanellopoulou C., Livingston D. M., Rao A., Rajewsky K. (2005). Aberrant T cell differentiation in the absence of Dicer. J. Exp. Med. 202, 261–269 10.1084/jem.20050678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensa S., Watson C. J., Poli V. (2009). Stat3 and the inflammation/acute phase response in involution and breast cancer. J. Mammary Gland Biol. Neoplasia 14, 121–129 10.1007/s10911-009-9124-x [DOI] [PubMed] [Google Scholar]
- Piao H. L., Ma L. (2012). Non-coding RNAs as regulators of mammary development and breast cancer. J. Mammary Gland Biol. Neoplasia 17, 33–42 10.1007/s10911-012-9245-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer T. S., Sanders L. K., Nathans D. (1995). Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proc. Natl. Acad. Sci. USA 92, 9097–9101 10.1073/pnas.92.20.9097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackleton M., Vaillant F., Simpson K. J., Stingl J., Smyth G. K., Asselin-Labat M. L., Wu L., Lindeman G. J., Visvader J. E. (2006). Generation of a functional mammary gland from a single stem cell. Nature 439, 84–88 10.1038/nature04372 [DOI] [PubMed] [Google Scholar]
- Siegel P. M., Muller W. J. (2010). Transcription factor regulatory networks in mammary epithelial development and tumorigenesis. Oncogene 29, 2753–2759 10.1038/onc.2010.43 [DOI] [PubMed] [Google Scholar]
- Stingl J., Eirew P., Ricketson I., Shackleton M., Vaillant F., Choi D., Li H. I., Eaves C. J. (2006). Purification and unique properties of mammary epithelial stem cells. Nature 439, 993–997 [DOI] [PubMed] [Google Scholar]
- Valdez K. E., Fan F., Smith W., Allred D. C., Medina D., Behbod F. (2011). Human primary ductal carcinoma in situ (DCIS) subtype-specific pathology is preserved in a mouse intraductal (MIND) xenograft model. J. Pathol. 225, 565–573 10.1002/path.2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan S., Tong Y., Steitz J. A. (2007). Switching from repression to activation: microRNAs can up-regulate translation. Science 318, 1931–1934 10.1126/science.1149460 [DOI] [PubMed] [Google Scholar]
- Watson C. J., Khaled W. T. (2008). Mammary development in the embryo and adult: a journey of morphogenesis and commitment. Development 135, 995–1003 10.1242/dev.005439 [DOI] [PubMed] [Google Scholar]
- Xia H., Qi Y., Ng S. S., Chen X., Li D., Chen S., Ge R., Jiang S., Li G., Chen Y. et al. (2009). microRNA-146b inhibits glioma cell migration and invasion by targeting MMPs. Brain Res. 1269, 158–165 10.1016/j.brainres.2009.02.037 [DOI] [PubMed] [Google Scholar]
- Zammarchi F., de Stanchina E., Bournazou E., Supakorndej T., Martires K., Riedel E., Corben A. D., Bromberg J. F., Cartegni L. (2011). Antitumorigenic potential of STAT3 alternative splicing modulation. Proc. Natl. Acad. Sci. USA 108, 17779–17784 10.1073/pnas.1108482108 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.