

Abstract
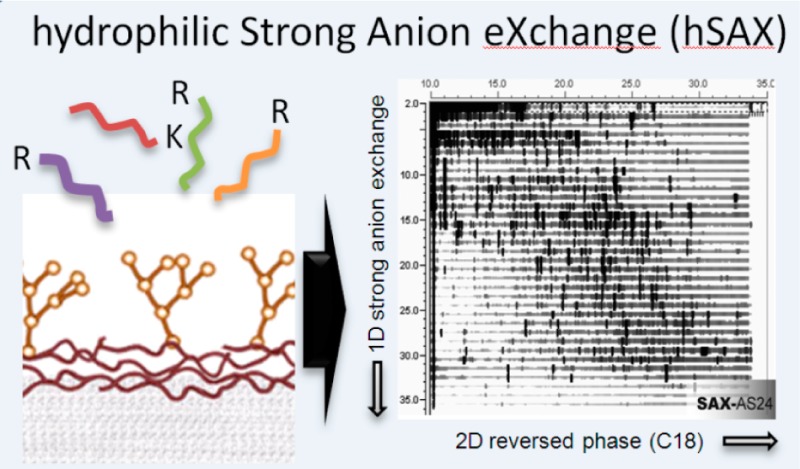
Due to its compatibility and orthogonality to reversed phase (RP) liquid chromatography (LC) separation, ion exchange chromatography, and mainly strong cation exchange (SCX), has often been the first choice in multidimensional LC experiments in proteomics. Here, we have tested the ability of three strong anion exchanger (SAX) columns differing in their hydrophobicity to fractionate RAW264.7 macrophage cell lysate. IonPac AS24, a strong anion exchange material with ultralow hydrophobicity, demonstrated to be superior to other materials by fractionation and separation of tryptic peptides from both a mixture of 6 proteins as well as mouse cell lysate. The chromatography displayed very high orthogonality and high robustness depending on the hydrophilicity of column chemistry, which we termed hydrophilic strong anion exchange (hSAX). Mass spectrometry analysis of 34 SAX fractions from RAW264.7 macrophage cell lysate digest resulted in an identification of 9469 unique proteins and 126318 distinct peptides in one week of instrument time. Moreover, when compared to an optimized high pH/low pH RP separation approach, the method presented here raised the identification of proteins and peptides by 10 and 28%, respectively. This novel hSAX approach provides robust, reproducible, and highly orthogonal separation of complex protein digest samples for deep coverage proteome analysis.
Keywords: ultralow hydrophobicity, chromatography, proteomics, strong anion exchange, hydrophilic strong anion exchange, HPLC, mouse macrophage, RAW264.7, orthogonality, mass spectrometry
Introduction
Mass spectrometric analysis of proteome samples from eukaryotic cell lines, tissues or biological fluids is still hampered by the vast complexity of samples and concentration differences of proteins within making comparisons of proteomics and transcriptomics data difficult. Online and off-line two-dimensional (2D) or multidimensional separation to reduce sample complexity is therefore still of high importance. The most widely used methods involve two subsequent steps, an off-line fractionation step followed by (low pH) reversed phase liquid chromatography (RP-LC) directly coupled to mass spectrometry. Ideally, the separation in the two-dimensions is highly complementary and uses different mechanisms, that is, they separate according to different properties of the analyte such as size, charge and hydrophobicity. Many approaches have shown decent orthogonality such as hydrophilic interaction liquid chromatography (HILIC),1 electrostatic repulsion hydrophilic interaction chromatography (ERLIC),2 strong cation exchange (SCX),3,4 isoelectric focusing (such as OFFGEL),5 SCX-weak-anion exchange (WAX) 3D6 and high-pH RP.7 Particularly, high pH RP chromatography has shown great potential as it is highly resolving and partly orthogonal to low pH RP.7 However, this setup requires the samples to be dried down after the first dimension resulting in sample loss, unless another, third dimension such as SAX is added,8,9 which increases the number of fractions and thus instrument time and reduces robustness of the system. Furthermore, the high pH of the first RP dimension is corroding most of the silica-based stationary phase, leading to reproducibility issues and short column lifetime.
While high-pH RP can never achieve full orthogonality to low-pH RP as they both separate according to hydrophobicity, ion exchange chromatography such as SCX or strong anion exchange (SAX) chromatography – at least theoretically – can. But while SCX has been widely used, SAX has been not widely considered for proteomic analysis. This is surprising as it was observed that the majority of tryptic peptides cluster between pI of 3 and 5, partly because many post-translational modifications such as phosphorylation, pyro-glutamination or acetylation decrease the pI.10 This suggests that the majority of the tryptic peptides in a total cell digest are likely acidic, thus negatively charged and should be well separable by SAX.
Some groups reported studies involving SAX fractionation, mostly for phospho-proteomics and glycomics.11−14 On the proteome level, an online mixed mode reversed phase anion exchange (MM, RP-AX),15 a SAX-SCX mixed bed ion exchange chromatography,16 a SAX microreactor17 or a SAX stage-tip approach were successfully used;18 however, the use of SAX for large-scale proteome analyses is still underdeveloped.
Here, we present a SAX column with a hyperbranched architecture,19 quaternary ammonium ion functionality and an ultralow hydrophobicity. While the column was originally designed for the separation of small organic and haloacetic acids, we show its remarkable separation power for proteome research. This column exhibits high reproducibility, high capacity, robustness and very high orthogonality when coupled to low pH RP, allowing the identification of more than 9000 proteins from a RAW264.7 macrophage cell lysate on an Orbitrap Velos Pro mass spectrometer within only one week of instrument time. We show that both hydrophilicity and the ion exchange properties of the column are important for the high orthogonality of this approach, which we term hydrophilic Strong Anion Exchange (hSAX) chromatography.
Experimental Section
Chemicals and Reagents
The standard proteins mixture consisted in bovine thyroglobulin, bovine serum albumin, chicken ovalbumin, bovine beta-casein, cytochrome C from horse heart, lysozyme from chicken eggs. All proteins were purchased from Sigma Aldrich (St. Louis, MO). Solvents for off-line systems were purchased from Rathburn Chemicals Ltd. (Walkerburn, Scotland), while those for online system were purchased from Merck KGaA (Darmstadt, Germany).
Cell Culture and Preparation of Cell Lysate Digests
The mouse macrophage cell line RAW264.7 was obtained from ATCC and grown in DMEM, 10% heat-inactivated fetal bovine serum (FBS, Sigma), 2 mM l-glutamine, 5000 U/mL penicillin and 5000 μg/mL streptomycin (Invitrogen).
RAW 264.7 cells were harvested, washed with ice-cold PBS and lysed in 8 M urea/50 mM Tris-HCl (pH 8.0)/10 mM DTT plus a phosphatase inhibitor cocktail containing 1.15 mM sodium molybdate, 1 mM sodium orthovanadate, 4 mM sodium tartrate dehydrate, 5 mM glycerophosphate (all Sigma Aldrich) and 1 mM sodium fluoride (AnalaR NormapuR, West Sussex, UK); finally, 1 μL of benzonase (Merck) was added and the lysate passed through a 26.5G needle. The solution was left to reduce for 60 min at 30 °C under shaking. Afterward, 15 mM iodoacetamide (IAA) was added and the sample was left to alkylate for 40 min at room temperature (RT) in the dark. The IAA was then deactivated with 20 mM DTT for 40 min at RT and at this point the protein concentration was assessed by RC/DC Protein Assay according to the manufacturer’s instructions (BioRad). Prior to digestion, the protein mixture was diluted 8 times with 50 mM Tris-HCl pH 8.0 to 1 M urea.
Trypsin TPCK (Worthington) was methylated as described previously,20 added to the protein mixture (1:100, trypsin: sample) and the digestion was performed at RT overnight. Another 1:100 was added the day after for 3 h followed by addition of 1% TFA to stop trypsin activity. Finally, the sample was desalted by SepPack Oasis solid phase extraction cartridges (Waters), dried down and stored at −80 °C.
The same procedure was applied to the standard protein mixture (“6-mix”).
Hydrophilic Strong Anion Exchange and Reversed-phase Chromatography
An off-line Thermo HPLC Ultimate 3000 system equipped with the WPS-3000T(B) FC autosampler, the DGP-3600BM pump system including the SRD-3600 system for solvent autodegasser, the VWD-3400RS UV/vis photometer and the TCC-3000SD thermo-controlled column compartment were used.
In this work, we compared three different strong anion exchange (SAX) columns: the AS24, the AS11-HC and the AS15 (IonPac series, Thermo-Fisher Scientific) that were chosen according to their chemical/physical characteristics.
The AS24 (2 × 250 mm, 2000 Å pore size) is a low bleed column primarily designed for the separation of environmental ions as well as for ion chromatography separation coupled with mass spectrometry. AS24 is a high capacity (140 μeq) analytical column and it is compatible with pH 0–14 eluents and samples (Table 1). It consists in a supermacroporous resin with alkanol quaternary ammonium ions as functional groups, which are ultralow hydrophobic. It uses hyperbranched chemistry with extremely hydrophilic architecture19 whereas AS11-HC and AS15 used aromatic monomers and are thus significantly more hydrophobic.
Table 1. Characteristics of SAX Columns Tested.
| characteristics | IonPac AS24a | IonPac AS11-HCb | IonPac AS15c |
|---|---|---|---|
| Size | 2 × 250 mm | 2 × 250 mm | 2 × 250 mm |
| Bead diameter | 7 μm | 9 μm | 9 μm |
| Pore size | 2000 Å | 2000 Å | 100 Å |
| Functional group | Alkanol quaternary ammonium ion | Alkanol quaternary ammonium ion | Alkanol quaternary ammonium ion |
| Capacity | 140 μeq | 72.5 μeq | 56.25 μeq |
| Hydrophobicity | Ultralow | Medium-low | Medium-high |
| pH range | 0–14 | 0–14 | 0–14 |
IonPac AS24, ThermoFisher Scientific (Part Nr: 064153).
IonPac AS11-HC, ThermoFisher Scientific (Part Nr: 052961).
IonPac AS15, ThermoFisher Scientific (Part Nr: 053941).
Finally, the AS25 is very similar to AS24 but it is the most hydrophilic member of the product family (IonPac AS25, 2 × 250 mm, 2000 Å, 87.5 μeq of capacity) but has a lower capacity. It was tested to confirm that the hydrophilic architecture of AS24 improves the orthogonal separation of tryptic peptides.
To compare the results of SAX/RP with (high pH) RP/ (low pH) RP, two reversed-phase columns were tested off-line in the first dimension, which differ in matrix composition. In this work, Acclaim 120 C18 (a conventional RP, 3 μm, 120 Ǻ, 2.1 × 150 mm, Thermo-Fisher Scientific) and Acclaim RSLC Polar Advantage II -PA2- (a polar-embedded RP, 2.2 μm, 120 Ǻ, 2.1 × 100 mm, Thermo-Fisher Scientific) columns were used to separate tryptic digested proteins from RAW264.7 cell lysate. While both C18 columns are based on ultrapure silica, Acclaim PA2 has the advantage to have specially designed amide-embedded ligands. Those features make the column compatible with 100% aqueous environments over a wide range of pH (1.5–10), exhibiting high polarity for selectivity complementary to conventional RP columns.
The Acclaim PA2 column provided higher efficiencies than the Acclaim 120 C18 column (Supporting Information, Figure S1) and therefore it was chosen for the comparison with the AS24.
For the off-line 2D LC, a highly resolving C18 column was used (Gemini-C18, 3 μm, 110 Ǻ, 3 × 250 mm, Phenomenex). This column uses ultrapure spherical silica particles and can operate at wide pH range (1–12) increasing method development flexibility.
Peptide separation on the off-line 1D separation system employing strong anion exchange was achieved with a flow rate of 0.25 mL/min (solvent A: 20 mM Tris-HCl pH 8.0; solvent B: 20 mM Tris-HCl pH 8.0, 1 M NaCl). A gradient (slope 7 for 6-mix, 8 for RAW264.7 cells) spanning 0–100% mobile phase B over 35 min was used. In case of high pH RP, separation was based on an acetonitrile (AcN) gradient (solvent A: 20 mM NH4OH/H2O, pH 10.0; solvent B: 20 mM NH4OH/AcN, pH 10.0) at the same flow rate (0.250 mL/min, 0–80% B in 35 min). When the Acclaim 120 was tested, the AcN gradient was set at 5–80% B for 35 min.
The off-line 2D was commonly performed at pH 2.5 (solvent A: 0.1% TFA/H2O; solvent B: 0.08% TFA/AcN) at a flow rate of 0.4 mL/min and a multistep gradient of 35 min: 5–15% B in 5 min, following by 30 min up to 90% B.
All chromatograms were recorded by a UV detector at 214 nm.
Mass Spectrometry Analysis
Off-line 1D-fractions (either SAX without further handling or high pH RP, after drying down and resuspension in 0.1% TFA) from the RAW264.7 cell lysate were run online on an Orbitrap Velos Pro (Thermo-Fisher Scientific). All nanoflow chromatography was performed on an Ultimate 3000 nano LC system (Thermo-Fisher Scientific), using an Acclaim PepMap 100 (75 μm ID × 500 mm, 3 μm C18) in conjunction with an Acclaim C18 PepMap trapping column (100 μm ID × 20 mm, 5 μm C18) (Thermo-Fisher Scientific). Linear gradient elution was performed using buffer A (0.1% formic acid) and buffer B (0.08% formic acid, 80% ACN) starting from 5% buffer B to 35% over 277 min at a flow rate of 300 nL/min. A coated silica tip (New Objective, Woburn, MA) was used for electrospraying the sample into the mass spectrometer, at an ion spray voltage of 1.45 kV. MS analysis was operated in data dependent mode, such that the top 20 most abundant precursors in each MS scan were subjected to MS/MS (CID in the linear trap, normalized collision energy = 35%, precursor isolation width = 2 Da, intensity threshold for precursor selection = 2000). As global parameters, precursor ions between m/z 400–2000 were selected, at resolution of 60000 collected in profile mode. Ions with charge state = 1 were rejected. Dynamic exclusion was enabled with a repeat count of 1 and exclusion duration set to 60 s. The lock mass feature was enabled for m/z = 445.120025 ([Si(CH3)2O]6) as the internal calibrant ion.
Data Analysis
The data analysis of all MS/MS proteomics data sets was performed through the Trans-Proteomic Pipeline (TPP) as previously described.21 Briefly, mass spectrometry raw output files were first converted to mzXML by ReAdW22 and afterward searched through the X!Tandem (CYCLONE 2011.12.01) search engine with K-score plug-in.23 Data was searched against a randomized forward and reverse IPI mouse database (version 3.87) and X!Tandem parameters included cysteine carbamylation as fixed modifications, methionine oxidation and asparagine/glutamine deamidation were set as variable modifications. Cyclization of glutamine/glutamic acid at the N-termini (pyroglutamate) was automatically searched, since Tandem automatically checks for formation of pyroglutamic acid, that is, the loss of water (E) or ammonia (Q), respectively, when a peptide starts with E or Q. This modification is considered to be an N-terminal modification only, so it does not affect any potential modifications specified for Q, E or C. Search parameters specified a precursor mass tolerance of ±25 ppm (required by TPP, typical mass accuracy was <2 ppm), a MS/MS tolerance at 0.4 Da and full trypsin specificity allowing for up to 2 missed cleavages.
All the results obtained were converted to the pep.xml file format. PeptideProphet24 was then used to validate the search engine results and to assign accurate probabilities to peptide–spectrum matches (PSMs). Peptide probabilities were then calculated with the iProphet tool.25 Finally, protein-level validation and protein inference were performed with ProteinProphet.26 Results were filtered at a calculated 1% FDR on the peptide level and then protein level. Data were imported into R (v2.11.0), an open source platform for statistical analysis (http://www.R-project.org) and graphs were plotted using the ggplot2 library.27
For identification via Andromeda,28 raw-files were loaded into MaxQuant 1.3.0.529 and searched against the same IPI mouse database (version 3.87). Variable and fixed modifications were the same as above; precursor mass accuracy was set to 6 ppm, MS/MS tolerance to 0.4 Da and Trypsin specificity allowing for up to 2 missed cleavages. False-discovery rate was set to 0.05 for peptides and 0.01 for proteins.
Results and Discussion
From a range of 12 high-capacity Hydroxide-Selective Anion-Exchange Columns (IonPac series, Thermo-Fisher Scientific) which were initially developed for the separation of small organic and haloacetic acids and inorganic anions,30,31 we tested three members (AS24, AS11-HC and AS15) for the use in off-line fractionation in a proteomics setting. These columns all contain the same functionality of alkanol quaternary ammonium ions, but differ in backbone hydrophobicity, with AS24 exhibiting ultralow hydrophobicity, AS11-HC medium hydrophobicity and AS15 high hydrophobicity.
In initial experiments we injected 180 μg of a tryptic digest of six proteins (“6-mix”) onto each of the three columns. Fractions were automatically collected and reinjected onto a C18 RP column using an Ultimate 3000 HPLC (Thermo-Fisher Scientific) and data acquired on a UV detector. With the use of Chromeleon software (v6.8), 2D retention maps were created by which the evaluation of the peptide distribution over the two dimensions was possible. Retention maps showed a highly orthogonal separation by AS24—as shown by the wide distribution over the two-dimensional space—some separation by AS11-HC and almost full retention of peptides on AS15 even when eluted with 1 M NaCl (Figure 1A). As the functionality in all three materials is identical, the difference of separation efficiency must depend on their differences in hydrophobicity. Indeed, addition of at least 25% AcN to the mobile phase allowed elution of tryptic peptides from AS15 and improved elution from AS11-HC (Figure 1B), showing that high hydrophobicity of the chemical backbones of these columns’ hampers the use for separation of peptides. Furthermore, the most hydrophilic member of the ion exchange materials in this line (AS25) performs similarly well as AS24 (Supporting Information, Figure S2) which led us to conclude that both the hydrophilicity in conjunction with the ion exchange functionality are important for the high orthogonality of this type of chromatography that we termed hydrophilic Strong Anion Exchange (hSAX) chromatography.
Figure 1.

IonPac A24 shows high orthogonality, reproducibility and linearity. (A) 2D retention maps of peptides of a 6-protein mix digest show that elution with up to 1 M NaCl in absence of organic solvent leads to high orthogonality for AS24 (ultralow hydrophobicity), but part and full retention of peptides by AS11-HC (medium hydrophobicity) and AS15 (high hydrophobicity), respectively. (B) Addition of 25% acetonitrile (AcN) into the elution buffer enhances elution from AS11-HC and AS15. (C) Three replicate injections on AS24 over the course of 48 h show high reproducibility. (D) Increasing amounts of protein digest loaded on AS24 show good linear behavior without peak broadening.
Next, we tested how pH affected AS24 performance. As the functional groups are primarily quaternary amines, changes of pH have no to little effect on column chemistry, but will change the available negative charge on peptides. When the mobile phase was adjusted to pH 3, a major breakthrough was observed in 2D retention maps, which was absent at pH 8. Furthermore, acidic pH in sample solution, while keeping the mobile phase at pH 8, or changing the mobile phase to pH 10 reduced retention of peptides or had no beneficial effect (Supporting Information, Figure S3). We therefore performed all following experiments in 20 mM Tris-HCl, at pH 8. This has actually the benefit that tryptic digestion of the cell lysate can be performed at 20 mM Tris-HCl, pH 8 and directly injected on the column, avoiding solid phase extraction or lyophilization and, therefore, minimizing sample loss.
Now that running conditions were optimized, we calculated the column efficiency of the AS24 on a digested peptide mixture from BSA (Supporting Information, Figure S4) and we analyzed the run-to-run reproducibility and linearity which is required for quantitative proteomics experiments, using replicate injections of digested 6-mix samples over a period of 48 h (Figure 1C) and injections of increasing material (Figure 1D). In both cases, AS24 showed very high reproducibility (Supporting Information, Figure S5) and good linearity (Supporting Information, Figure S6).
Separation of a Complex Proteome and Comparison to RP/RP
After optimized parameters were established, the method was applied to a very complex mixture, a total cell lysate of the mouse macrophage cell line RAW264.7. Tryptic peptides of 200 μg of cell lysate were separated on the AS24 column in a 35 min gradient from 0 to 1 M NaCl (34 fractions) which were automatically collected and injected onto a Gemini C18 column (Phenomenex). 2D retention maps show a very high orthogonality of separation between the two chromatographic dimensions (Figure 2A), displayed by the elution of peptides over the entire 2D map.
Figure 2.

Separation of total cell lysate digests by hSAX and a high pH/low pH reversed phase approach show differences in orthogonality. Tryptic RAW264.7 macrophage cell lysate digests were separated on an AS24 SAX column (A) or an Acclaim PA2 RP column at pH 10 (B). Thirty-four fractions of both first dimensions were separated in the second dimension by reversed phase chromatography at pH 3.
Furthermore, we separated the same cell lysate digest on two different columns using high pH (pH 10)/ low pH (pH 3) RP/RP chromatography (Supporting Information, Figure S1) which has shown good orthogonality in the past and is currently seen as the gold standard in two-dimensional separation due to its high resolution.32,33 We then optimized elution parameters and chose the Acclaim PA2 (Thermo-Fisher Scientific) as it performed best, likely due to its higher polarity than the conventional C18 (Acclaim 120, Thermo-Fisher Scientific). Then we compared its orthogonality to the AS24 ion exchange column using 2D peptide maps using identical run time and numbers of fractions (Figure 2B).
2D retention maps show that the hSAX/RP approach performs considerably better than the RP/RP approach, as a much greater area in the 2D maps is covered with peaks, while peptides in the RP/RP approach are scattered around the diagonal. This provides further evidence that high pH RP/low pH RP cannot be fully orthogonal, as both dimensions separate according to hydrophobicity. Recent approaches to concatenate early and late fractions of high pH RP to increase orthogonality have improved results;32 however, concatenation involves user interference and might provide problems in quantitative proteomics experiments when the same peptides are then present in non-neighboring fractions as quantitative proteomics software such as MaxQuant requires peptides to be in neighboring fractions for accurate quantitation.
In order to test both hSAX and high pH RP approaches for their performance in a proteomics experiment, we injected aliquots (3–6%, corresponding to ∼300–400 ng of protein digest; ∼1 × 108 base peak chromatogram) of each of the 34 fractions of the RAW264.7 cell lysate separated by hSAX or high pH RP onto an Orbitrap Velos Pro mass spectrometer, using 300 min gradients on a 75 μm × 50 cm Pepmap C18 column (Thermo-Fisher Scientific), corresponding to one week of instrument time each. Online separation of hSAX and high pH RP fractions showed a similar degree of orthogonality comparable to the offline 2D chromatography (Supporting Information, Figure S7A and B).
MS/MS spectra were searched against the mouse IPI database v3.87 using X!Tandem. Peptide and protein probabilities were calculated using the TPP. Results were filtered at Peptide- and ProteinProphet estimated 1% FDR. While we identified with the RP/RP approach 99110 peptides and 8627 proteins (1% FDR), the hSAX approach resulted in identification of 126318 unique peptides and 9469 proteins, representing 28% and 10% more identifications respectively (Table 2 and Supporting Information, Table S1), while keeping the average sequence coverage equal (31% respectively 32%) (Supporting Information, Figure S8). Combining both approaches, we identified 159847 peptides and 9871 proteins showing that we could increase the number of peptides and proteins identified by only 20% and 4% respectively compared to hSAX alone (Table 2). In addition, we analyzed the data using the Andromeda search engine included in MaxQuant28,29 which identified overall 6–8% less proteins and peptides in each experiment, but confirmed the superiority of our SAX approach (Table 2 and Supporting Information, Table S1). This data compares well to two recent publications in which about similar number of proteins were identified from a mammalian cell line using 3 different proteases and 2 weeks of instrument time34 or in which samples were analyzed repeatedly with inclusion lists.35
Table 2. Proteins and Unique Peptides Identified by X!Tandem and Andromeda in the hSAX and High pH RP LC–MS Experiments from RAW264.7 Cell Lysate.
| separation | search engine | unique proteinsa | unique peptidesb |
|---|---|---|---|
| 1D-SAX | X!Tandem | 9469 | 126318 |
| 1D high pH RP | X!Tandem | 8627 | 99110 |
| combined | X!Tandem | 9871 | 159847c |
| 1D-SAX | Andromeda | 8830 | 116093 |
| 1D high pH RP | Andromeda | 8131 | 95721 |
| combined | Andromeda | 9613 | 154553 |
Unique proteins (FDR ≤ 1%) according to ProteinProphet (X!Tandem) and MaxQuant (Andromeda).
Unique nonstripped peptides (FDR ≤ 1%) according to PeptideProphet (X!Tandem) and MaxQuant (Andromeda).
Number differing from Figure 4A due to FDR recalculation on combined pep.xml (PeptideProphet).
The high orthogonality of the hSAX approach is not only shown by the broad distribution in the 2D peptide maps, but also exemplified by the fact that we identified more than 3000 proteins in 16 out of 34 fractions, while the RP/RP approach achieved this for only 6 fractions (Figure 3A). Data also indicated that we could have saved instrument time by combining the last 5 fractions, which could have reduced run time by almost another day. The hSAX approach also showed a considerably higher number of proteins (7292 vs 6315 in RP/RP, +15%) that were identified with 3 and more peptides which considerably improves identification and quantitation in proteomics experiments.
Figure 3.

Comparison of protein and peptide identifications from off-line separation by either hSAX/RP or RP/RP and LC–MS experiments. (A) The high orthogonality of the hSAX approach is exemplified by the identification of more than 3000 proteins in 16 out of 34 fractions, outperforming RP/RP approach (6 out of 34 fractions). (B) Moreover, the hSAX was comparable to the RP/RP approach in separating peptides in a single fraction (69% of RP peptides were present in one fraction compared to 55% for the hSAX approach), which is beneficial for quantitative proteomics experiments.
As quantitative proteomics performs best when peptides are only present in one fraction, we tested how many peptides were only identified in a single fraction. Here, the RP/RP approach expectedly outperformed the hSAX approach, showing a higher resolution as 69% of peptides were present in one fraction compared to 55% for the hSAX approach (Figure 3B). Only 18.5% (hSAX) and 11.6% (RP) of all peptides respectively were identified in 3 or more fractions. Nevertheless, separation on the AS24 showed good resolution for quantitative proteomics experiments. In addition, separate experiments with dimethyl-labeled36 proteome samples indicated that deuterated samples do not change retention time in hSAX chromatography as they can do in RP experiments (data not shown), which can potentially separate light and heavy forms of isotopically labeled peptides into neighboring fractions complicating peptide quantitation.
Interestingly, the two techniques seem to favor certain subsets of peptides as only 58679 peptides (46% for hSAX and 60% for RP) are shared among the two approaches (Figure 4A). To further identify the characteristics of these subsets, we analyzed which amino acids were over-represented in the data sets unique to each approach (Supporting Information, Figure S9) and identified peptides with a higher number of acidic residues to be the major contributor to the difference (Figure 4B). Furthermore, the peptides identified by the hSAX approach matched much better to an in silico digest of all proteins of the mouse IPI database (Figure 4B), suggesting that the hSAX approach allows for the identification of more representative peptides of the total proteome than the RP/RP approach.
Figure 4.

hSAX/RP and RP/RP lead to the identification of different peptide subsets. (A) Both hSAX and RP approaches lead to the identification of different subsets of peptides with only about half the peptides identified in the two approaches (please note: number of combined peptides higher than in Table 2 due to FDR recalculation) (proportional Venn diagram has been produced using BioInfoRx). (B) In-depth analysis of the amino acid distribution in the subset of unique peptides revealed that hSAX favored more acidic peptides than RP, a trend that is more representative to the in-silico tryptic digestion of the whole IPI database. (C) Moreover, both hSAX and RP approaches showed similar peptide charge state distribution over the whole gradient, favoring doubly and triply charged peptide ions.
Finally, we tested how separation via hSAX affected the charge-state distribution of peptide ions in mass spectrometry over the gradient. Unlike SCX, peptides fractionated by SAX should not show a charge-distribution as the negative charges by which the peptides were separated in the first dimension at pH 8 do not affect electrospray ionization (ESI) after separation at pH 3 in the second dimension, where basic residues are critical. Evaluation of peptide charge states showed that peptides separated by both hSAX and RP/RP showed a relatively even charge state distribution over the whole gradient (Figure 4C). This is of great advantage as the identification of doubly and triply charged peptide ions is highly favored in ESI–MS/MS compared to singly and highly charged ions which populate, for example, early and late fractions of SCX experiments.37
Conclusion
In this work we demonstrate that hydrophilic strong anion exchange chromatography provides a robust and reproducible separation of complex protein digest samples for proteomics analysis. Its very high orthogonality proved to be superior to an optimized high pH RP/low pH RP approach and enabled us to identify >9,000 proteins from RAW264.7 mouse macrophage cell lysate in just one week of mass spectrometry instrument time which will allow a better comparison of proteomics and transcriptomics data. In the future, we will test the applicability of the hSAX material for online 2D LC-MS and the separation of complex mammalian proteomes for quantitative proteomics as well as phosphopeptide analysis.
Acknowledgments
This work was supported by the Medical Research Council (MRC) UK and in part by a grant from the Scottish Government to the Scottish Institute for Cell Signaling (SCILLS). We thank Brian Dill and Manman Guo for help with mass spectrometric analysis, Christopher Pohl for discussions, Van Kelly for help with TPP data mining, and David Campbell and Robert Gourlay for technical advice.
Glossary
Abbreviations
- CID
collision-induced dissociation
- DTT
dithiothreitol
- IAA
2-iodoacetamide
- LC
liquid chromatography
- RP
reversed phase
- TPP
trans-proteomic pipeline
- SAX
strong anion exchange
- SCX
strong cation exchange
Supporting Information Available
Table S1: List of identified proteins. Figure S1: Evaluation of two C18 RP columns for 2D RP/RP LC. Figure S2: Chromatography analysis of 6-mix and total cell lysate digests on the IonPac AS25. Figure S3: Performance of peptide separation by AS24 at different pH. Figure S4: AS24 column efficiency evaluation. Figure S5: Reproducibility of the AS24 SAX column. Figure S6: Linearity of AS24 SAX column. Figure S7: Selected base peak chromatograms of SAX and RP fractions analyzed by LC–MS. Figure S8: Sequence coverage of identified proteins in SAX/RP and RP/RP approaches. Figure S9: Characteristics of peptides identified uniquely in hSAX/RP or RP/RP approaches. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Alpert A. J. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. J. Chromatogr. 1990, 499, 177–96. [DOI] [PubMed] [Google Scholar]
- Alpert A. J. Electrostatic repulsion hydrophilic interaction chromatography for isocratic separation of charged solutes and selective isolation of phosphopeptides. Anal. Chem. 2008, 80162–76. [DOI] [PubMed] [Google Scholar]
- Wolters D. A.; Washburn M. P.; Yates J. R. 3rd An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 2001, 73235683–90. [DOI] [PubMed] [Google Scholar]
- Davis M. T.; Beierle J.; Bures E. T.; McGinley M. D.; Mort J.; Robinson J. H.; Spahr C. S.; Yu W.; Luethy R.; Patterson S. D. Automated LC-LC-MS-MS platform using binary ion-exchange and gradient reversed-phase chromatography for improved proteomic analyses. J. Chromatogr., B: Biomed. Sci. Appl. 2001, 7522281–91. [DOI] [PubMed] [Google Scholar]
- Horth P.; Miller C. A.; Preckel T.; Wenz C. Efficient fractionation and improved protein identification by peptide OFFGEL electrophoresis. Mol. Cell. Proteomics 2006, 5101968–74. [DOI] [PubMed] [Google Scholar]
- Hennrich M. L.; Groenewold V.; Kops G. J.; Heck A. J.; Mohammed S. Improving depth in phosphoproteomics by using a strong cation exchange-weak anion exchange-reversed phase multidimensional separation approach. Anal. Chem. 2011, 83187137–43. [DOI] [PubMed] [Google Scholar]
- Gilar M.; Olivova P.; Daly A. E.; Gebler J. C. Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J. Sep. Sci. 2005, 28141694–703. [DOI] [PubMed] [Google Scholar]
- Zhou F.; Sikorski T. W.; Ficarro S. B.; Webber J. T.; Marto J. A. Online nanoflow reversed phase-strong anion exchange-reversed phase liquid chromatography-tandem mass spectrometry platform for efficient and in-depth proteome sequence analysis of complex organisms. Anal. Chem. 2011, 83186996–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong R. P.; Siu S. O.; Lee S. S.; Lo C.; Chu I. K. Development of online high-/low-pH reversed-phase-reversed-phase two-dimensional liquid chromatography for shotgun proteomics: a reversed-phase-strong cation exchange-reversed-phase approach. J. Chromatogr., A 2011, 1218233681–8. [DOI] [PubMed] [Google Scholar]
- Cargile B. J.; Sevinsky J. R.; Essader A. S.; Stephenson J. L. Jr.; Bundy J. L. Immobilized pH gradient isoelectric focusing as a first-dimension separation in shotgun proteomics. J. Biomol. Tech. 2005, 163181–9. [PMC free article] [PubMed] [Google Scholar]
- Nuhse T. S.; Stensballe A.; Jensen O. N.; Peck S. C. Large-scale analysis of in vivo phosphorylated membrane proteins by immobilized metal ion affinity chromatography and mass spectrometry. Mol. Cell. Proteomics 2003, 2111234–43. [DOI] [PubMed] [Google Scholar]
- Trost M.; Bridon G.; Desjardins M.; Thibault P. Subcellular phosphoproteomics. Mass Spectrom. Rev. 2010, 296962–90. [DOI] [PubMed] [Google Scholar]
- Han G.; Ye M.; Zhou H.; Jiang X.; Feng S.; Tian R.; Wan D.; Zou H.; Gu J. Large-scale phosphoproteome analysis of human liver tissue by enrichment and fractionation of phosphopeptides with strong anion exchange chromatography. Proteomics 2008, 871346–61. [DOI] [PubMed] [Google Scholar]
- Wang F.; Han G.; Yu Z.; Jiang X.; Sun S.; Chen R.; Ye M.; Zou H. Fractionation of phosphopeptides on strong anion-exchange capillary trap column for large-scale phosphoproteome analysis of microgram samples. J. Sep. Sci. 2010, 33131879–87. [DOI] [PubMed] [Google Scholar]
- Phillips H. L.; Williamson J. C.; van Elburg K. A.; Snijders A. P.; Wright P. C.; Dickman M. J. Shotgun proteome analysis utilising mixed mode (reversed phase-anion exchange chromatography) in conjunction with reversed phase liquid chromatography mass spectrometry analysis. Proteomics 2010, 10162950–60. [DOI] [PubMed] [Google Scholar]
- Motoyama A.; Xu T.; Ruse C. I.; Wohlschlegel J. A.; Yates J. R. 3rd Anion and cation mixed-bed ion exchange for enhanced multidimensional separations of peptides and phosphopeptides. Anal. Chem. 2007, 79103623–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.; Hou W.; Lambert J. P.; Figeys D. New ammunition for the proteomic reactor: strong anion exchange beads and multiple enzymes enhance protein identification and sequence coverage. Anal. Bioanal. Chem. 2010, 39783421–30. [DOI] [PubMed] [Google Scholar]
- Wisniewski J. R.; Ostasiewicz P.; Mann M. High recovery FASP applied to the proteomic analysis of microdissected formalin fixed paraffin embedded cancer tissues retrieves known colon cancer markers. J. Proteome Res. 2011, 1073040–9. [DOI] [PubMed] [Google Scholar]
- Pohl C.; Saini C. New developments in the preparation of anion exchange media based on hyperbranched condensation polymers. J. Chromatogr., A 2008, 1213137–44. [DOI] [PubMed] [Google Scholar]
- Taylor I. A.; Webb M. Chemical modification of lysine by reductive methylation. A probe for residues involved in DNA binding. Methods Mol. Biol. 2001, 148, 301–14. [DOI] [PubMed] [Google Scholar]
- Pedrioli P. G. Trans-proteomic pipeline: a pipeline for proteomic analysis. Methods Mol. Biol. 2010, 604, 213–38. [DOI] [PubMed] [Google Scholar]
- Pedrioli P. G.; Eng J. K.; Hubley R.; Vogelzang M.; Deutsch E. W.; Raught B.; Pratt B.; Nilsson E.; Angeletti R. H.; Apweiler R.; Cheung K.; Costello C. E.; Hermjakob H.; Huang S.; Julian R. K.; Kapp E.; McComb M. E.; Oliver S. G.; Omenn G.; Paton N. W.; Simpson R.; Smith R.; Taylor C. F.; Zhu W.; Aebersold R. A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 2004, 22111459–66. [DOI] [PubMed] [Google Scholar]
- MacLean B.; Eng J. K.; Beavis R. C.; McIntosh M. General framework for developing and evaluating database scoring algorithms using the TANDEM search engine. Bioinformatics 2006, 22222830–2. [DOI] [PubMed] [Google Scholar]
- Keller A.; Nesvizhskii A. I.; Kolker E.; Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74205383–92. [DOI] [PubMed] [Google Scholar]
- Shteynberg D.; Deutsch E. W.; Lam H.; Eng J. K.; Sun Z.; Tasman N.; Mendoza L.; Moritz R. L.; Aebersold R.; Nesvizhskii A. I. iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics 2011, 1012M111 007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii A. I.; Keller A.; Kolker E.; Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75174646–58. [DOI] [PubMed] [Google Scholar]
- Wickham H.ggplot2: Elegant Graphics for Data Analysis, Springer, New York, 2009, 1–212. [Google Scholar]
- Cox J.; Neuhauser N.; Michalski A.; Scheltema R. A.; Olsen J. V.; Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 1041794–805. [DOI] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26121367–72. [DOI] [PubMed] [Google Scholar]
- Zakaria P.; Bloomfield C.; Shellie R. A.; Haddad P. R.; Dicinoski G. W. Determination of bromate in sea water using multi-dimensional matrix-elimination ion chromatography. J. Chromatogr., A 2011, 1218509080–5. [DOI] [PubMed] [Google Scholar]
- Liang C.; Lucy C. A. Characterization of ion chromatography columns based on hydrophobicity and hydroxide eluent strength. J. Chromatogr., A 2010, 1217528154–60. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Yang F.; Gritsenko M. A.; Clauss T.; Liu T.; Shen Y.; Monroe M. E.; Lopez-Ferrer D.; Reno T.; Moore R. J.; Klemke R. L.; Camp D. G. 2nd; Smith R. D. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 2011, 11102019–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F.; Shen Y.; Camp D. G.; Smith R. D. High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev. Proteomics 2012, 92129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj N.; Wisniewski J. R.; Geiger T.; Cox J.; Kircher M.; Kelso J.; Paabo S.; Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011, 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M.; Schmidt A.; Malmstroem J.; Claassen M.; Ori A.; Szymborska A.; Herzog F.; Rinner O.; Ellenberg J.; Aebersold R. The quantitative proteome of a human cell line. Mol. Syst. Biol. 2011, 7, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersema P. J.; Raijmakers R.; Lemeer S.; Mohammed S.; Heck A. J. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 2009, 44484–94. [DOI] [PubMed] [Google Scholar]
- Trinidad J. C.; Specht C. G.; Thalhammer A.; Schoepfer R.; Burlingame A. L. Comprehensive identification of phosphorylation sites in postsynaptic density preparations. Mol. Cell. Proteomics 2006, 55914–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.