

Abstract
Immunohistochemistry (IHC) is a valuable tool to visualize the distribution and localization of specific cellular components within morphologically preserved tissue sections or cell preparations. It combines the histologic morphology of tissues for detecting the actual antigen distribution, specificity of antibody–antigen interaction for optimal detection, and sensitivity of immunochemical methods for assessing the amount of antigen in tissues. It is routinely used clinically to diagnose type (benign or malignant), stage, and grade of cancer using specific tumor markers. The application of IHC ranges from disease diagnosis and prognosis to drug development and analysis of the pathobiological roles of various molecular players during disease development. Due to better availability of highly specific antibodies and optimal methodologies for performing immunohistochemical studies, IHC is being used at an expanding rate to understand pancreatic tumor biology as well as to study the fate of various molecular markers during the initiation, progression, and metastasis of pancreatic neoplasia. Herein, we describe the detailed protocol for IHC analyses of pancreatic intraepithelial neoplasia in tissues and fine needle aspirates from both human and mouse samples.
Keywords: Immunohistochemistry, Mucins, Pancreatic cancer, Antibodies, Fixation
1. Introduction
Histopathological analysis of tumor tissue is a fast, simple, and inexpensive method for classifying the type, stage, and grade of neoplastic disease. It forms the basis for clinical research as well as therapeutic decision making. Immunohistochemistry (IHC) as the name implies uses the specificity of antigen–antibody interaction for assessing the localization, distribution, and expression of specific antigen(s) in a histological tissue section or cytological preparation via an antibody specific to that antigen. The distribution of the primary antibody is then visualized microscopically via an appropriate enzymatic or fluorescent detection system.
The origin of IHC goes back to the early 1940s when fluorescein-conjugated antibodies were used to demonstrate the expression of bacterial antigens in infected tissues (1). The developments in technology and usage of paraffin blocks in the 1970s further enhanced the application of IHC in various biochemical fields. Since then, IHC has undergone many modifications to improve its applicability and efficiency. The applicability of IHC can be ascertained from the fact that it can be performed on a vast variety of samples. After their collection, samples are immediately fixed and processed for embedding and sectioning. Once fixed and embedded, tissue samples can be preserved for years with no significant changes in tissue architecture, stability, and antigenicity of targeted epitopes. Unfortunately, many potentially important antigens become altered during tissue fixation and processing, interfering with their immunohistochemical detection. Due to this, fixation and processing of samples must be standardized for each antigen to achieve optimal preservation of tissue architecture while simultaneously maintaining maximum antigenicity. For this, advancement in IHC strategy has led to the development of various methods for antigen fixation, which includes paraffin-embedding, freezing, free-floating, and traditional smears of cytological specimens as well as monolayer preparation of cytological specimens. Further, various antigen retrieval methods have been evolved to expose epitopic structures hidden during the process of fixation (i.e., heat-induced antigen retrieval and enzymatic antigen retrieval). Further, the growing list of available antibodies and a better understanding of biology have contributed to the broader utility of IHC for solving everyday diagnostic problems in pathology.
Today, physicians use IHC to diagnose tumors as benign or malignant, determine the grade of a tumor, and identify the cell type of a metastasis to find the site of primary tumor. IHC is also used in drug development to test drug efficacy by detecting either the activity and/or the up- or down-regulation of therapeutic targets. In the case of breast cancer, IHC has been routinely used to distinguish the usual ductal hyperplasia (UDH) from atypical ductal hyperplasia (ADH)/low-grade carcinoma in situ, subtyping lesions as ductal versus lobular or basal versus luminal, helping to distinguish true microinvasion from mimics (“pseudoinvasion”), predicting the likelihood of response to anti-hormonal and other therapeutic agents, improving sentinel node staging, and finally, helping to recognize metastatic carcinoma of an unknown primary site as originating in the breast (1). Further, IHC has been commonly used to check the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) status in breast cancer patients to classify patient subtype (2). Similarly, attempts have been made to identify IHC-based molecular markers for identifying prognostic subtypes of pancreatic ductal adenocarcinoma (PDAC), to accurately predict clinical and therapeutic outcomes, and to define novel therapeutic targets. An ideal example of its utility is that analyses of 76 independent prognostic and predictive molecular markers implicated in pancreatic tumor growth, apoptosis, angiogenesis, and invasion and resistance to chemotherapy yielded 11 markers (Ki-67, p27, p53, transforming growth factor β1, Bcl-2, survivin, vascular endothelial growth factor, cyclo-oxygenase 2, CD34, S100A4, and human equilibrative nucleoside transporter 1) that provide independent prognostic or predictive information in two or more IHC-based studies (3). Further, IHC has played a critical role in understanding the distribution, localization, and fate of various molecular markers at very early stages of PDAC (i.e., pancreatic intraepithelial neoplasias (PanINs)) (4–6).
2. Materials
All the solutions should be prepared in double-distilled or Millipore water (obtained from a MILLIPORE water purifying system Elix 3, conductivity>18.0 MΩ cm). Chemicals used for IHC are of analytical grade.
2.1. Specimen Fixation Reagent
10% Neutral buffer Formalin (Fisher Scientific, Cat. # SF100-4).
Superfrost Plus Slides (Fisher Scientific, Cat. # 22-035813).
2.2. Specimen Processing Reagent
Liquid paraffin.
Tissue cassette (Fisher brand, True-tissue cassettes, Cat. # 15-200-403E).
Molds.
2.3. Deparaffinization and Tissue Rehydration Reagents
100% Xylene (Fisher Scientific, Cat. # X5P-1 Gal).
Graded Ethanol (Decon Laboratories, Cat. # DSP-MD. 43) (100, 95, 90, 80, 70, 50, 30, 20% in DDW).
3% Hydrogen peroxide methanolic solution: Add 30 mL of 3% hydrogen peroxide (Sigma, Cat. # H3410) in 270 mL methanol (Fisher Scientific, Cat. # A412P-4). Working solution is prepared fresh.
2.4. Antigen-Retrieving Reagents
Buffers for antigen/epitope retrieval: Both composition and pH of the buffer used for antigen retrieval play critical roles in unmasking antigens during IHC. Table 1 shows the composition of buffers used during antigen retrieval.
Table 1.
Composition of buffers used for heat-induced epitope retrieval (HIER) and enzymatic antigen retrieval
| Buffer | Composition (1×) | pH range | Notes |
|---|---|---|---|
| Buffers used for heat-induced epitope retrieval (HIER) | |||
| Citrate buffer | 10 mM Citric Acid (Sigma), 0.05% Tween 20 | Adjust pH to 6.0 with 1 N NaOH, then add 0.5 ml of Tween 20, and mix well. (10× solution can be prepared and stored at 4°C for long-term use) | Cross-links made by formalin or aldehyde fixers mask the antigen sites in tissue specimens leading false negative staining to some proteins, citrate buffers by breaking protein cross-links unmask the antigenic epitopes and enhance staining intensity of antibodies |
| EDTA buffer | 1 mM EDTA (Sigma), 0.05% Tween 20 | Adjust pH to 6.0 with 1 N NaOH, then add 0.5 ml of Tween 20, and mix well | Works optimal for many antibodies; however, often results in high background staining (might be due to endogenous biotin revealed after this pretreatment). Buffer of choice for low-affinity antibodies and expression of antigen is quite low |
| Tris–EDTA buffer | 10 mM Tris Base, 1 mM EDTA solution, 0.05% Tween 20 | Adjust pH to 9.0 using 1 N NaOH, then add 0.5 ml of Tween 20, and mix well | |
|
| |||
| Buffers used for enzymatic antigen retrieval (HIER) | |||
| Trypsin | 0.05% Trypsin, 0.1% CaCl2 | Adjust pH to 7.8 with 1 N NaOH | Enzymes at high concentration destroy epitope and tissue morphology; therefore optimal enzyme concentration needs to be standardized for the antigen of interest. Standardize the time for enzymatic retrieval according to the antigen of choice Apart from trypsin, pepsin and chymotrypsin should be checked for enzyme-based antigen retrieval |
2.5. IHC-Staining Reagents
Phosphate-buffered saline (PBS; 10×): 32 mM Na2HPO4, 5 mM KH2PO4, 13 mM KCl, 1.35 M NaCl, pH 7.4.
1× PBS containing 0.05% Tween-20 (PBST).
Blocking buffer: 2.5% horse serum in 1× PBS or blocking buffer from immunoPEROXIDASE kit (Vector Laboratories).
Primary antibody against desired antigen. Dilute the primary antibody in 0.1% horse serum in 1× PBS at the appropriate dilution. The dilution must be standardized for each antibody.
Isotype control antibody at a dilution similar to the primary antibody.
Secondary antibodies: Anti-mouse or anti-rabbit secondary antibody conjugated to peroxidase (ImmPRESS reagent kit, Peroxidase, UNIVERSAL Cat. # MP-7500, Vector Laboratories) or alkaline phosphatase or Biotinylated secondary antibody (Sigma).
Enzyme Substrate/Detection Reagent: This substrate will depend upon the enzyme tagged to secondary antibody and color needed. For peroxidase: Peroxidase substrate kit (Cat. # SK-4100, Vector), and for alkaline phosphatase: Fast Red TR/Naphtol AS-MX (Sigma, Cat. #. B5655).
2.6. Counterstaining Reagents Following IHC Staining
Hematoxylin (Vector, Cat. # H-3401). Staining jars (Sigma, Cat. # S5641).
2.7. IHC Slide-Mounting Reagents
Permount (Fisher Cat. # SP15-100).
Coverslips (Fisher Cat. # 12-548-5P).
2.8. Equipment
56°C incubator for baking slides.
IHC jar.
PAP Pen (Vector, Cat. # H-4000).
Humidifying chamber for incubation of slides.
Microtome for cutting 3–5 μM thick sections.
Microwave or pressure cooker or water bath (depending upon the protocol used for antigen retrieval).
Microscope (Leica Microsystems).
3. Methodology
Perform all the steps at room temperature unless otherwise specified.
3.1. Preparation of Tissue Samples for IHC
Proper preparation of tissue samples is critical for optimal IHC (Note 1). After removal from their in vivo host environment, tissues rapidly undergo multiple changes usually caused by hypoxia, lysosomal enzymes, and putrefactive changes due to bacterial and mold growth. Thus, once collected, samples should be preserved immediately to maintain tissue architecture, prevent degradation of proteins, and maintain the antigenicity of the targeted probe. Sample preparation includes fixation, embedding, and specimen sectioning.
3.1.1. Fixation of Tissues
Fixation of tissue or cells is usually the first stage in a multistep process to prepare a sample of biological material for IHC analyses. Routinely performed by experts, fixation can be done through multiple ways. Various methods (fixation, snap freezing, and free-floating section) have been used to preserve and process tissue sections or fine needle aspirates for IHC. Various kinds of fixative (10% neutral buffer formalin (NBF); 10% formalin in tap water; 10% formal saline; 10% neutral buffered formalin with saline (NBFS); 10% formal acetic acid; 10% zinc formalin; Carson’s fixative; and Bouin’s fixative) have been used for preserving tissue morphology (7). The ideal fixative (i) protects from autolysis and bacterial decomposition, (ii) preserves tissue in its natural state and fixes all components, (iii) makes the cellular components insoluble to the reagents used in tissue processing, (iv) preserves tissue volume, (v) should harden the tissue while avoiding excessive tissue hardness, (vi) allows enhanced staining of tissue, (vii) should be nontoxic and nonallergic to the user, (viii) should be inexpensive, and (ix) penetrates rapidly and kills the tissue to prevent postmortem changes. Although there is no universal method for ideal fixation of all antigens, various studies have revealed that formalin fixation followed by paraffin embedment (FFPE) successfully localizes the distribution of many antigens with high specificity and minimal artifacts. For IHC, samples from biopsies, excisions, or resections or animals are perfused, or rinsed of blood with sterile saline, prior to preservation to prevent the detection of hematologic antigens that may interfere with the detection of target antigens. Following fixation, protocols are employed routinely for biological specimens.
Fixation by perfusion: Preserve the tissues by vascular perfusion with 500–700 mL of 10% NBF (formalin fixes the tissues by forming cross-links, phosphate salts prevent damage to erythrocytes, and neutral pH prevents the formation of cross-links) (Note 8 and 9).
Fixation by immersion: For autopsy and biopsy sections, immerse the tissue block (5–10 mm thick) in 15–20 volumes of fixative (NBF) (Note 12).
Fixation by alcohol: In case of cytology sections (fine needle aspirates, cystic fluid, and endoscopic brushing sections), formalin fixation is not appropriate as it alters cellular morphology and interferes with staining. For small-volume specimens (FNA, cyst fluids), mix with equal amounts of 50% ethanol. For smears of tissues on slides, dip the slides in 95% ethanol or pipette the fixative directly onto the slides. (If slides are already air-dried it should not be put in alcohol fixative.)
In addition to the nature of fixative and type of fixation, other factors which play an important role during specimen fixation include time, temperature, and pH of the fixative as well as the size of the specimen (Note 2). Although most fixative performs at a rate of 1 mm/h, tissue fixation time should be standardized for each tissue. Routinely, tissues are fixed for 18–24 h followed by dehydration and embedment in paraffin (Note 2–4). Further, in order to achieve adequate and consistent fixation, it is essential that tissue specimens be sliced to a thickness of 5 mm or less (Note 3 and 7). Tissue fixation can be carried out at room temperature. To avoid formation of ice-crystal, tissue should not be frozen once it has been placed in the fixative solution (Note 13).
3.1.2. Washing Tissue After Fixation
Before proceeding to tissue embedding, the aqueous phase of the tissue fixative must be replaced with an organic phase compatible with paraffin wax (tissue embedding material).
As phosphate salts of formalin have limited solubility in high concentrations of ethanol, wash the fixed tissue in 70% ethanol three times for 30 min each at room temperature.
Wash the fixed tissue in 90% ethanol at room temperature two times for 30 min each followed by three washings with 100% ethanol for 30 min each.
Since the paraffin wax used for tissue embedding in the following step is immiscible with ethanol, replace ethanol with xylene (clearing agent) by immersing the tissue in xylene for 20 min at room temperature.
To completely replace ethanol, give two more washes of xylene for 20 min each at room temperature.
3.1.3. Tissue Embedding
Tissue embedding is routinely done with specialized automated tissue processing systems. For this:
After the final wash with xylene, tissues are immersed in 25–30 volumes of melted paraffin wax (55–60°C) for 2–4 h for processing.
Paraffin wax is changed two to three times to remove traces of xylene from tissues. Processed tissue is placed into a tissue cassette. Excessive wax is removed from the tissue by heating the cassette at 58°C for 15 min.
Finally, the tissue is placed in appropriate sized molds and processed for making tissue block.
3.1.4. Tissue Sectioning and Mounting
Before sectioning, keep tissue blocks in ice-cold water for 30 min to allow for further hardening of the wax as it helps to cut finer and smoother sections.
Cut 3 μm thick tissue sections from the paraffin block using a rotary microtome.
Pick finely cut tissue sections from the microtome using a toothpick and float them in a 44°C water bath until the section is fully expanded.
Pick up the fully stretched tissue section using forceps and mount it onto the adhesive side of a Superfrost Plus Slide.
Remove the associated water by tilting the slide.
Dry the tissue mounted on the Superfrost slides for 20 min in a 65°C oven followed by overnight drying at room temperature.
Store the paraffin-embedded sections at room temperature or at 2–8°C in slide storage boxes until further use.
3.2. Deparaffinization and Hydration of Tissue Blocks
Deparaffinize the slides by baking overnight at 58°C overnight or 2 h at 65°C.
Take the slides out of the oven and let cool to room temperature (Note 5).
Remove wax by dipping slides in four consecutive jars of xylene for 10 min each (Note 6).
After the final wash with xylene, shake off excess xylene and rehydrate slides with decreasing grades of ethanol for 5 min each (100, 95, 90, 70, 50, 30, 20%) (Note 14).
Wash the slides with running tap water for 10 min to remove traces of ethanol.
3.3. Quenching of Endogenous Peroxidases
Endogenous peroxidase and biotin react with secondary reagents, causing nonspecific background staining. A saturating amount of hydrogen peroxide inactivates endogenous peroxidases and helps in reducing the nonspecific background.
For quenching of internal peroxidases, incubate the slides in 3% hydrogen peroxide solution in methanol for 30 min at room temperature under dark conditions. In the case of mouse tissues, incubate the slides for 1 h in 3% hydrogen peroxide solution in methanol (Note 15–16).
Wash the slides with running tap water for 10 min.
3.4. Antigen Retrieval
The majority of fixatives fix tissues by forming methylene bridges, often leading to the masking of antigenic epitopes and causing weak to negative staining for many antigens. In order to retrieve masked epitopes, multiple methods can be employed depending upon the antigen and the antibody.
3.4.1. Heat-Induced Epitope Retrieval
Heat-induced epitope retrieval (HIER) is routinely utilized by our group as a prime method for epitope retrieval. It has worked optimally for retrieving mucinous epitopes (Fig. 1) from lesions as early as PanIN-1 (pancreatic intraepithelial neoplasias).
Fig. 1.
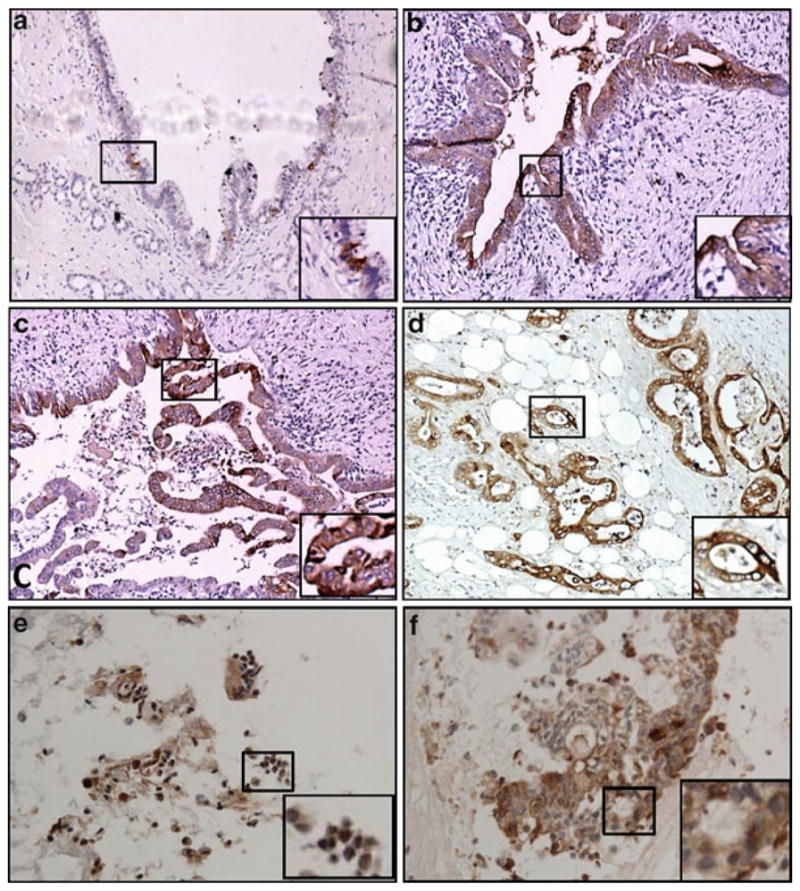
Expression of MUC4 mucin assessed by IHC after heat-induced epitope retrieval (HIER) at different stages of pancreatic ductal adenocarcinoma (PDAC) and in fine needle aspirate (FNA) sections from pancreatic cancer patients. Paraffin-embedded, formalin-fixed pancreatic tissue sections were quenched with 3% methanolic solution of hydrogen peroxide followed by antigen retrieval using citrate buffer following the HIER method. The antigen-retrieved sections were stained for MUC4 using a mouse anti-MUC4 monoclonal antibody (8G7). The sections were developed using DAB as substrate. As seen in various panels, MUC4 is absent in normal exocrine pancreatic ducts (Panel a), while de novo expression was observed in PanIN-1 (Panel a), PanIN-2 lesions (Panel b), PanIN-3 lesions (Panel c), and PDAC (Panel d). High MUC4 expression was even observed in cells from FNA after HIER (Panels e, f).
Preheating of epitope retrieval buffer: Fill the empty plastic IHC jar with 0.05 M citrate buffer (pH = 6.0) and place it into a bigger plastic box which is filled 3/4 full with distilled water. Preheat the assembly in a microwave oven until the solution starts boiling.
Immerse the racked slides into the preheated solution of citrate retrieval buffer and heat for 15 more minutes.
After heating, replace the water in the outer jar with cold water and let the retrieval buffer cool to room temperature.
Wash the slides with running tap water for 10 min.
Wash the slides with two changes of 1× PBST for 5 min each at room temperature (Note 10 and 11).
In addition to heating in the microwave, heating in an autoclave or pressure cooker is also employed by various groups for HIER. In various studies, we have observed that the microwave-based method of heating has worked as a reliable and inexpensive source of heating (4, 6, 8).
In addition to citrate buffer, Tris and EDTA buffers can also be employed for HIER (Table 1). Studies have found no significant difference between microwave and autoclave treatment, but choice of buffer does make significant difference for antigen retrieval. The optimal buffer for heat-induced epitope retrieval should be determined experimentally.
3.4.2. Enzymatic Antigen Retrieval
Apart from HIER, enzyme-based epitope retrieval is carried out either alone or in combination with HIER to unmask difficult antigenic epitopes. Two methods are routinely employed for applying the enzyme solution to the tissue: directly pipetting the solution onto the tissue on the slide, or placing a rack of tissue slides into a container of enzyme solution. By placing the slide into the jar of enzyme solution, uniform digestion is carried out for all the tissue sections and consequently is the preferred method for enzymatic antigenic epitope retrieval.
For enzymatic digestion, remove the racked slides from 1× PBST solution and place them into 1× PBS.
Prepare the working stock solution of the chosen enzyme in warm water and place the racked slides into a jar containing enzymatic solution.
Incubate at 37°C for 30 min with intermittent shaking.
Remove the slides and wash with running tap water for 10 min to remove traces of enzyme.
Wash the slides twice with PBST for 5 min each at room temperature.
3.5. Blocking Nonspecific Sites
Nonspecific antibody reactivity is blocked by incubating the tissue with blocking buffer (2.5% horse serum in PBS).
Prepare a humidifying chamber and put wet tissue paper on the sides of the chamber.
Remove the slides from the jar and rinse off excess PBST.
Encircle the tissue with a PAP pen and put the slide on the wet tissue paper in the IHC chamber. Add two to three drops of blocking buffer from ImmPRESS reagent kit (Vector Laboratories), enough to cover the tissues completely.
Cover the chamber with the lid.
Incubate for 1–2 h at room temperature.
3.6. Immunodetection
Drain the blocking solution by tilting the slide.
Add the appropriate dilution of primary antibody diluted in 0.1% horse serum in 1× PBS, enough to cover the tissue section completely. Add isotype control antibody in different section at the same dilution as a negative control.
Put the slide back into the chamber and cover the chamber with a lid.
Incubate the tissue section with primary antibody overnight at 4°C or 2 hrs at 37°C.
After incubation, remove the primary antibody solution and place the slides in an IHC jar.
Wash the slides four times with PBST for 10 min each at room temperature while shaking.
After the final wash, rinse off excess washing solution and again place the slides in the IHC chamber (Note 17).
Add two to three drops of the appropriate secondary antibody conjugated to HRP from an ImmPRESS reagent kit (Vector Laboratories) and incubate the slides in the humidified chamber under dark conditions for 30 min – 1 hr.
After secondary antibody incubation, remove the secondary antibody solution and place the slides in the IHC jar.
Wash the slides four times with PBST for 10 min each at room temperature while shaking.
After the final wash, just swipe the excess washing solution off the slide with a kimwipe without touching the tissue and place the slides in the IHC chamber again.
3.7. Colorimetric Detection
Preparation of Substrate Solution: To 5 ml of DDW, add two drops of substrate buffer, four drops of DAB, and two drops of hydrogen peroxide solution (Peroxidase substrate kit, DAB, SK-4100 (Vector)).
Add two to three drops of the substrate solution to the PAP pen-encircled tissue section and incubate for 3–6 min until the signal can be observed under a microscope.
Wash the slides with tap water after the desired signal strength.
3.8. Counterstaining (Hematoxylin)
Remove the slides from the water and put back in the IHC chamber. Add two to three drops of filtered hematoxylin and incubate for 2–3 min at room temperature.
Wash the slides with tap water.
Dip the rack of slides into a jar containing 0.1% HCl solution three times, followed by three washes with tap water.
Dip the rack of slides into 0.1% ammonium hydroxide three times followed by three dips into the tap water jar.
Dehydrate the slides by successive 5-min washes in graded ethanol (30, 50, 70, 90, 95% and absolute ethanol).
Wash the slides three times with xylene, with each wash of 5 min.
Dry the slides at 37°C for 1 h.
Add two to three drops of paramount onto the slides and put a coverslip on the top of mounting medium.
Let the paramount spread uniformly onto slides and dry the slides for 1 h at room temperature.
Visualize the slide under the microscope.
Acknowledgments
The authors on this work are supported, in part, by grants from the National Institutes of Health (RO1 CA78590, EDRN UO1 CA111294, RO1 CA 131944, RO1 CA133774, SPORE P50 CA127297, and UO1 CA163120).
Footnotes
After collection, tissue should be rapidly preserved to avoid breakdown of cellular proteins and tissue architecture.
At no point during initial processing of a tissue section, should the temperature be higher than 65°C as it causes loss of tissue antigenicity, which might not be recovered.
Tissue sections for IHC should be 3–5 μm thick as tissues thicker than this have multiple layers of cells which make interpretation of IHC staining difficult.
The length of fixations should be appropriate as both inadequate and longer fixation leads to masking of antigenic epitopes and low-intensity IHC.
After baking slides at 56°C overnight or 65°C for 2 h, IHC slides should be cooled down to room temperature before proceeding for dewaxing and rehydration.
While dewaxing the slides, use an optimal amount of xylene (for 50 slides use 250 ml) as the efficacy of xylene for dewaxing drastically reduces below this and residual wax causes artifact development during staining.
Tissue blocks used for fixation should be of required thickness but not thicker than 5 mm.
Presence of alcohols insoluble salts in neutral buffer saline leads to tissue precipitation or tearing upon direct exposure of these to absolute or 95% alcohol. To avoid the issues, tissue should be first incubated with ≤60% alcohol and processing machines should be routinely flushed with water to avois precipiates from phosphonates. If transferred directly to 95% or absolute ethanol, the phosphates are likely to precipitate on and in the tissue, causing difficulties in sectioning such as tearing and scoring. Processing machines should be flushed periodically with water to remove accumulated salts.
NBF is carcinogenic, corrosive, and a severe eye and skin irritant, and affects the respiratory system. Due to this high toxicity, wear gloves and a lab coat and work in a well-ventilated area while fixing tissues.
Plastic racks should be used for boiling during the antigen retrieval step as standard glass-made IHC jars could crack.
Due to uneven heating in domestic microwaves, their usage should be avoided during HIER-based antigen retrieval. Further, enough retrieval buffers should be added as boilover or evaporation of retrieval buffer might result in tissue drying.
When it is not possible to fix by perfusion, dissected tissue may be fixed by immersion in a 10% formalin solution for 4–8 h at room temperature. Volume of fixative should be 50 times greater than the size of the immersed tissue. Further, it is emphasized that fixation length of >24 hrs should be avoided as it masks or destroy the epitope present on antigen. Avoid fixing the tissue for greater than 24 h since tissue antigens may either be masked or destroyed.
Fixation by chemically cross-linking of proteins, fixation masks the epitopes of antigen and make them unnavigable for antibody reactivity. In order to get proper antibody reactivity, the right fixation method must always be optimized based on the application and the target antigen to be stained.
During hydration of tissue sections, make sure to add ethanol in consecutive decreasing grades.
Hydrogen peroxide used for quenching should be stored in the refrigerator well protected from sunlight in order to prevent its thermal decomposition.
Hydrogen peroxide can be diluted in methanol, water, or PBS. Since aqueous hydrogen peroxide solution damages blood smears and peroxidase-rich tissues, aqueous hydrogen peroxide should be avoided for IHC analyses of blood and tissues rich in peroxidases. Conversely, surface markers are more sensitive to methanolic hydrogen peroxide solutions, so aqueous solutions of hydrogen peroxide are recommended in these cases.
Do not let the section dry at any step of IHC.
References
- 1.Linnoila I, Petrusz P. Immunohistochemical techniques and their applications in the histopathology of the respiratory system. Environ Health Perspect. 1984;56:131–148. doi: 10.1289/ehp.8456131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Onitilo AA, Engel JM, Greenlee RT, Mukesh BN. Breast cancer subtypes based on ER/PR and Her2 expression: comparison of clinicopathologic features and survival. Clin Med Res. 2009;7:4–13. doi: 10.3121/cmr.2009.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ansari D, Rosendahl A, Elebro J, Andersson R. Systematic review of immunohistochemical biomarkers to identify prognostic subgroups of patients with pancreatic cancer. Br J Surg. 2011;98:1041–1055. doi: 10.1002/bjs.7574. [DOI] [PubMed] [Google Scholar]
- 4.Moniaux N, Chakraborty S, Yalniz M, Gonzalez J, Shostrom VK, Standop J, Lele SM, Ouellette M, Pour PM, Sasson AR, Brand RE, Hollingsworth MA, Jain M, Batra SK. Early diagnosis of pancreatic cancer: neutrophil gelatinase-associated lipocalin as a marker of pancreatic intraepithelial neoplasia. Br J Cancer. 2008;98:1540–1547. doi: 10.1038/sj.bjc.6604329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chakraborty S, Baine MJ, Sasson AR, Batra SK. Current status of molecular markers for early detection of sporadic pancreatic cancer. Biochim Biophys Acta. 2011;1815:44–64. doi: 10.1016/j.bbcan.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swartz MJ, Batra SK, Varshney GC, Hollingsworth MA, Yeo CJ, Cameron JL, Wilentz RE, Hruban RH, Argani P. MUC4 expression increases progressively in pancreatic intraepithelial neoplasia. Am J Clin Pathol. 2002;117:791–796. doi: 10.1309/7Y7N-M1WM-R0YK-M2VA. [DOI] [PubMed] [Google Scholar]
- 7.Williams JH, Mepham BL, Wright DH. Tissue preparation for immunocytochemistry. J Clin Pathol. 1997;50:422–428. doi: 10.1136/jcp.50.5.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Senapati S, Ho SB, Sharma P, Das S, Chakraborty S, Kaur S, Niehans G, Batra SK. Expression of intestinal MUC17 membrane-bound mucin in inflammatory and neoplastic diseases of the colon. J Clin Pathol. 2010;63:702–707. doi: 10.1136/jcp.2010.078717. [DOI] [PMC free article] [PubMed] [Google Scholar]