

Abstract
Background
Paris polyphylla var. yunnanensis is an important medicinal plant. Seed dormancy is one of the main factors restricting artificial cultivation. The molecular mechanisms of seed dormancy remain unclear, and little genomic or transcriptome data are available for this plant.
Results
In this study, massive parallel pyrosequencing on the Roche 454-GS FLX Titanium platform was used to generate a substantial sequence dataset for the P. polyphylla embryo. 369,496 high quality reads were obtained, ranging from 50 to 1146 bp, with a mean of 219 bp. These reads were assembled into 47,768 unigenes, which included 16,069 contigs and 31,699 singletons. Using BLASTX searches of public databases, 15,757 (32.3%) unique transcripts were identified. Gene Ontology and Cluster of Orthologous Groups of proteins annotations revealed that these transcripts were broadly representative of the P. polyphylla embryo transcriptome. The Kyoto Encyclopedia of Genes and Genomes assigned 5961 of the unique sequences to specific metabolic pathways. Relative expression levels analysis showed that eleven phytohormone-related genes and five other genes have different expression patterns in the embryo and endosperm in the seed stratification process.
Conclusions
Gene annotation and quantitative RT-PCR expression analysis identified 464 transcripts that may be involved in phytohormone catabolism and biosynthesis, hormone signal, seed dormancy, seed maturation, cell wall growth and circadian rhythms. In particular, the relative expression analysis of sixteen genes (CYP707A, NCED, GA20ox2, GA20ox3, ABI2, PP2C, ARP3, ARP7, IAAH, IAAS, BRRK, DRM, ELF1, ELF2, SFR6, and SUS) in embryo and endosperm and at two temperatures indicated that these related genes may be candidates for clarifying the molecular basis of seed dormancy in P. polyphlla var. yunnanensis.
Keywords: Embryo, Stratification, Seed dormancy, High-throughput sequencing, Paris polyphylla
Background
Paris polyphylla var. yunnanensis (named “Chonglou” in Chinese) is one of the most famous medicinal plants in China. It is a perennial herbaceous plant of the Trilliaceae family and is found in damp, shady woodlands, forests, and bamboo forests. The rhizome of this plant has been developed into traditional Chinese medicines such as “Yunnan BaiYao” and “GongXueNing”, which are used to treat dispersing blood stasis and hemostasis, to activate blood circulation, to alleviate pain, for detoxification, and to reduce swelling, stop bleeding and reduce inflammation [1-3]. P. polyphylla var. yunnanensis is easily grown in moist, humus-rich soil in woodland conditions, in full or partial shade. However, its cultivation is difficult because of long seeds dormancy and very slow growth from seed. At present, the wild plant is the only source of the rhizome. However, the wild plant has become rare and endangered because of over collection in recent decades. To preserve the natural resources and ensure a stable and renewable source of P. polyphylla var. yunnanensis for medical purposes, successful cultivation of seedlings and planting is imperative.
There are two key factors limiting the extended cultivation of P. polyphylla var. yunnanensis. One is the difficulty of obtaining enough seedlings. Development of the seed embryo stops at the globular stage, about 120 days after fertilization [4]. Thus, seed germination requires a long period of embryo development and release from dormancy. Under natural conditions, the seeds are dormant for 18 months (some are dormant for over 2 years) with about 40% of them germinating [5,6]. Some studies indicated that stratification could speed up the breaking of seed dormancy to about 6 months [7,8]. The other factor is that this plant grows slowly, taking four years from seed to flowering and another three or four years to develop enough for herb harvesting.
Freshly harvested P. polyphylla var. yunnanensis seeds consist of a mesophyll outer layer coat (bright red), an inner hardy coat, a large endosperm and a very small, undeveloped embryo [5]. According to Baskin and Baskin [9,10] and Huang et al. [7], the seeds are of the morphophysiological dormancy (MPD) type and need a long stratification period. To date, research on P. polyphylla has focused on seed dormancy release and changes in seed phytohormone content [7,8], seed stratification [4], cultivation methods [11] and phylogeny and classification [12]. There is little research on molecular mechanisms, especially functional gene studies of seed development and dormancy release. In this work, a high-throughput gene mining method using the 454 Genome Sequencer FLX platform was used for embryo transcriptome sequencing of P. polyphylla var. yunnanensis. Karin et al. [13] suggested that a combination of high-throughput sequencing with more classical methods could greatly advance our knowledge of plant developmental processes. Recently, some studies on plant development using 454 sequencing were successfully conducted [14,15], showing the efficacy of 454 transcriptome sequencing for rapid gene discovery. The aim of this study was to identify transcripts of the embryo that are involved in seed development and dormancy release in stratification process and attempt to explore its molecular mechanism. qRT-PCR analysis was used to compare the expression differences of identified genes between embryo and endosperm to gain more insight into the stratification process. The increased genomic information produced in this study will aid our understanding of the molecular basis of seed development and dormancy release in P. polyphylla var. yunnanensis.
Results
Sequencing and assembly
We obtained 393,805 reads totaling 88,618,152 bases using the 454 GS FLX Titanium platform. After filtering out the adaptors sequences and removing the short sequences of less than 50 bases, 369,496 (93.8%) high-quality (HQ) reads with lengths ranging from 50 bp to 1146 bp were obtained. The average read length was 219 bp. Using paired-end joining and gap-filling, these reads were assembled into 47,768 unigenes, including 16,069 contigs and 31,699 singletons. The size distribution of these reads and contigs are shown in Figure 1. There were 2,451 (15.3%) contigs with lengths longer than 500 bp, which are considered large contigs. All HQ reads were deposited in the National Center for Biotechnology Information (NCBI) and can be accessed in the Sequence Read Archive (SRA) under the accession number SRX155369. An overview of the sequencing and assembly is given in Table 1.
Figure 1.

Size distribution of the high quality (HQ) 454 reads and the contigs assembly (A: 454 HQ reads, B: contigs).
Table 1.
Summary of sequencing and assembly
| Sequence | |
|---|---|
| Sequencing |
|
| Total bases |
80,978,250 |
| Hight-quality (HQ) reads |
369,496 |
| Average HQ reads length |
219 ±101(bases) |
| Contigs |
|
| Number of contigs |
16069 |
| Contigs larger than 500 bases |
2451(15.3%) |
| Average length of contigs |
402 ±160 (bases) |
| Range of contig lengths |
88–1778 (bases) |
| Singletons |
|
| Number of singletons |
31,699 |
| Average length of singletons |
237 ±168 (bases) |
| Range of singleton lengths |
50–1146 (bases) |
| Unigenes | 47,768 |
Annotation of unigenes
To capture the most informative and complete annotations, all unigenes were first used in BLASTX searches against non-redundant protein database at NCBI. This yielded 15,757 well-identified sequences with E values <10-5, which accounted for 32.9% of the total unique sequences. All unique putative transcripts were then subjected to a BLASTX search against the Arabidopsis database. Approximately 32.8% of the unique putative transcripts (15,673) were annotated. Subjecting these sequences to a BLASTN search against the NCBI non-redundant nucleotide database yielded 12,663 well-identified sequences with E values <10-5, accounting for 26.5% of the total unique sequences. Among the contigs, 30 contained more than 1,000 reads, representing the most abundant transcripts in the 454 cDNA library. The 30 most abundant transcripts included one that encoded early flowering protein. Some transcripts were related with phytochromone and seed ripening, including gibberellic acid stimulated-like (GAST-like), gibberellin oxidase, auxin-repressed protein, ABA 8'-hydroxylase (CYP707A), 9-cis-epoxycarotenoid dioxygenase (NCED), seed maturation protein, late embryogenesis abundant (LEA) protein, xyloglucan endotransglucosylase/hydrolase, dormancy-associated protein (DRM). A number of unique sequences related to seed development and ripening were selected and shown in Table 2. Highly expressed transcripts included cytochrome P450 (60 unigenes), metallothionein-like proteins (105 unigenes), glutathione S-transferase (71 unigenes), and PR10 proteins (22 unigenes).
Table 2.
Selected genes related to seed embryo development in stratification seeds
| Biological process | Gene bank description (gene name) | No. of unigene | Length (bp) | |
|---|---|---|---|---|
| Phytohormone related |
GA |
Gibberellin 2-oxidase (GA2ox) |
3 |
124–357 |
| Gibberellin 20 oxidase (GA20ox) |
3 |
147–339 |
||
| GA oxidase (GAox) |
5 |
232–522 |
||
| GAST-like gene product (GAST-Like) |
7 |
345–626 |
||
| Gibberellin regulated protein; GRAS family transcription factor |
8 |
207–684 |
||
| Kaurene oxidase (KO) |
2 |
130–213 |
||
| ABA |
ABA 8′-hydroxylase CYP707A1(CYP707A1) |
2 |
256,439 |
|
| 9-cis-epoxycarotenoid dioxygenase (NCED) |
2 |
144,336 |
||
| Abscisic stress ripening (ASR) |
5 |
176–772 |
||
| ABA INSENSITIVE 2 (ABI2); Protein phosphatase 2C (PP2C) |
20 |
103–669 |
||
| Auxin |
Auxin-repressed protein (ARP) |
8 |
312–806 |
|
| SAUR family protein (SAUR); Auxin response factor (ARF);Auxin influx carrier component (AUX) |
18 |
119–727 |
||
| IAA |
IAA hydrolase (IAAH); IAA type protein; Indole-3-acetic acid-amido synthetase (IAAS) |
13 |
273–593 |
|
| BR |
BRASSINOSTEROID INSENSITIVE 1-associated receptor kinase 1 precursor (BRRK) |
3 |
121–309 |
|
| CPD brassinosteroid C-23 hydroxylase |
|
|
||
| Ethylene |
Ethylene-responsive protein (ERP) |
7 |
171–391 |
|
| |
Ethylene insensitive (EIN3/EIL)-like transcription regulator 1-aminocyclopropane-1-carboxylic acid oxidase (ACC) |
3 |
258–387 |
|
| Seed maturation related |
Late embryogenesis abundant protein (LEA) |
14 |
150–709 |
|
| Dehydrin (Dhn) |
13 |
174–836 |
||
| Seed maturation protein (SMP) |
13 |
178–1304 |
||
| Seed specific protein Bn15D1B (SCP) |
5 |
311–646 |
||
| Lipid transfer protein precursor (LTP) |
6 |
372–727 |
||
| Lipid binding protein (LBP) |
5 |
126–738 |
||
| Somatic embryogenesis receptor-like kinase1 (SERK) |
6 |
166–474 |
||
| Ripening regulated protein DDTFR19 (TPS) |
10 |
221–776 |
||
| Trehalose-6-phosphate synthase (TrPS) |
7 |
248–494 |
||
| Cell wall growth related |
Xyloglucan endotransglucosylase/hydrolase (XET) |
19 |
96–756 |
|
| Cell wall invertase (CIN) |
4 |
144–654 |
||
| Endo-beta mannanase (MAN) |
5 |
136–560 |
||
| Beta 1,3 glucanase |
14 |
200–647 |
||
| Pectin methylesterase (PME) |
18 |
109–603 |
||
| Expansins (EXP) |
8 |
186–639 |
||
| Polygalacturonase/inhibitor protein (PG/PGIP) |
36 |
184–826 |
||
| Circadian rhythms |
Early flowering protein (ELF) |
21 |
256–691 |
|
| Sensitive to freezing 6 (SFR^) |
20 |
235–542 |
||
| Sucrose synthase (SUS) |
22 |
122–1313 |
||
| Flavonol biosynthesis related |
Chalcone-flavanone isomerase (CHI) |
11 |
98–468 |
|
| Chalcone synthase (CHS) |
17 |
159–831 |
||
| Cytochrome P450 |
Cytochrome P450 (CYP) |
60 |
93–525 |
|
| Obtusifoliol 14 alpha-demethylase (CYP51) |
4 |
152–555 |
||
| Others |
Cell elongation protein (CEP) |
4 |
306–721 |
|
| Dormancy-associated protein (DRM) |
3 |
439–653 |
||
| Vaculoar H+-translocating inoraganic pyrophosphatase (VPP) |
10 |
107–848 |
||
| Total number of unigenes | 464 | |||
Gene Ontology (GO) and Cluster of Orthologous Groups of Proteins (COG) assignments
The unique putative transcripts were compared with the Universal Protein Resource database (UniPort), formed by uniting the Swiss-Prot, TrEMBL and PIR protein database activities. The BLASTX and the Gene Ontology (GO) assignments of the UniPort proteome produced assignments for 14,601 (30.6%) sequences, matching 9,749 unigenes, and then assigned them to 43 major GO categories for molecular functions, biological processes and cellular components (Additional file 1). The overall distributions suggested that our library sampled widely across sub-categories and provided a good representation of the embryo transcriptome of P. polyphylla.
To further evaluate the completeness of our transcriptome library and the effectiveness of our annotation process, we searched the annotated sequences for genes involved in the Cluster of Orthologous Groups of Proteins (COG) classifications. Out of 30,500 nr hits, 5,339 sequences had a COG classification (Additional file 2). Among the 25 COG categories, the cluster for ‘translation, ribosomal structure and biogenesis’ represented the largest group (852, 16.0%), followed by ‘post-translational modification, protein turnover, and chaperones’ (630, 11.8%) and ‘transcription’, which we focused heavily on (207, 3.9%), which were particularly interested in. The categories of extracellular structures (0, 0%), nuclear structures (3, 0.06%) and cell motility (11, 0.2%), represented the smallest groups.
Functional classification by KEGG
The 47,768 unigenes were compared with the KEGG database using BLASTX with an E-value cutoff of < 10-5. Of these unigenes, 15,628 (32.7%) had significant matches in the database. Among those, 5,962 unigenes were assigned to metabolic pathways with an enzyme commission (EC) number; however, 9,666 unigenes were not assigned to any pathways. Figure 2 shows the features of the pathway assignment based on KEGG. Most of the assigned unigenes are involved in the primary metabolites, such as carbohydrate metabolism, amino acid metabolism, energy metabolism and cell growth. Few unigenes participated in secondary metabolites. In particular, 127 unigenes related to phytohormone, sucrose biosynthesis and catabolism were assigned in the KEGG pathway. Some metabolite pathways related with sugar and phytohormone or phytohormone precursor metabolism, including starch and sucrose metabolism, steroid, terpenoid backbone, brassinosteroid and carotenoid biosynthesis are shown in Additional file 3.
Figure 2.
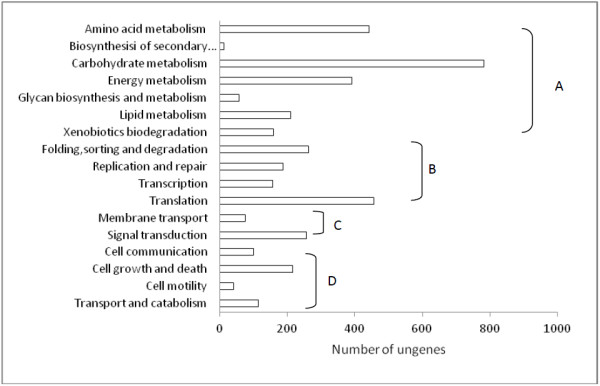
Pathway assignment based on KEGG (A: metabolism; B: genetic information processing; C: environmental information processing; D: cellular processes).
Gene expression analyses of P. polyphylla seeds during stratification using qRT-PCR
Using the method of Livak and Schmittgen [16], the level of GAPDH mRNA expression was selected as the internal control from four candidate genes (data not shown). The relative expression levels were calculated by comparing the CTs (cycle thresholds) of the target genes with that of the housekeeping GAPDH gene, using the 2-ΔΔCt method. Using the Student’s T-test, differences in relative transcript expression levels were compared at P < 0.05 level between the embryo and the endosperm at two temperature treatments during seeds stratification. Sixteen primer pairs out of the thirty-six designed primers were successfully amplified a product (the primers shown in Additional file 4).
The hormones abscisic acid (ABA) and gibberellic (GA) are considered central to dormancy and control of germination completion, while auxins are important for plant development and growth. In this work, six ABA and GA related genes (CYP707A, NCED, GA20ox2, GA20ox3, ABI2, PP2C) and five auxins-related genes (IAAH, IAAS, ARP3, ARP7, BRRK) were studied (Figure 3 and Figure 4). According to the previous studies [17,18], CYP707A, GA20ox2, GA20ox3, IAAH, IAAS and BRRK might be positively related to dormancy release, in contrast to NCED, ABI2, PP2C, ARP3 and ARP7 negatively related with dormancy release.
Figure 3.
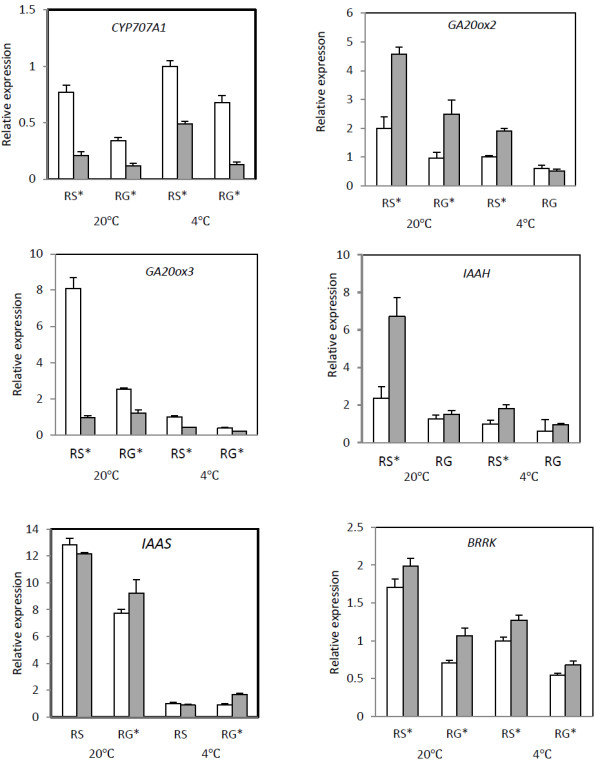
Relative expression levels analysis of transcripts positively associated with dormancy release (CYP707A,GA20ox2, GA20ox3, IAAH, IAAS, BRRK,) in the Embryo (Em□) and endosperm (En■) under warm (20°C)/cold (4°C) seed stratification processes. RS, radical just sprouted; RG, radical growing up to 1.5 cm. Expression is shown relative to GAPDH as the reference gene. RNA was extracted from three biological samples and qRT-PCR was performed. Data represent means ± SD. * indicate a significant difference between Em and En at P < 0.05.
Figure 4.
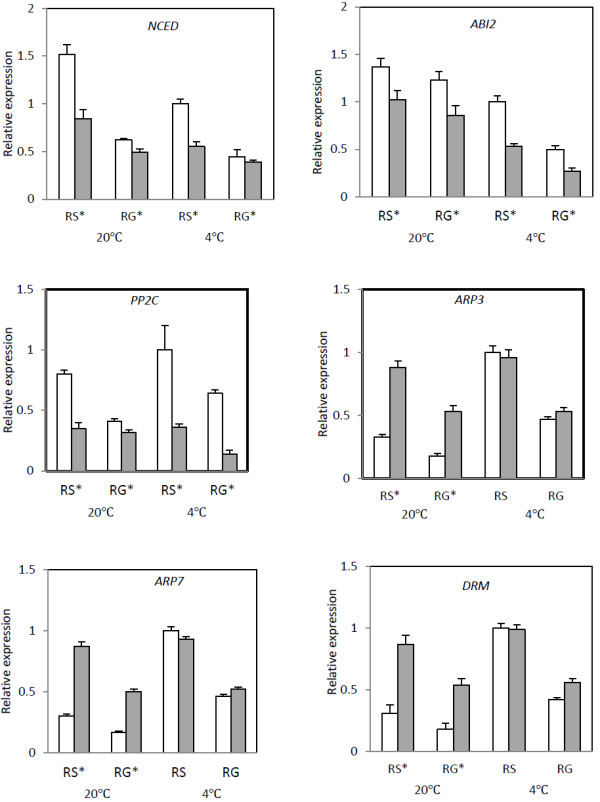
Relative expression levels analysis of transcripts negatively associated with dormancy release (NCED, ABI2, PP2C, ARP3, ARP7, DRM) levels in the Embryo (Em□) and endosperm (En■) under warm (20°C)/cold (4°C) seed stratification process. RS, radical just sprouted; RG, radical growing up to 2 cm. Expression is shown relative to GAPDH as the reference gene. RNA was extracted from three biological samples and qRT-PCR was performed. Data represent means ± SD. * indicate a significant difference between Em and En at P < 0.05.
Figure 3 shows genes that are positively associated with dormancy release. CYP707A, which participates in ABA catabolism, showed higher expression levels in the embryo at two stages (radical just sprout (RS) and radical growing up to 1.5 cm (RG)) and at two temperature treatments than within the endosperm (P < 0.05). This indicates that ABA catabolism is active at low temperature in both the embryo and endosperm. GA20ox2 and GA20ox3, which are involved in GA biosynthesis, were expressed significantly differently (P < 0.05) between the embryo and the endosperm at the two temperature treatments. The mRNA level of the GA20ox2 in the endosperm was higher than that in the embryo, but the GA20ox3 expression was higher in the embryo than the endosperm. The expression levels of five auxin related transcripts (IAA hydrolase (IAAH), IAA synthetase (IAAS), auxin-repressed protein (ARP), and brassinosteroid insensitive 1-associated receptor kinase (BRRK)) were successfully determined using qRT-PCR. IAA is a major plant growth hormone that is important for numerous processes throughout plant growth and development. Both IAAH and IAAS participate in increasing plant IAA levels. In this work, the mRNA levels of IAAH and IAAS were significantly higher (P < 0.05) in both the embryo and endosperm under warm stratification compared with cold stratification. In addition, the IAAH expression level in endosperms was significantly higher (P < 0.05) than in embryos at the two temperatures. The difference in IAAS expression level was negligible between the two plant tissues. The BRRK gene expression level was positively correlated with temperature, but it expressed higher in endosperms than in embryos (P < 0.05).
Figure 4 shows genes that are negatively associated with dormancy release. NCED, an ABA biosynthesis key gene, was expressed at an increased level in the embryo relative to the endosperm. It showed decreased expression at the RG stage compared with the RS stage at both temperatures (P < 0.05). Among the many protein phosphatease 2C (PP2C) family members in plants, the ABA insensitive 2 (ABI2) belong to the subgroup A and acts as a global negative regulator of ABA signaling. In the presented study, the expression of ABI2 and a PP2C gene were determined by qRT-PCR. The results showed that expression of the two genes had a similar mode, being significantly higher (P < 0.05) in the embryo than in the endosperm. PP2C showed higher expression in the embryo at 4°C. The relative expression levels of ARP3 and ARP7 in the embryo were significantly lower (P < 0.05) than in the endosperm at 20°C, but there was no significant difference under cold treatment. These results indicate that the expressions of ARP3 and ARP7 were inhibited by warm stratification. In this work, three unigenes were annotated to the same dormancy-associated protein (DRM; AAW02792) and the mRNA level of the DRM gene in embryos was significantly inhibited (P < 0.05) compared with in the endosperm, under warm stratification, but not under cold stratification. The warm stratification stage is a morphological dormancy release period when the radical, hypocotyls and cotyledon are formed (Figure 5D). Therefore, this result indicates that DRM activity may be highly correlated with morphological dormancy release in P. polyphylla seeds.
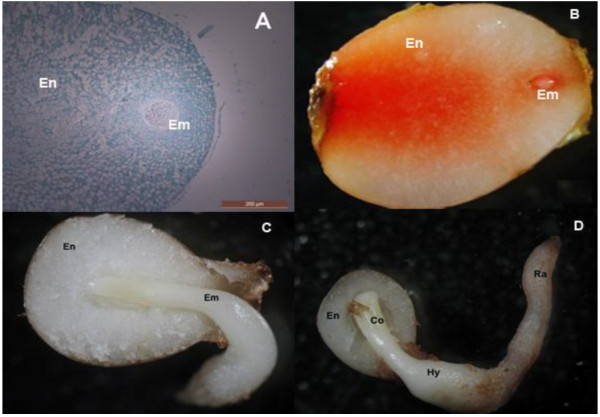
Figure 5.
Paris polyphylla var. yunnannsis seed (Em, embryo; En, endosperm; Co, cotyledon; Hy, hypocotyls; Ra, radical). A, the ovule 130d after fertilization; B, Freshly harvested seed not at full development (dyed with tetrazole); C, D, Radicle sprout testa after about 60d stratification.
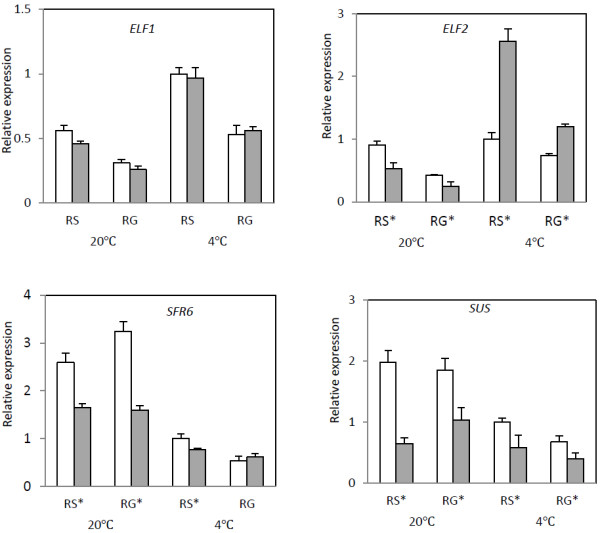
Figure 6 shows the qRT-PCR analysis of four plant circadian rhythms related genes, ELF1 (Early flowering protein), ELF2, SFR6 (SENSITIVIE TO FREEZING-6) and Sus (Sucrose) [19-21]. The relative expression levels of ELF1 and ELF2 were significantly higher (P < 0.05) under cold stratification than under warm stratification, whether in the embryo and endosperm or at RS and RG stages. There were no differences in the relative expression of ELF1 in the embryo and endosperm across RNA samples; however, the expression of ELF2 showed a particular pattern: it was significantly higher expressed (P < 0.05) in the endosperm at 4°C stratification but was expressed at a low level in the endosperm at 20°C stratification. The expression levels of SFR6 and Sus were positively correlated with temperature treatments and they both showed higher expressions in embryos than in endosperms (P < 0.05). These results indicated that ELF1 and ELF2 might participate in different biological clock regulator processes, while SFR6 and Sucrose might have coincident regulator functions.
Figure 6.
Relative expression levels analysis of four circadian rhythms related transcipts (ELF1, ELF2, SFR6, SUS) in the Embryo (Em□) and endosperm (En■) under warm (20°C)/cold (4°C) seed stratification process. RS, radical just sprouted; RG, radical growing up to 2 cm. Expression is shown relative to GAPDH as the reference gene. RNA was extracted from three biological samples and qRT-PCR was performed. Data represent means ± SD. * indicate a significant difference between Em and En at P < 0.05.
Discussion
High-throughput transcriptome sequencing is an effectively gene mining method
Freshly matured seeds of P. polyphylla var. yunnanensis have embryos that are at an undeveloped globular stage. The embryos are very small relative to size of seed, which has a large endosperm (Figure 5A, B). Baskin and Baskin [9] characterized these plant seeds as the Morphophysiological dormancy (MPD) type with a temperature requirement for breaking dormancy and embryo growth. Although cold and/or warm stratification can break dormancy in seeds of many species [9,10], the molecular mechanism of seed dormancy release remains unclear. Nambara and Nonogaki [22] and Mochida and Shinozaki [23] suggested that new methodologies including analysis of transcriptomes, proteomes and metabolomes, might advance our knowledge and understanding of seeds development. Here, we obtained 47,768 unigenes, including 16,069 contigs and 31,699 singletons from the embryo of Paris polyphylla var. yunnanensis seeds that had undergone a stratification process, using the Roche 454-GS FLX Titanium platform. Using BLASTX searches of public databases, 15,757 unique transcripts were annotated with specific biological functions. Some transcripts were assigned to important plant physiological and biochemical processes, such as phytohormone biosynthesis and catabolism, seed maturation, cell wall growth, and circadian rhythms regulator, many of which might participate in seed development and dormancy release (Table 2 and Additional file 5). These results demonstrated that de novo sequencing and analysis of the P. polyphylla seeds during stratification were effective and informative in reflecting the transcript levels in the seed embryo.
However, although we obtained abundant transcripts information, there were two aspects that require further study in future research: (1) Among the annotated 15,757 unigenes, some had functions that were obviously related to seed development and dormancy, others had no specific functions related to any particular biological processes; (2) 32,011 (67%) unigenes were not assigned to any specific functions. To further discover valuable genes in our data base, we consider that combinations of high-throughput sequencing, gene-chip and microarray analysis, and digit gene expression tag profiling with more classical methods (for example gene clone and Southern blotting) could greatly advance our knowledge of plant developmental processes. Recently such combinations were used to study seed-specific transcription factors and gene expression [24,25], and determine the roles of genes in seed dormancy in Arabidopsis[26].
Phytohormone related genes involved in P. polyphylla var. yunnanese seeds stratification
Compared with orthodox seeds, recalcitrant seeds like P. polyphylla undergo little or no maturation dehydration and remain desiccation sensitive during development. However, as in orthodox seeds, Huang et al. [7] and Chen et al. [8] indicated that the ABA levels increased during P. polyphylla seed development. According to previous studies [27-29], seed dormancy and germination are controlled primarily by the balance of ABA and GA. ABA is a negative regulator of seed germination, while GA, BR, cytokinins, and ethylene are positively associated with dormancy release. In this work, there were six types of phytohormone-related genes annotated in the database including GA2ox, GA20ox, GA3ox, CYP707A1, NCED, BR, ACC, ACO, and IAA. GA3ox (GA 3-oxidase) and GA20ox (GA 20-oxidase) participated in GA biosynthesis, while GA2ox (GA 2-oxidase) catalyzes the catabolism of biologically active GA and its precursors [29-32]. ABA 8′-hydroxylation (encoded by the CYP707A gene family) was shown to play the predominant role in ABA catabolism [33], while NCEDs (9-cis-epoxycarotenoid dioxygenase genes) are rate-limiting enzymes in ABA biosynthesis [34,35]. Therefore, GA3ox, GA20ox and CYP707A are positively associated with dormancy release, while GA3ox and NCEDs are negatively related. The qRT-PCR analysis indicated that GA20ox2 and GA20ox3 expression patterns were very dissimilar in embryos and endosperm even at both temperatures, suggesting they might be different kinds of GA20ox genes. These observations are consistent with our annotation results for GA20ox2 and GA20ox3 from Zea mays (GenBank: ACG35782) and Glycine max (GenBank: ACJ76438) (Additional file 5). The GA2ox and GAST-like (gibberellic acid stimulated-like gene) [36] are also important GA regulator genes; however, qRT-PCR analysis using the designed primers was not successful. In our cDNA library, four unigenes (two contigs for CYP707A1 and two singletons for NCED) associated with ABA metabolism were obtained. Relative expression analysis indicated that CYP707A and NCED are both expressed both in embryos and endosperm, and are expressed at higher levels in embryos than in endosperm. These result demonstrated that a shift in ABA levels in embryos might be an important factor during seed stratification. However, we only found four transcripts related to ABA metabolism among 47,768 unigenes, which indicated that ABA metabolism is low in the embryos. Footitt et al. [28] suggested that ABA signaling and sensitivity were more likely regulators of dormancy than the absolute level of ABA. We found 22 transcripts of PP2Cs, which are the ABA signaling molecules [37], in our sequence data set. PP2Cs, including ABA-INSENSITIVE1-2 (ABI1, ABI2), are negative regulators of seed germination [27,37]. The expressions of PP2C and ABI2 genes in this work suggested that ABA signals are more active in embryos than in the endosperm and their expression patterns are consistent with the expressions of CYP707A and NCED.
IAA hydrolase contributes free IAA to the auxin pool during germination in Arabidopsis[38], while IAA synthetase may catalyze the entire pathway of biosynthesis of the major plant growth hormone [39]. The relative genes expression ratios of IAAH (IAA hydrolase gene) and IAAS (IAA synthetase gene) were compared between embryos and endosperm in P. polyphylla at two temperatures. The results indicate that higher IAAH and IAAS relative expression levels were correlated with stratification temperature and also related to seed morphological dormancy release. We also found that the two expressions of two ARP genes were decreased in embryos during warm stratification and were negatively related with IAAH and IAAS expressions. Park and Han [40] demonstrated that cold treatment abolished the auxin-mediated repression of RpARP gene expression and that its expression was negatively associated with hypocotyl elongation. The Arabidopsis thaliana SERKs (Somatic embryogenesis receptor kinases) are essential for the early events of BR signaling pathway [41,42]. In the present study, two transcripts of brassinosteroid insensitive 1-associated receptor kinase gene (BRRK) and six transcripts of SERK gene were found. Relative expression analysis of the BRRK indicated differences in transcription levels between embryos and endosperm at two temperatures.
Other genes associated with P. polyphylla var. yunnanensis seeds stratification
Dyer [43] identified two dormancy-associated mRNAs with over 3-fold higher expressions in dormant compared with non-dormant embryos of Avena fatua. Expression of the dormancy-associated genes ATS2 and ATS4 was high in the dry seed and decreased during germination [44]. Using qRT-PCR method, we analyzed a DRM gene that is very similar to the dormancy-associated protein gene (GenBank: AAW02792). The expression pattern of DRM in this work is similar to that reported by Dyer [43]. Toorop et al. [44] reported high expression in dormant seeds and low expression during germination. However, many similar genes were also found, such as Oryza sativa dormancy-associated protein (GenBank: AF467730) and Zea mays auxin-repressed 12.5 kDa protein (Eu967389), in nucleotide database. These findings indicated that although only one dormancy related gene (three unigens with same accession no. GenBank: AAW02792) was sequenced in this work, its specific physiological function must be further investigated in the future.
Several genes, such as CCA1 (Circadian Clock Associated 1), LHY (Late Elongated Hypocotyl), TOC1 (Timing of CAB1), and GI (GIGANTEA) have been identified as major components of the circadian clock [17,45]. In this work, we obtained two genes, ELF (Early Flowering Protein gene) and SFR6 (Sensitive to Freezing 6 gene) that are related to circadian rhythms. However we did not find CCA1, LHY, TOC1, and GI. ELF3 is a novel protein associated with control of plant morphology, flowering time, and circadian rhythms in Arabidopsis. Some studies showed that elf3 mutations cause a light-dependent circadian dysfunction, elongated hypocotyls, and early flowering [20,46]. In this research, 21 unigenes were annotated to ELF, which were found in monocots Elaeis guineensis (16 unigenes matching GenBank: ACF06553) and Asparagus officinalis (five unigenes matching GenBank: AAB09084). Knight et al. [21] indicated that SFR6 is a component of the photoperiodic regulatory pathway. They also observed that clock gene expression and sucrose might act as a regulator of clock function and interacts with SFR6. In the present study, we obtained 20 SFR6 unigenes, which all annotated to the same Arabidopsis SFR6 (NP-192401.5) gene, and 22 unigenes that matched sucrose synthase genes. A decline curve of sucrose content variation was found in the P. polyphylla seeds stratification process from zero to 100 days and this was accompanied by seed dormancy release (data not shown). In this study, the mRNA expression levels of ELF1, ELF2, SFR6 and SUS varied in the embryo and endosperm and at both temperatures. These results suggest that ELF, SFR6 and sucrose, as clock components, may participate in the germination of P. polyphylla seeds during warm/cold temperature stratification.
LEA proteins might be involved in embryo development or genetic diversity [47,48]. The deduced LEA protein sequences are classified into at least five groups according to their conserved motifs or sequence similarity [49]. Group I to IV LEA proteins are highly hydrophilic, and the remainders are hydrophobic. Dehydrins are group II LEA proteins that contain high glycine levels and are highly hydrophilic [50]. Some dehydrins are constitutively present in vegetative tissues during normal growth [51], but others are induced by tissue water-deficits, such as drought, salinity, low temperature, and seed maturation [52,53]. Recently, in wheat, it was demonstrated that Dehydrin is associated with the maintenance and breaking of seed dormancy, and that ABA affected the expression of Dehydrin gene at the transcript level [54]. Sixteen dehydrins transcripts (Additional file 5), which were annotated to seven species of plant dehydrins, were found in this research in our cDNA library.
Cell wall remodeling enzyme, including endo-β-mannanase, β—1, 3- glucanases, expansins, xyloglucan endotransglycosylase, pectin methylesterase, polygalacturonase and cell wall invertase may participate in endosperm weakening and seed germination [55-62]. In the present study, we found 104 unigenes with high similarity to these proteins (Table 2 and Additional file 5). We also found many genes, such as CHS (chalcone synthase), CHI (chalcone-flavanone isomerase), F3H (flavanone 3-hydroxylase), and DFR (dihydroflavonol reductase), which were expressed abundantly in the embryo of stratified seeds. Recent research [63] showed that these genes are expressed in the embryo and in the endosperm at germination stages of Arabidopsis seeds. However, the physiological role of flavonols in the endosperm is still unclear, though such flavonols might protect the embryo or control seed germination by regulating auxin transport or by acting as scavengers [64].
The relative expression levels of analyzed genes in seed stratification
In this study, the expression levels of mRNA in different stratification stages or different treatments were altered by less than twofold. We considered that there might be two explanations. (1) Although these genes (GA2ox, GA20ox, CYP707A1, and NCED) are key enzymes involved in a metabolic network, we cannot determine the amount of the final product. Some studies [7,8] showed that ABA content decreased while GA content increased, and the GA/ABA increased by over tenfold when seed stratification was completed. (2) The embryo and the endosperm are two tissues, but they exist in one organ, the seed. Comparisons of gene expression between the embryo and endosperm might show only subtle differences under the same temperature treatment.
Conclusion
Despite considerable progress in seed biology, the major factors affecting seed dormancy, seed stratification and drying after-ripening remain unclear. Finkelstein et al. [17] and Kucera et al. [18]) have reviewed the role of plant hormones during seed dormancy release and germination, and the molecular aspects of seed dormancy. However, unlike orthodox seeds, the dormancy release of P. polyphylla seed is different because of its undeveloped embryo, which requires morphological (cotyledon, hypocotyls and radicle) establishment and physiological dormancy release during the stratification process.
Post-genome methodologies, such as analysis of transcriptomes, proteomes, metabolomes and bioinformatics have advanced our understanding of seed germination. The present study further demonstrates that the high-throughput embryos transcriptome sequencing of P. polyphylla seeds is a highly effective method for mining genes that may be involved in seed stratification and dormancy release. A large number of transcriptome data of P. Polyphylla seeds have been recently released in the NCBI SRA database under the series identifier SRX155369, which provides an additional resource for the discovery of genes in P. polyphylla. Bioinformatics and qRT-PCR analysis help us to identify many important genes, such as CYP707A, NCED, GA20ox, PP2C, SFR6, ELF, DRM, which should be studied further to clarify their specific function in seed dormancy release. In addition, there were 67.01% (32,011) unique putative transcripts that were not annotated during BLAST searches against NCBI non-reduntant protein database. This large amount of unidentified sequences should also be studied with a view to annotating new transcripts.
Methods
Plant material and RNA preparation
Seeds of P. polyphylla var. yunnanensis were harvested from plants growing in Wuding county, Yunnan Province, China in October 2010. The freshly matured seeds were stored in wet sand in a temperature-controlled incubator at 20°C (warm stratification treatment) for 2 months and then at 4°C (cold stratification treatment) for 2 months. The pulpy outer layer of the seeds coat was removed, and then soaked in water for 24 h prior to the temperature stratification. For the construction of the 454-sequencing cDNA library, three embryo samples at different development stages (radical just sprout testa, radical growing to length 0.5 and 1.5 cm) at two temperature treatments (4 and 20°C) were excised, frozen in liquid nitrogen immediately, and then stored at −80°C until RNA isolation. For qRT-PCR analysis, the embryo and endosperm were sampled at two stages RS (Figure 5C) and RG (Figure 5D), and at the two temperature stratifications.
Total RNA was isolated from 2 mg each of the two temperature treatments using the RNeasy plant kit (BioTeke, Beijing, China). The RNA quality was assessed using a 1% agarose gel and quantified by a GeneQuant100 spectrophotometer (GE Healthcare, Chalfont St Giles, UK) before proceeding. After pollination (13d), the ovules were fixed using FAA (95% alcohol: acetic acid: formalin: water = 10:1:2:7) and made into tablets for microscopic inspection.
cDNA library construction and pyrosequencing
Approximately, 2 ug of poly(A) RNA was isolated from equal mixtures of the three total RNAs using an Oligotex mRNA Midi Kit (Qiagen, Shanghai China). The long poly(A/T) tails in cDNA may lead to low-quality sequencing reads from the GS FLX system. To overcome this limitation, we designed a modified poly (T) primer with a BsgI site between the adaptor and the poly(T) 5′-AAGCAGTGGTATCAACGCAGAGTACT(20)VN-3′) [65]. For cDNA synthesis, this poly(T) primer was used in combination with the Clontech SMART IV primer. The cDNA was then treated overnight with BsgI (NEB, MA, USA) at a concentration of 5 units/μg of cDNA. This restriction enzyme cut within the poly(A) tail, and greatly increased the quantity and quality of the sequencing reads. For the library, digested cDNA was amplified using PCR Advantage II polymerase (Clontech, Dalian, Chian) and the following thermal profiles were used: 1 min at 95°C; followed by 16 cycles of 95°C for 15 s, 65°C for 30s, and 68°C for 6 min. PCR products (5 ul) were determined by electrophoresis. Approximately 3 ug ds cDNA was sent to the Beijing Institute of Genomics of the Chinese Academy of Sciences (Beijing, China) for pyrosequencing using the 454-GS FLX Titanium Kit.
Unigene assembly
Using the GS FLX pyrosequencing software, high-quality sequences (> 99.5% accuracy on single base reads) were selected for further processing and assembly. A subsequent filtering step, which included the masking of SMART PCR primer sequences and the removal of sequences shorter than 50 bp, was performed before assembly. The Newbler software 2.3 (provided with the GS FLX sequencer) was used for sequence assembly using the default parameters.
Sequence annotation
The sequence annotation was based on a set of BLAST searches. The sequences were searched using BLASTX against the NCBI non- redundant protein (nr) database with an E-value cut off of 10-5, then the NCBI non- redundant nucleotide (nt) database was searched. The remaining no hit sequences that putatively encoded proteins were searched against the Arabidopsis protein database in the Arabidopsis Information Resource (TAIR 2.2.8); a typical cutoff value of E < 1.0-5 was used. Pathway assignments were carried out according to KEGG mapping. Enzyme commission (EC) numbers were assigned to unique sequences that had BLASTX scores with cutoff values of E < 1.0e5, as determined upon searching the protein databases. The sequences were mapped to the KEGG biochemical pathways according to the EC distribution in the pathway database. We performed a functional classification of the unigenes following the Gene Ontology (GO) scheme. The sequences were annotated by a BLAST search against a series of protein and nucleotide databases, including the curated protein database of Uniprot/SwissProt. The transcripts were classified into 45 GO categories under the major categories of Cellular Component, Molecular Function and Biological Process. A unigene blast was performed using the uniprot database and the annotated GO function. The unigene (congtig and singletons) blast was performed on reference canonical pathways and annotated KEGG pathway.
Real-time PCR analysis
Total RNA was extracted from embryos and endosperms from warm and cold seed stratification stages using the RNeasy Plant kit (BioTeke, Beijing China). Approximately 1 μg of Dnase I-treated total RNA from each was converted into single-stranded cDNA using a PrimeScript™ RT reagent Kit (Takara, Dalian, China). The cDNA products were then diluted 10-fold with deionized water before use as a template for real-time PCR. Each reaction contained 10 μl 2× SYBR Premix DimerEraser (Takala, Dalian, China), 1 μM each of the forward and reverse primers, and 1 μl of template cDNA. The total reaction volume was 20 μl. PCR amplification was performed under the following conditions: 95°C for 30 s, followed by 40 cycles of 95°C for 5 s and 60°C for 30 s, using CFX96TM Real-Time system (Bio-Rad, USA). Thirty-six unigenes from the 454 data were selected to design qRT-PCR primers and ten housekeeping gene transcripts were selected for design internal control primers (Additional file 4). Each embryo sample in 4°C stratification was selected as a calibrator and the data were presented as the fold change in gene expression normalized to an endogenous reference gene and relative to the calibrator [16]. The qRT-PCR analyses were performed three times with independent RNA samples.
Abbreviations
ABA: Abscisic acid; BR: Brassinosteroid; COG: Cluster of orthologous groups of proteins; EST: Expression sequence tags; GA: Gibberellins; GO: Gene ontology; IAA: Indo-3 acetic acid; KEGG: Kyoto Encyclopedia of Genes and Genomes.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JQ conceived this study, designed and built the cDNA library, participated in data analysis, and drafted the manuscript. NZ participated in seeds stratification, RNA extraction, library construction and data analysis. BZ and SH performed the sequence assembly, alignment and annotations. PS and WX participated in sequence data analysis, the real-time qPCR and the corresponding data analysis. QM and TZ carried out the field management, seed collection and seed stratification. LZ and MQ helped to conceive the study and performed the statistical analysis. XL initiated the project, helped to conceive the study, and participated in the design and coordination. All authors read and approved the final manuscript.
Supplementary Material
Functional annotation of putative unique transcripts from P. polyphylla based on GO categories.
COG annotations of putative proteins. All putative proteins were aligned to the COG database and could be classified functionally into at least 25 molecular families.
KEGG pathway involved in phytohormone synthesis such as steroid, terpenoid backbone, brassinosteroid and carotenoid biosynthesis (numbers in red indicate expressed genes).
Primers used in qRT-PCR analysis.
Possible transcripts involved in seed stratification of P. polyphylla var. yunnanensis.
Contributor Information
Jianjun Qi, Email: jjqi@implad.ac.cn.
Na Zheng, Email: zhengna08@163.com.
Bing Zhang, Email: zhangbing@big.ac.cn.
Peng Sun, Email: psun@implad.ac.cn.
Songnian Hu, Email: husn@big.ac.cn.
Wenjuan Xu, Email: xwjxuwenjuan@163.com.
Qin Ma, Email: maqin3680@163.com.
Tingzhou Zhao, Email: zhaotz3686@163.com.
Lili Zhou, Email: llzhou@implad.ac.cn.
Mingjian Qin, Email: mingjianqin@163.com.
Xianen Li, Email: xeli@implad.ac.cn.
Acknowledgements
We would like to thank Dr. A. Egrinya Eneji of the University of Calabar, Nigeria for helpful comments on the manuscript. This work was supported by the National Natural Science Foundation of China (Grant number 31171623) and Traditional Chinese Medicine Support project (2008) of Ministry of Industry and Information technology of PR China.
References
- He J, Zhang S, Wang H, Chen CX, Chen SF. Advances in studies on and uses of Paris polyphylla var. yunnanensis. Acta Botanica Yunnanica. 2006;28:271–276. [Google Scholar]
- Fan YM, Li YM, Cheng QM. Introduction and domestication research of Paris polyphylla var. yunnanensis. Chinese Chemistry. 2005;36:1102–1104. [Google Scholar]
- Liu GZ, Xu QP, Wang T. The essentials of traditional Chinese herbal medicine. Beijing: Foreign Languages Press; 2003. p. P171. [Google Scholar]
- Liang HX, Zhang XL. Development of seed and aril of two species of genus Paris. Acta Botanica Yunnanica. 1987;9(3):319–324. [Google Scholar]
- Li H. The Genus Paris (Trilliaceae) Beijing: Science Press; 1998. [Google Scholar]
- Chen C, Yang LY, Lv LF, Zhao Q, Yuan LC. Study on seedling techniques of Paris polyphylla var. yunnanensis. China Journal of Chinese Materia Medica. 2007;32:1979–1983. [PubMed] [Google Scholar]
- Huang W, Meng FY, Zhang WS, Wang YY. Study on seed dormancy mechanism of Paris polyphylla var. yunnanensis. Chinese Agricultural Science Bulletin. 2008;24(12):242–246. [Google Scholar]
- Chen SY, Yin PX, YAN YQ, Wang L, Ye Y, Shen Y. Rule of breaking Paris polyphylla var. yunnanensis seed dormancy under fluctuating temperature stratification and content changes of endogenous hormone. Chinese Traditional and Herbal Drugs. 2011;42:793–795. [Google Scholar]
- Baskin CC, Baskin JM. Seeds, Ecology, Biogeography and Evolution of Dormancy, and Germination. San Diego: Academic Press; 1998. [Google Scholar]
- Baskin JM, Baskin CC. A classification system for seed dormancy. Seed Sci Res. 2004;14:1–16. [Google Scholar]
- Yang B, Li SP, Wang X, Li LY, Yang LY, Yan SW, Gu AY. On cultivation and rational utilization of Paris. Chinese Wild Plant Resource. 2008;27(6):70–73. [Google Scholar]
- Ji YH, Fritsch PW, Li H, Xiao TJ, Zhou ZK. Phylogeny and classification of Paris (Melanthiaceae) inferred from DNA sequence data. Ann Bot. 2006;98:245–256. doi: 10.1093/aob/mcl095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin W, Kerstin M, Gerhard LM. First off the mark: early seed germination. J Exp Bot. 2011;62(10):3289–3309. doi: 10.1093/jxb/err030. [DOI] [PubMed] [Google Scholar]
- Edwards CE, Parchman TL, Weekley CW. Assembly, Gene annotation and marker development using 454 floral transcriptome sequences in Ziziphus celata (Rhamnaceae), a highly endangered, Florida endemic plant. DNA Res. 2012;19:1–9. doi: 10.1093/dnares/dsr037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Xu RH, Wang RL, Liu AZ. Transcriptome analysis of Sacha Inchi (Plukenetia volubilis L.) seeds at two developmental stages. BMC Genomics. 2012;13:716–63. doi: 10.1186/1471-2164-13-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-△△CT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Finkelstein R, Reeves W, Ariizumi T, Steber C. Molecular aspects of seed dormancy. Annu Rev Plant Biol. 2008;59:387–415. doi: 10.1146/annurev.arplant.59.032607.092740. [DOI] [PubMed] [Google Scholar]
- Kucera B, Cohn MA, Leubner-Metzger G. Plant hormone interactions during seed dormancy release and germination. Seed Sci Res. 2005;15:281–307. doi: 10.1079/SSR2005218. [DOI] [Google Scholar]
- Dalchau N, Baek SJ, Briggs HM, Robertson FC, Dodd AN, Gardner MJ, Stancombe MA, Haydon MJ, Stan GB, Goncalves JM, Webb AA. The circadian oscillator gene GIGANTEA mediates a long-term response of the Arabidopsis thaliana circadian clock to sucrose. Proc Natl Acd Sci USA. 2011;108:5104–5109. doi: 10.1073/pnas.1015452108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WY, Hicks KA, Somers D. Independent roles for Early Flowering 3 and Zeitlupe in the control of circadian timing, hypocotyls length, and flowering time. Plant Physiol. 2005;139:1557–1569. doi: 10.1104/pp.105.067173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight H, Thomson AJW, McWatters HG. SENSENTIVE TO FREEZING6 integrates cellular and environmental inputs to the plant circadian clock. Plant Physiol. 2008;148:293–303. doi: 10.1104/pp.108.123901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambara E, Nonogaki H. Seed biology in the 21st century: perspectives and new directions. Plant Cell Physiol. 2012;53:1–4. doi: 10.1093/pcp/pcr184. [DOI] [PubMed] [Google Scholar]
- Mochida K, Shinozaki K. Advances in omics and bioinformatics tools for systems analysis of plant functions. Plant Cell Physiol. 2011;52:2017–2038. doi: 10.1093/pcp/pcr153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le BH, Cheng C, Bui AQ, Wagmaister JA, Henry KF, Pelletier J, Kwong L, Belmonte M, Kirkbride R, Horvath S, Drews GN, Fischer RL, Okamuro JK, Harada JJ, Goldberg RB. Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc Natl Acad Sci USA. 2010;107:8063–8070. doi: 10.1073/pnas.1003530107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang D, Venglat P, Tibiche C, Yang H, Risseeuw E, Cao YG, Babic V, Cloutier M, Keller W, Wang E, Selvaraj G, Datla R. Genome-wide analysis reveals gene expression and metabolic network dynamics during embryo development in Arabidopsis. Plant Physiol. 2011;156:346–356. doi: 10.1104/pp.110.171702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths J, Barrero JM, Taylor J, Helliwell CA, Gubler F. ALTERED MERISTEM PROGRAM 1 is involved in development of seed dormancy in Arabidopsis. PLoS One. 2011;6(5):e20408. doi: 10.1371/journal.pone.0020408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo M, Nambara E, Choi G, Yamaguchi S. Interaction of light and hormone signals in germination seeds. Plant Mol Biol. 2009;69:463–472. doi: 10.1007/s11103-008-9429-y. [DOI] [PubMed] [Google Scholar]
- Footitt S, Douterelo-Soler I, Clay H, Finch-Savage WE. Dormancy cycling in Arabidopsis seeds is controlled by seasonally distinct hormone-signaling pathways. Proc Natl Acd Sci USA. 2011;108:20236–20241. doi: 10.1073/pnas.1116325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HG, Jun SH, Kim J, Kawaide H, Kamiya Y, An G. Cloning and molecular analyses of a gibberellin 20-oxidase gene expressed specifically in developing seeds of watermelon. Plant Physiol. 1999;121:373–382. doi: 10.1104/pp.121.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchum MG, Yamaguchi S, Hanada A, Kuwahara A, Yoshioka Y, Kato T, Tabata S, Kamiya Y, Sun TP. Distinct and overlapping roles of two gibberellins 3-oxidases in Arabidopsis development. Plant J. 2006;45:804–818. doi: 10.1111/j.1365-313X.2005.02642.x. [DOI] [PubMed] [Google Scholar]
- Sakai M, Sakamoto T, Saito T, Matsuoka M, Tanaka H, Kobayashi M. Expression of novel rice gibberellins 2-oxidase gene is under homeostatic regulation by biologically active gibberellins. J Plant Res. 2003;116:161–164. doi: 10.1007/s10265-003-0080-z. [DOI] [PubMed] [Google Scholar]
- Yamauchi Y, Ogawa M, Kuwahara A, Hanada A, Kamiya Y, Yamaguchi S. Activation of gibberellins biosynthesis and response pathways by low temperature during imbibitions of Arabidopsis thaliana seeds. Plant Cell. 2004;16:367–378. doi: 10.1105/tpc.018143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdsworth MJ, Bentsink L, Soppe WJJ. Molecular networks regulating Arabidopsis seed maturation, after-ripening, dormancy and germination. New Phytol. 2008;179:33–54. doi: 10.1111/j.1469-8137.2008.02437.x. [DOI] [PubMed] [Google Scholar]
- Schwartz SH, Tan BC, Gage DA, Zeevaart JA, McCarty DR. Specific oxidative cleavage of carotenoid by vp14 of maize. Science. 1997;276:1872–1874. doi: 10.1126/science.276.5320.1872. [DOI] [PubMed] [Google Scholar]
- Seo M, Peeters AJ, Koiwai H, Oritani T, Marion-poll A, Zeevaart JA, Koomneef M, Kamiya Y, Koshiba T. The Arabidopsis aldehyde oxidase 3 (AAO3) gene product catalyzes the final step in abscisic acid biosynthesis in leaves. Proc Natl Acd Sci USA. 2000;97:12908–12913. doi: 10.1073/pnas.220426197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann R, Sakai H, Hochholdinger F. The gibberellic acid stimulated-like gene family in maize and its role in lateral root development. Plant Physiol. 2010;152:356–365. doi: 10.1104/pp.109.149054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura N, Yoshida T, Kitahaa N, Asami T, Shinozaki K, Hirayama T. ABA-Hypersensitive Germination1 encodes a protein phosphatase 2C, an essential component of abscisic acid signaling in Arabidopsis seed. Plant J. 2007;50:935–949. doi: 10.1111/j.1365-313X.2007.03107.x. [DOI] [PubMed] [Google Scholar]
- Rampey RA, LeCleve S, Kowalczyk M, Ljung K, Bartel B. Sandberg. A family of auxin-conjugate hydrolases that contributes to free indole-3-acetic acid levels during Arabidopsis germination. Plant Physiol. 2004;135:978–988. doi: 10.1104/pp.104.039677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller A, Weiler EW. AII-synthase, an enzyme complex from Arabidopsis thaliana catalyzing the formation of indole-3-acetic acid from (S)-tryptophan. Biol Chem. 2000;381:679–686. doi: 10.1515/BC.2000.088. [DOI] [PubMed] [Google Scholar]
- Park SC, Han KH. Anauxin-repressed gene (RpARP) from black locust is posttranscriptionally regulated and negatively associated with shoot elongation. Tree Physiol. 2003;23:815–823. doi: 10.1093/treephys/23.12.815. [DOI] [PubMed] [Google Scholar]
- Gou XP, Yin HJ, He K, Du JB, Yi J, Xu SB, Lin H, Clouse SD, Li J. Genetic evidence for an indispensable role of somatic embryogenesis receptor kinases in brassinostroid signaling. PLoS Genet. 2012;8(1):e1002452. doi: 10.1371/journal.pgen.1002452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlova R, Boeren S, Russinova E, Aker J, Vervoort J, Vries S. The Arabidopsis SOMATIC EMBRYOGENESIS RECEPTR-LIKE KINASE1 protein complex includes BRASSINOSTEROID-INSENSITIVE1. Plant Cell. 2006;18:626–638. doi: 10.1105/tpc.105.039412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer WE. Dormancy-associated embryonic mRNA and proteins in imbibing Avena fatua caryopses. Physiol Plant. 1993;88:201–211. doi: 10.1111/j.1399-3054.1993.tb05490.x. [DOI] [Google Scholar]
- Toorop PE, Barroco RM, Engler G, Groot SP, Hilhorst HW. Differentially expressed genes associated with dormancy or germination of Arabidopsis thaliana seeds. Planta. 2005;221:637–647. doi: 10.1007/s00425-004-1477-1. [DOI] [PubMed] [Google Scholar]
- McClung CR. Plant circadian rhythms. Plant Cell. 2006;18:792–803. doi: 10.1105/tpc.106.040980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nefissi R, Natsui Y, Miyata K, Oda A, Hase Y, Nakagawa M, Ghorbel A, Mizoguchi T. Double loss-of function mutation in Early Flowering 3 and Cryptochrome 2 genes delays flowering under continuous light but accelerates it under long days and short days: an important role for Arabidopsis CRY2 to accelerate flowering time in continuous light. J Exp Bot. 2011;62:2731–2744. doi: 10.1093/jxb/erq450. [DOI] [PubMed] [Google Scholar]
- Manfre AJ, Lanni LM, Marcotte WR Jr. The Arabidopsis group 1 LATE EMBRYOGENESIS ABUNDANT protein ATEM6 is required for normal seed development. Plant Physiol. 2006;140:140–149. doi: 10.1104/pp.105.072967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouchkina-Stantcheva NN, McGee BM, Boschetti C, Tolleter D, Chakrabortee S, Popova AV, Meersman F, Macherel D, Hincha DK, Tunnacliffe A. Functional divergence of former alleles in an ancient asexual invertebrate. Science. 2007;318:268–271. doi: 10.1126/science.1144363. [DOI] [PubMed] [Google Scholar]
- Shih MD, Hoekstra FA, Hsing YI. Late embryogenesis abundant proteins. Adv Bot Res. 2008;48:211–255. [Google Scholar]
- Ismail FA, Nitsch LM, Wolters-Arts MM, Mariani C, Derksen JW. Semi-viviparous embryo development and dehydrin expression in the mangrove Rhizophora mucronata Lam. Sex Plant Reprod. 2010;23(2):95–103. doi: 10.1007/s00497-009-0127-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, He M, Zhu Z, Li S, Xu Y, Zhang C, Singer SD, Wang Y. Identification of the dehydrin gene family from grapevine species and analysis of their responsiveness to various forms of abiotic and biotic stress. BMC Plant Biol. 2012;12:140. doi: 10.1186/1471-2229-12-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Wang Z, Du H, Huang B. Differential accumulation of dehydrins in response to water stress for hybrid and common Bermuda grass genotypes differing in drought tolerance. J Plant Physiol. 2010;167(2):103–109. doi: 10.1016/j.jplph.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Lin CH, Peng PH, Ko CY, Markhart AH, Lin TY. Characterization of a novel Y2K-type dehydrin VrDhn1 from Vigna radiate. Plant Cell Physiol. 2012;53(5):930–942. doi: 10.1093/pcp/pcs040. [DOI] [PubMed] [Google Scholar]
- Zhang HP, Chang C, Zhang XY, Yan CS, Xiao SH. Isolation and expression analysis of dehydrin gene involved in ABA-regulated seed embryo dormancy in wheat. Molecular Plant Breeding. 2008;6:1175–1181. [Google Scholar]
- Nonogaki H, Gee OH, Bradford KJ. A germination-specific endo-β-mannanase gene is expressed in the micropylar endosperm cap of tomato seeds. Plant Physiol. 2000;23(4):1235–1246. doi: 10.1104/pp.123.4.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons CR. The physiology and molecular biology of plant 1, 3-β-glucanases and 1, 3; 1, 4–glucanases. Critical Reviews in Plant Sci. 1994;13:325–387. [Google Scholar]
- Cosgrove DJ. Loosening of plant cell walls by expansins. Nature. 2000;407:321–326. doi: 10.1038/35030000. [DOI] [PubMed] [Google Scholar]
- de Silva J, Jarman CD, Arrowsmith DA, Stronach MS, Chengappa S, Sidebottom C, Reid JS. Molecular characterization of a xyloglucan-specific endo-(1–4)-β-D-glucanase (xyloglucan endotransglycosylase) from nasturtium seeds. Plant J. 1993;3(5):701–711. doi: 10.1111/j.1365-313X.1993.00701.x. [DOI] [PubMed] [Google Scholar]
- Tine MAS, Cortelazzo AL, Buckeridge MS. Xyloglucan mobilization in cotyledons of developing plantlets of Hymenaea courbaril L. (Leguminoseae-Caesalpinoideae) Plant Sci. 2000;154:117–126. doi: 10.1016/S0168-9452(99)00245-9. [DOI] [PubMed] [Google Scholar]
- Ren C, Kermode AR. An increase in pectin methyl esterase activity accompanies dormancy breakage and germination of yellow cedar seeds. Plant Physiol. 2000;124:231–22. doi: 10.1104/pp.124.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourey PS, Jain M, Li QB, Carlson SJ. Genetic control of cell wall invertases in developing endosperm of maize. Planta. 2006;223:159–167. doi: 10.1007/s00425-005-0039-5. [DOI] [PubMed] [Google Scholar]
- Jain M, Chourey PS, Li QB, Pring DR. Expression of cell wall invertase and several other genes of sugar metabolism in relation to seed development in sorghum (Sorghum bicolor) J Plant Physiol. 2008;165(3):331–344. doi: 10.1016/j.jplph.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Endo A, Tatematsu K, Hanada K, Duermeyer L, Okamoto M, Yonekura-Sakakibara K, Saito K, Toyoda T, Kamiya Y, Seki M, Nambara E. Tissue-specific transcriptome analysis reveals cell wall metabolism, flavonol biosynthsis, and defense responses are activated in the endosperm of germinating Arabidopsis thaliana seeds. Plant Cell Physiol. 2012;53(1):16–27. doi: 10.1093/pcp/pcr171. [DOI] [PubMed] [Google Scholar]
- Murphy A, Peer WA, Taiz L. Regulation of auxin transport by aminopeptidases and endogenous flavonoids. Planta. 2000;211:315–324. doi: 10.1007/s004250000300. [DOI] [PubMed] [Google Scholar]
- Sun C, Li Y, Wu Q, Luo HM, Sun YZ, Song JY, Lui MK, Chen S. De novo sequencing and analysis of the American ginseng root transcriptome using a GS FLX Titanium platform to discover putative genes involved in ginsenoside biosynthesis. BMC Genomics. 2010;11:262–273. doi: 10.1186/1471-2164-11-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Functional annotation of putative unique transcripts from P. polyphylla based on GO categories.
COG annotations of putative proteins. All putative proteins were aligned to the COG database and could be classified functionally into at least 25 molecular families.
KEGG pathway involved in phytohormone synthesis such as steroid, terpenoid backbone, brassinosteroid and carotenoid biosynthesis (numbers in red indicate expressed genes).
Primers used in qRT-PCR analysis.
Possible transcripts involved in seed stratification of P. polyphylla var. yunnanensis.