

Abstract
In schistosomiasis, the severity of CD4+ T-cell-mediated hepatic granulomatous inflammation against parasite eggs varies considerably in humans and among mouse strains. In C57BL/6 mice, pronounced exacerbation of immunopathology induced by immunization with schistosome egg Ag in CFA (SEA/CFA) substantially recapitulates the natural high pathology seen in CBA mice; both are associated with a significant elevation of Th17- and Th1-cell-derived proinflammatory cytokines. We now investigated the relative contribution of the effector cytokines IL-17 and IFN-γ in pathology development of 7 wk-infected, SEA/CFA-immunized, IL-17−/−, IFN-γ−/−, and IL-17/IFN-γ−/− mice. In IL-17−/− mice there was significant reduction of immunopathology despite increased levels of IFN-γ, whereas in IFN-γ−/− mice, markedly exacerbated immunopathology correlated with an increase in IL-17. In IL-17/IFN-γ−/− mice, complete resistance to SEA/CFA-induced disease exacerbation was associated with a reduction in IL-23p19, IL-1β, CXCL1 and iNOS, and with an increase in IL-5, IL-10 and Relmα. IL-17 and IFN-γ were derived from distinct CD4+ T cells in which production of each cytokine was suppressed by the other. Our results indicate that severe immunopathology in murine schistosomiasis is mainly driven by IL-17 and regulated by IFN-γ; however, in the absence of IL-17, IFN-γ is capable of exerting a limited, yet significant, pathogenic function.
Keywords: CD4+ T cells, Cytokines, Host/pathogen interactions, Inflammation
Introduction
The main immunopathology in infection with the helminth Schistosoma mansoni is characterized by granulomatous and fibrosing inflammation around parasite eggs in the liver and intestines [1, 2]. The extent of disease varies greatly, both in humans as well as among mouse strains. In mice, the CBA strain develops severe inflammation, whereas in C57BL/6 (BL/6) mice the lesions are significantly milder [3, 4]. Granuloma formation is mediated by CD4+ T cells specific for egg Ags, as these lesions fail to develop in athymic, MHC class II−/−, TCRαβ−/− or Rag-1−/− mice [5–7], however, it is still not clear how the cytokine environment modulates the prevailing inflammatory process. Previous studies in BL/6 mice have demonstrated that during the course of the schistosome infection there is an initial Th1-polarized proinflammatory response, marked by IFN-γ, which following parasite oviposition at 5 wk of infection, is gradually replaced by a Th2-dominated environment characterized by the rise of IL-4, IL-5, IL-10 and IL-13. The Th1 to Th2 cytokine switch is critical for the modulation of immunopathology and host survival of the acute infection [1, 8], although the Th2 milieu can potentially be detrimental in the chronic disease, mainly by promoting liver fibrosis through IL-13 [9, 10].
A distinctive form of immunopathology develops in schistosome-infected BL/6 mice following immunization with schistosome egg Ags (SEA) in CFA (SEA/CFA). Under these circumstances, marked exacerbation of hepatic inflammation and early death correlates with the persistence of a proinflammatory state and the failure of the Th2 response to materialize. The severe immunopathology in these mice was originally attributed to uncontrolled Th1 cell activity [11]. However, this paradigm had to be revised following the detection of high levels of IL-17A (henceforth referred as IL-17), alongside with IFN-γ in supernatants from SEA-stimulated mesenteric lymph node cells (mLNCs) and granuloma cells (GrCs) from SEA/CFA-immunized mice [12]. A similar reinterpretation occurred in a number of other CD4+ T-cell-mediated conditions, including EAE [13] and collagen-induced arthritis (CIA) [14], in which IL-17, rather than IFN-γ, was shown to be the main cytokine associated with the autoimmune inflammatory process. IL-17 was demonstrated to be the signature cytokine and largely the product of a novel and distinct proinflammatory CD4+ T-helper (Th17) cell population induced by a combination of innate immune cell-derived cytokines including IL-6, TGF-β, IL-23, IL-1β and IL-21 [15–20].
We have previously shown that in schistosome-infected (BL/6) IL-12p40−/− mice, which are deficient in IL-12 and IL-23 and cannot produce normal levels of either IFN-γ or IL-17, there is a complete failure to develop the exacerbated form of egg-induced immunopathology in response to SEA/CFA immunization. In contrast, in IL-12p35−/− mice, which are deficient in IL-12 but not in IL-23 and can produce IL-17 but not IFN-γ, the augmented pathology is not different from that observed in BL/6 mice [12]. Interestingly, IL-23p19−/− mice, which produce suboptimal amounts of IL-17 in lymphoid tissues and none in the hepatic lesions, and in which IFN-γ production is regulated by IL-10, the resulting immunopathology is modest and significantly below that observed in the BL/6 WT [21]. These studies strongly implicated IL-17 in the development of severe inflammation but did not specifically address the contribution of IFN-γ towards lesional exacerbation or regulation.
The present study was designed to formally examine the role of the effector cytokines IL-17 and IFN-γ in the development of severe schistosome egg-induced immunopathology. This is a topic of considerable general interest particularly in view of the differing roles of these cytokines in the pathogenesis of a number of autoimmune and infectious diseases [19, 22–26]. We now show that in the absence of IL-17 there was a significantly reduced immunopathology associated with the increased levels of IFN-γ, whereas in the absence of IFN-γ there was a marked enhancement in immunopathology as well as in the levels of IL-17. Mice deficient in both IL-17 and IFN-γ were completely refractory to pathology exacerbation. Altogether, these findings indicate that in this model of high pathology, IL-17 exerts a powerful pathogenic function that normally is regulated by IFN-γ.
Results
IL-17−/− mice develop reduced immunopathology despite higher levels of IFN-γ
We previously demonstrated that in IL12-p40−/− and IL-23p19−/− mice, a markedly diminished immunopathology in response to SEA/CFA immunization is associated with a significant reduction in IL-17 and IFN-γ [12, 21]. We now investigated the role of IL-17 and IFN-γ in immunopathology directly using mice lacking either one or both cytokines. In schistosome-infected IL-17−/− mice, the SEA/CFA immunization elicited an increase in hepatic perioval inflammation, which was significantly lower than that seen in BL/6 mice and only slightly above the unimmunized controls (Fig. 1A). Similarly, the recruitment of Gr-1+ cells to the egg granulomas following SEA/CFA immunization was markedly impaired in the IL-17−/− mice; this decrease was more pronounced in the Gr-1+ CCR3− cell population, consistent with neutrophils, than in Gr-1+ cells expressing CCR3, the receptor for CCL11 (eotaxin), consistent with eosinophils [27] (Fig. 1B). SEA/CFA immunization induced strong in vitro production of both IFN-γ and IL-17 by SEA-stimulated mLNCs as well as GrCs from infected BL/6 mice (Fig. 1C and D). Interestingly, in similarly treated IL-17−/− mice, there was a marked increase in IFN-γ production by both mLNCs and GrCs in comparison with the BL/6 controls, while IL-17 was predictably absent (Fig. 1C and D). These findings demonstrated that in the absence of IL-17 there was a significant reduction in immunopathology despite the marked increase in IFN-γ.
Figure 1.
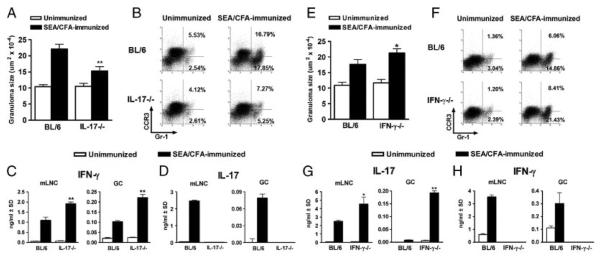
Immunopathology and cytokine production in infected IL-17−/− and IFN-γ−/− mice. (A and E) Hepatic egg granuloma size at 7 wk of infection (with or without SEA/CFA immunization) comparing BL/6 mice with (A) IL-17−/− or (E) IFN-γ−/− mice was measured by computer-assisted morphometric analysis. Data represent the mean+SEM of 3–5 mice per group from one experiment representative of three. (B and F) GrCs were purified from the livers of BL/6 and (B) IL-17−/− or (F) IFN-γ−/− mice and stained ex vivo to determine the percentage of Gr-1+CCR3+ and Gr-1+CCR3− cells by flow cytometry. Data are from one experiment representative of three, using pooled GrCs from 3 to 5 mice from each group. (C and H) IFN-γ and (D and G) IL-17 levels in 48-h supernatants from SEA-stimulated mLNCs and GrCs were measured by ELISA. Bars represent the mean+SD of triplicate determinations. Data are from one experiment representative of three or four. All statistical analyses are between SEA/CFA-immunized mouse groups and were determined by ANOVA followed by Tukey's multiple comparison test. **p<0.01 and *p<0.05. All gating strategies for flow cytometric analysis are shown in Supporting Information Fig. 1–3.
IFN-γ−/− mice develop increased immunopathology which correlates with higher levels of IL-17
In contrast to the IL-17−/− mice, schistosome-infected IFN-γ−/− mice displayed an even greater increase in egg-induced immunopathology than did the BL/6 mice in response to SEA/CFA immunization (Fig. 1E). The large, granulocyte-rich perioval granulomas in the IFN-γ−/− mice were poorly circumscribed, with inflammatory cells infiltrating deep into the surrounding liver parenchyma. Flow cytometric analysis of GrCs corroborated that those from the SEA/CFA-immunized IFN-γ−/− mice contained a significantly higher proportion of Gr-1+ cells in comparison with the BL/6 controls (Fig. 1F). The increase in Gr-1+ cells observed in the SEA/CFA-immunized IFN-γ−/− mice was more pronounced in the Gr-1+CCR3− than in the Gr-1+CCR3+ population (Fig. 1F). SEA/CFA immunization again resulted in strong in vitro production of both IL-17 and IFN-γ by SEA-stimulated mLNCs as well as GrCs from infected BL/6 mice (Fig. 1G and H). However, IL-17 production by SEA-stimulated mLNCs and GrCs was significantly higher in the IFN-γ−/− mice than in BL/6 controls, whereas IFN-γ was predictably absent (Fig. 1G and H). These findings again demonstrated a clear correlation between the intensity of immunopathology and the levels of IL-17, and suggested that under normal circumstances both are restrained by IFN-γ.
IL-17/IFN-γ−/− mice are completely resistant to SEA/CFA-induced pathology exacerbation
To more precisely assess the role of high levels of IFN-γ in the diminished but still higher hepatic egg-induced immunopathology elicited by SEA/CFA-immunization in IL-17−/− mice, we generated mice lacking both of these cytokines. Strikingly, the IL-17/IFN-γ−/− mice were completely refractory to SEA/CFA-induced exacerbation of immunopathology (Fig. 2A). The smaller granulomas, with fewer granulocytes and little parenchymal spillover, approached those typically seen in the absence of immunization. Flow cytometric analysis confirmed the failure to recruit both CCR3− as well as CCR3+, Gr-1+ cells to the granulomas (Fig. 2B). Furthermore, the pronounced activation of T cells and DCs in the granulomas normally induced by SEA/CFA immunization did not materialize in the IL-17/IFN-γ−/− mice. As such, in the CD4+ T cells there was a failure to up-regulate CD69 (Fig. 2C), and in the CD11c+ DC population there was a marked inhibition of CD80 (Fig. 2D) and CD86 (Fig. 2E) costimulatory molecule expression. Cytokine analysis of supernatants from SEA-stimulated mLNCs and GrCs again demonstrated a marked up-regulation of IL-17 and IFN-γ in the infected, SEA/CFA-immunized BL/6 mice, whereas in similarly treated IL-17/IFN-γ−/− mice, both cytokines were predictably absent (Fig. 3A and B). By comparison, the production of IL-5 by mLNCs and GrCs, normally downregulated in BL/6 mice following SEA/CFA immunization, was significantly higher in the IL-17/IFN-γ−/− mice (Fig. 3C). Similarly, IL-10 was downregulated in the immunized BL/6 mice but markedly increased in their IL-17/IFN-γ−/− counterparts (Fig. 3D). These findings demonstrated that IL-17 and IFN-γ by themselves largely account for immunocyte activation, granulocyte recruitment and pathology exacerbation, and that in their absence, an overall anti-inflammatory milieu associated with milder pathology, correlated with higher levels of IL-5 and IL-10.
Figure 2.

Immunopathology and cell marker expression in infected IL-17/IFN-γ−/− mice. (A) Hepatic egg granuloma size at 7 wk of infection (with or without SEA/CFA immunization) was measured by computer assisted morphometric analysis. Data represent the mean+SEM of 3–5 mice per group from one experiment representative of five. (B–E) GrCs were purified from the livers and stained ex vivo to determine by flow cytometry the percentage of (B) Gr-1+CCR3+ and Gr-1+CCR3− cells, (C) CD4+CD69+ T cells, and (D and E) CD80 and CD86 expression on CD11c+ DCs. Data represent (B) the percentages of ungated GrCs; (C) the percentage of CD4+ T cells that express CD69; (D) the percentage of CD11c+ DCs that express CD80; and (E) the percentage of CD11c+ DCs that express CD86. Data are from one experiment representative of five with similar results, using pooled GrCs from 3 to 5 mice from each group.
Figure 3.

Cytokine production by mLNCs and GrCs from infected IL-17/IFN-γ−/− mice. (A–D) IL-17, IFN-γ, IL-5 and IL-10 levels in 48-h supernatants from SEA-stimulated mLNCs and GrC from 7 wk-infected mice were measured by ELISA. Bars represent the mean+SD of triplicate determinations. Data are from one experiment representative of five with similar results. **p<0.01 and *p<0.05 were determined by ANOVA followed by Tukey's multiple comparison test.
Resistance to severe immunopathology correlates with low IL-23p19, IL-1β and CXCL1
To further address the mechanisms affecting the outcome of hepatic immunopathology, we measured gene expression of molecules involved in the induction and effector function of Th17 and Th1 cell subsets under conditions where their signature cytokines are absent. Expression of IL-23p19 in mesenteric lymph nodes (mLNs) was higher in SEA/CFA-immunized BL/6 and IFN-γ−/− than in IL-17/IFN-γ−/− and IL-17−/− mice and thus closely mirrored the level of immunopathology (Fig. 4A); such a correlation was not seen in the case of IL-12p40 and IL-12p35, although their level of expression was generally higher in response to SEA/CFA-immunization (Fig. 4B and C). IL-1β is also necessary for the induction of pathogenic Th17 responses in a number of conditions [18, 28]; in fact, we previously demonstrated that it is a key mediator of schistosome egg-induced Th17 cell development in vitro [29]. We now found a significantly lower mLN expression of IL-1β in SEA/CFA-immunized IL-17/IFN-γ−/− and IL-17−/− mice, which corresponded with their reduced pathology in the absence of IL-17 (Fig. 4D). Additionally, expression of the Th17-related neutrophil chemoattractant CXCL1 [19] was also markedly reduced in the SEA/CFA-immunized mouse groups lacking IL-17 (Fig. 4E), which correlated with the decreased number of neutrophils recruited to the granulomas (Figs. 1B and 2B).
Figure 4.

mLN cytokine expression and liver macrophage marker expression in infected IL-17−/−, IFN-γ−/− and IL-17/IFN-γ−/− mice. Cytokine mRNA expression of (A) IL-23p19, (B) IL-12p40, (C) IL-12p35, (D) IL-1β (E) CXCL1, and (F) IL-17F in mLNs, and macrophage marker mRNA expression of (G) Relmα and (H) iNOS in livers was measured by real time RT-PCR. Data represent mean mRNA levels+SD of 3–5 mice per group. The indicated statistically significant differences in all panels (**p<0.01 and *p<0.05) are between SEA/CFA-immunized cytokine-deficient and BL/6 mice, and were determined by ANOVA followed by Tukey's multiple comparison test. Additional statistically significant differences were as follows: (D and H) IFN-γ−/− versus IL-17−/− or IL-17/IFN-γ−/−, p<0.01, and (G) IFN-γ−/− versus IL-17/IFN-γ−/−, p<0.05.
IL-17F is a member of the IL-17 family of cytokines most closely related to IL-17, with a demonstrated role in a number of inflammatory responses [30]. We investigated IL-17F expression in the SEA/CFA-immunized mice since there was a possibility that IL-17F may exert some pathogenic function in the absence of IL-17. However, a higher mLN IL-17F expression in SEA/CFA-immunized IL-17/IFN-γ−/−, IFN-γ−/− and IL-17−/− mice than in BL/6 controls (Fig. 4F) in the face of markedly dissimilar levels of pathology and IL-17, suggested that IL-17F does not significantly contribute to pathology in schistosomiasis.
In the absence of IL-17, the reduced immunopathology correlates with high Relmα and low iNOS expression
The level of inflammation and the resulting immunopathology depend to a great extent on the nature of accessory cells that activate CD4+ T-cell subsets with different pathogenic potentials. In contrast to classically activated macrophages (CAMs), which promote proinflammatory T cells, alternatively activated macrophages (AAMs) are APCs that arise in a Th2-type environment and regulate immune responses [31]. In schistosomiasis, deletion of AAM function is associated with increased hepato-intestinal egg-induced immunopathology and death in a Th1-polarized environment [32]. SEA/CFA immunization caused a general drop in AAM activity as measured by the decrease in Relmα expression in the liver [31] (Fig. 4G). The lowest levels of Relmα were observed in BL/6 and IFN-γ−/− mice displaying the most severe immunopathology, whereas higher Relmα expression in IL-17/IFN-γ−/− and IL-17−/− mice correlated with little to no pathology increase with respect to the unimmunized controls. In contrast, high expression of the CAM-associated marker iNOS (Fig. 4H) correlated with severe immunopathology in the SEA/CFA-immunized BL/6 and IFN-γ−/− mice, and there was no increase in iNOS in the livers of low-pathology IL-17/IFN-γ−/− and IL-17−/− mouse groups. These findings demonstrated elevated AAM activity in low-pathology settings, whereas CAM activity is dominant in the context of exacerbated immunopathology.
IL-17 and IFN-γ are derived from distinct CD4+ T cells and suppress each other's production
Our findings so far indicated that the absence of IL-17, IFN-γ, or both, profoundly affects the overall cytokine balance and development of severe immunopathology in infection with schistosomes. Flow cytometric analysis demonstrated that in infected BL/6 mice, IL-17 and IFN-γ are associated with two small, discrete CD4+ T-cell populations, of which the IL-17-producing cells double, and the IFN-γ-producing cells increase by <50% upon SEA/CFA immunization (Fig. 5A). Based on the significant increase in IFN-γ in IL-17−/− mice (Fig. 1C) and of IL-17 in IFN-γ−/− mice (Fig. 1G), we further tested for the ability of each of these cytokines by themselves to regulate the production of the other by T cells from infected, SEA/CFA-immunized mice. As shown in Fig. 5B, the addition of rIFN-γ to SEA-stimulated mLNCs from SEA/CFA-immunized BL/6 mice significantly inhibited their production of IL-17; this effect was more pronounced in the case of mLNCs from SEA/CFA-immunized IFN-γ−/− mice, in which there is no endogenous source of IFN-γ. Similarly, the addition of rIL-17 equally inhibited the production of IFN-γ by mLNCs from SEA/CFA-immunized BL/6 and IL-17−/− mice (Fig. 5C), demonstrating that IFN-γ and IL-17 per se are capable of inhibiting each other's production, regardless of the opposite outcome of immunopathology in each case.
Figure 5.

Intracellular CD4+ T-cell cytokine expression and in vitro cytokine cross-regulation. (A) mLNCs from 7 wk-infected unimmunized and SEA/CFA-immunized BL/6 mice were stimulated in vitro with SEA and stained with anti-CD4, anti-IL-17 and anti-IFN-γ mAb as described in the Materials and Methods. Data represent the percentage of CD4+ T cells that express IL-17 and IFN-γ from one experiment representative of two with similar results, using pooled mLNCs from 3 to 5 mice from each group. (B and C) mLNCs from SEA/CFA-immunized BL/6, IFN-γ−/− and IL-17−/− mice were cultured in the absence or presence of SEA plus rIFN-γ or rIL-17 at the indicated concentrations. (B) IL-17 and (C) IFN-γ levels were measured after 48 h of culture by ELISA. Bars represent the mean+SD of triplicate determinations. **p<0.01 are between cultures containing recombinant cytokines and those with SEA alone and were determined by ANOVA followed by Tukey's multiple comparison test. Data are from one experiment representative of three with similar results.
Discussion
Severe immunopathology in murine schistosomiasis is precipitated by specific parasite–immunocyte interactions ultimately resulting in the development, activation and expansion of the proinflammatory Th17 and Th1 cell subsets. In this study we specifically examined the contribution of their respective effector signature cytokines, IL-17 and IFN-γ, to the development of immunopathology using mice deficient in either one or both cytokines. Given that these mice are on a BL/6 background, we took advantage of the SEA/CFA immunization model, which results in pathology exacerbation that bears considerable resemblance to the natural high pathology observed in CBA mice [11, 12].
We now demonstrate that both IL-17 and IFN-γ, produced by small, distinct, non-overlapping CD4+ T-cell populations, can by themselves account for the exacerbation and regulation of schistosome egg-induced immunopathology afforded by immunization of BL/6 mice with SEA/CFA. In these mice, the immunopathology mediated by IL-17 is restrained by IFN-γ as evidenced by the even greater pathology and IL-17 levels in mice lacking IFN-γ. On the other hand, the relatively smaller increase in pathology in IL-17−/− mice, which does not materialize in IL-17/IFN-γ−/− mice, is consistent with the notion that in the absence of IL-17, IFN-γ can take on a limited pathogenic function. These observations imply that in the absence of both IL-17 and IFN-γ, the “background” pathology, similar to that seen in unimmunized mice, is linked to a Th2-dominated cytokine environment, even though in the IL-17/IFN-γ−/− mice there was no lesional increase despite higher levels of IL-5.
We have previously observed an increase in pathology in mice lacking the Th1 lineage-specific T-box transcription factor T-bet [33]. These mice are incapable of making IFN-γ and of mounting Th1 immunity [34]. However, not until the present study has a direct effect of IFN-γ or IL-17 been tested on the ensuing schistosome egg-induced immunopathology. Several reports indicate that the absence of T-bet is not tantamount to the absence of IFN-γ, nor is T-bet invariably coupled to IFN-γ. For example, T-bet is not required for IFN-γ production by CD8+ T cells [35], and in humans, high T-bet expression does not necessarily correlate with IFN-γ production [36]. Moreover, IFN-γ-mediated class switching in B cells from IgG1 to IgG2a or IgG2b can be T-bet independent [37] and, importantly, T-bet has been shown to directly repress Th17 differentiation by preventing Runx1-mediated activation of the lineage-specific transcription factor RORγt [38].
Since the discovery of the Th17 cells, there have been numerous studies investigating the relative contribution of Th17 versus Th1 cells in the pathogenesis of various autoimmune and infectious diseases. Although it has been demonstrated that there is cross-regulation between the two subsets [39, 40], their individual contribution to the type, intensity and localization of immunopathology varies considerably among different experimental models. In agreement with our findings, increased severity of disease in CIA has been linked to a high IL-17/IFN-γ ratio, and in the absence of IFN-γ, enhanced pathology correlated with increased Th17 cell differentiation [14]. In two other arthritis models, Ag-induced and adjuvant-induced arthritis, IFN-γ similarly acted as a protective factor [41, 42] by restraining IL-17-stimulated granulopoiesis, neutrophil infiltration and bone destruction [43]; however, in a model of proteoglycan-induced arthritis in BALB/c mice, both IFN-γ and IL-17 contributed to disease, with severe disease dependent on IFN-γ, and IL-17 pathogenic only in the absence of IFN-γ [44]. Experimental autoimmune myocarditis (EAM) is another model in which IL-17 drives severe inflammation controlled by IFN-γ, with IFN-γ capable of causing milder disease in IL-17−/− mice [45–47]. In EAE, myelin-specific pathogenic Th1 versus Th17 cell responses were reported to mediate lesions in different anatomic locations [23], or to elicit inflammatory infiltrates rich in either macrophages or neutrophils respectively [22]. In a model of inflammatory bowel disease, Th17 cells synergized with Th1 cells to induce maximal colitis in IL-10−/− mice infected with Helicobacter hepaticus [48], while in experimental autoimmune uveitis (EAU), IL-23-dependent IL-17 played a dominant role in disease pathogenesis unaffected by IFN-γ [49]. Both Th17 or Th1 cells were instrumental in mediating protective immunity to Klebsiella pneumoniae [50], Mycobacterium tuberculosis [51] and Bordetella pertussis [52].
While it is difficult to reconcile the aforementioned differences among the various experimental models, our studies of the immunization-exacerbated form of schistosomiasis clearly demonstrate that severe hepatic egg-induced immunopathology is primarily mediated by IL-17, with IFN-γ acting as a regulatory element. Reciprocal APC and CD4+ T-cell activation following egg Ag stimulation leads to increased CD80 and CD86 costimulatory molecule expression, CD69 expression and proinflammatory cytokine/chemokine secretion resulting in increased lesional recruitment of mostly Gr-1+CCR3− neutrophils, and, ultimately, in pathology exacerbation. Using an in vitro system, we have previously demonstrated that egg-induced Th17 cell development is mainly driven by IL-23 and IL-1b [29]. SEA/CFA immunization also causes an increase in IL-12-dependent IFN-γ production, however, the precise means by which IFN-γ regulates IL-17 production are not clear and may indeed be manifold. One likely mechanism is through inhibition of IL-1β, as suggested in murine autoimmune disease models involving the use of mycobacterial products [53]. Such a mechanism is supported by the significant increase in IL-1β (Fig. 4D) seen in association with the most severe expression of immunopathology developed by IFN-γ−/− mice. On the other hand, in the absence of IL-17 and IFN-γ immunization with SEA/CFA resulted in decreased immunocyte activation, costimulatory molecule expression and proinflammatory cytokine production with consequent reduction in pathology. Under these conditions, IL-5 as well as IL-10, of demonstrated capacity to regulate schistosome egg-induced inflammation [54], are significantly elevated, particularly in secretions from GrCs isolated from the lesional environment. Moreover, the high expression of Relma (Fig. 2G), together with the significant decrease in iNOS expression (Fig. 4H) denotes a local state of alternative macrophage activation, which is critical to avert severe disease [32] and which we previously showed to be associated with low schistosome egg-induced immunopathology [33, 55]. However, there were no significant differences between the mouse groups in the expression of FoxP3, suggesting that in this setting T regulatory cells expressing this marker played no significant role in the outcome of pathology (data not shown).
The generation of the IL-17/IFN-γ−/− mouse was a critical step in demonstrating that in the absence of IL-17, IFN-γ can by itself also mediate weaker egg-induced inflammation resulting from a more limited leukocyte recruitment. The lack of correlation between levels of IL-17F and immunopathology suggested that IL-17F does not significantly affect the magnitude of inflammation in this system; neither did IL-6 or IL-22 (data not shown), both potentially associated with Th17 cell development or function [56–58]. Although our study was carried out in a model of severe pathology induced by immunization with SEA/CFA, the relevance of our findings is underscored by the significant inhibition of egg-induced immunopathology in novel schistosome-infected IL-12p40−/− mice on the natural high-pathology CBA background, which are unable to produce normal levels of either IL-17 or IFN-γ (our unpublished observation).
In summary, we show that the severe form of schistosome egg-induced immunopathology is primarily mediated by IL-17 and regulated by IFN-γ with each cytokine produced by distinct populations of CD4+ T cells, and each capable per se of regulating the production of the other. A similar scenario also applies to a number of other models. However, the considerable disparities in the reported pathogenic versus regulatory functions of these cytokines suggest that they can adopt unpredictable roles that depend on peculiarities of the different systems which include genetic host variation, nature of immunogen and type of adjuvant if any, and category of pathogen or, in the case of autoimmune disease, anatomic location and characteristics of target cells or tissues. Such individual variations need to be taken into consideration when designing immune-mediated strategies for control of severe immunopathology.
Materials and methods
Mice, infection and immunization
C57BL/6 (BL/6) and IFN-γ−/− mice, five to six-wk-old, were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). IL-17−/− mice (with a targeted deletion of IL-17A) were obtained from Dr. Yoichiro Iwakura, University of Tokyo, Japan [59]. We generated novel BL/6 IL-17 and IFN-γ double deficient (IL-17/IFN-γ−/−) mice in house by crossing IL-17−/−and IFN-γ−/− mice and selecting by PCR. All mice were housed at the Animal Facility at Tufts University School of Medicine in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) guidelines. Mice were infected by i.p. injection with 80 cercariae of S. mansoni (Puerto Rico strain), which were obtained from infected Biomphalaria glabrata snails, provided to us by Dr. Fred Lewis, Biomedical Research Institute (Rockville, MD, USA), through NIH/NIAID contract NO1-AI-55270. Some mice were immunized by s.c. injection with 50 μg of SEA emulsified in CFA (SEA/CFA), as previously described. Treatment of BL/6 mice with SEA/CFA causes marked exacerbation of their egg-induced immunopathology; either SEA or CFA by themselves are ineffective [11]. A soluble preparation of SEA was prepared as previously described [60].
Real-time quantitative RT-PCR
Total RNA from individual livers and mesenteric lymph nodes (mLN) of 7 wk-infected mice was isolated using the TRIzol reagent as per the manufacturer's instructions (Invitrogen). RNA (1–5 μg) was reverse-transcribed using High Capacity cDNA Reverse Transcription kit according to the manufacturer's instructions (Applied Biosystems). Real-time quantitative RT-PCR on 10 ng of cDNA from each sample was performed by TAQMAN™ analysis in a real-time quantitative PCR reaction on an ABI 7700 sequence detection system (Applied Biosystems). GAPDH levels were measured in a separate reaction and used to normalize the data. All reagents and protocols for real-time quantitative RT-PCR were obtained from Applied Biosystems. Using the average mean cycle threshold (Ct) value for GAPDH and the gene of interest for each sample, Equation 1.8e (Ct GAPDH–Ct gene of interest) × 104 was used to obtain normalized values [61].
Cell preparations, cell cultures and cytokine determinations
Livers and mLN were removed aseptically from 7-wk-infected mice. GrCs and mLNCs were prepared in complete RPMI-1640 medium (cRPMI) supplemented with 10% fetal calf serum (Aleken Biologicals), as described [11]. Bulk cell suspensions (5 × 106 cells/mL) from mLN and hepatic granulomas were incubated in the presence or absence of 15 μg/mL of SEA. Some mLNCs cultures received mouse rIFN-γ (BD-Pharmingen) or rIL-17 (R&D Systems) at 4 and 20 ng/mL. After 48 h, cytokine levels in the culture supernatants were measured in triplicate determinations by ELISA. For the detection of IL-17 and IFN-γ, mAb, standard cytokines and protocols were obtained from R&D Systems, and for the detection of IL-5 and IL-10, from BD-Pharmingen.
Flow cytometry
GrCs were stained ex vivo for flow cytometry analysis using allophycocyanin-conjugated anti-CCR3 mAb (clone 83101, R&D Systems.) and FITC-conjugated anti-Gr-1 mAb (clone RB6-8C5); allophycocyanin-conjugated anti-CD4 mAb (clone RM4-5) and FITC-conjugated anti-CD69 mAb (clone H1.2F3) or allophycocyanin-conjugated anti-CD11c mAb (clone HL3) and FITC-conjugated anti-CD80 mAb (clone 16-10A1) or anti-CD86 mAb (clone GL1) (all from BD-Pharmingen) following a protocol described before [62]. Propidium iodide (Sigma) was added before acquisition in the flow cytometer to exclude dead cells. For intracellular cytokine staining, mLNCs were stimulated with 15 mg/mL of SEA for 24 h and re-stimulated with PMA (50 ng/mL), ionomycin (500 ng/mL) and monensin (2 μg/mL) (all from Sigma) for 4 h to boost cytokine production and inhibit its secretion. After that time cells were washed, surface stained with allophycocyanin-conjugated anti-CD4 mAb and fixed overnight in 2% paraformaldehyde. The next day cells were washed once, permeabilized with 0.1% saponin buffer for 15 min at RT and further incubated with PE-conjugated anti-IL-17 (clone TC11-18H10) and FITC-conjugated anti-IFN-γ (clone XMG1.2) mAb (all from BD-Pharmingen) for 30 min at 4°C. Labeled cells were acquired on a FACSCalibur flow cytometer using the CELLQuest Pro software (Becton Dickinson). Data were analyzed using the WinList 5.0 Software (Verity Software House). Unstained cells and cells stained with irrelevant isotype-matched Ab were included as controls to assess the amount of non-specific staining.
Histopathology and morphometric analysis
Liver samples were fixed in 10% buffered formalin and processed for routine histopathologic analysis. Five-μm sections were stained with H&E and the extent of granulomatous inflammation around schistosome eggs was measured by computer-assisted morphometric analysis using Image-Pro Plus Software (Media Cybernetics) by an observer unaware of the experimental setting. To reflect more accurately the true shape and dimension of the granulomas, only those with a visible central egg were counted. A minimum of 10 granulomas were scored per liver section. Granuloma sizes are expressed as mean of areas measured in μm2+SEM.
Statistical analysis
ANOVA with post-test analysis was used to determine the statistical analysis of the differences among mouse groups. p-Values<0.05 were considered significant and were calculated with GraphPad Prism 4 (GraphPad Software). Each individual experiment was conducted with groups of 3–5 mice and repeated at least three times.
Acknowledgements
The authors thank the Tufts Laser Cytometry core facility for the assistance with the flow cytometry data analysis. This work was supported by US Public Health Service Grant RO1-18919.
Abbreviations
- AAM
alternative activated macrophage
- BL/6
C57BL/6
- CAM
classically activated macrophage
- GrC
granuloma cell
- mLNC
mesenteric lymph node cell
- SEA
schistosome egg Ag
- SEA/CFA
schistosome egg Ag (SEA) in CFA
Footnotes
Supporting Information available online
Conflict of interest: The authors declare no financial or commercial conflict of interest.
References
- 1.Pearce EJ, MacDonald AS. The immunobiology of schistosomiasis. Nat. Rev. Immunol. 2002;2:499–511. doi: 10.1038/nri843. [DOI] [PubMed] [Google Scholar]
- 2.Gause WC, Urban JF, Stadecker MJ. The immune response to parasitic helminths: insights from murine models. Trends Immunol. 2003;24:269–277. doi: 10.1016/s1471-4906(03)00101-7. [DOI] [PubMed] [Google Scholar]
- 3.Cheever A, Duvall R, Hallack T, Jr., Minker R, Malley J, Malley K. Variation of hepatic fibrosis and granuloma size among mouse strains infected with Schistosoma mansoni. Am. J. Trop. Med. Hyg. 1987;37:85–97. doi: 10.4269/ajtmh.1987.37.85. [DOI] [PubMed] [Google Scholar]
- 4.Fanning M, Peters P, Davis R, Kazura J, Mahmoud A. Immunopathology of murine infection with Schistosoma mansoni: relationship of genetic background to hepatosplenic disease and modulation. J. Infect. Dis. 1981;144:148–153. doi: 10.1093/infdis/144.2.148. [DOI] [PubMed] [Google Scholar]
- 5.Phillips SM, DiConza JJ, Gold JA, Reid WA. Schistosomiasis in the congenitally athymic (nude) mouse. I. Thymic dependency of eosinophilia, granuloma formation, and host morbidity. J. Immunol. 1977;118:594–599. [PubMed] [Google Scholar]
- 6.Hernandez HJ, Wang Y, Tzellas N, Stadecker MJ. Expression of class II, but not class I, major histocompatibility complex molecules is required for granuloma formation in infection with Schistosoma mansoni. Eur. J. Immunol. 1997;27:1170–1176. doi: 10.1002/eji.1830270518. [DOI] [PubMed] [Google Scholar]
- 7.Iacomini J, Ricklan D, Stadecker M. T cells expressing the γδ T cell receptor are not required for egg granuloma formation in schistosomiasis. Eur. J. Immunol. 1995;25:884–888. doi: 10.1002/eji.1830250404. [DOI] [PubMed] [Google Scholar]
- 8.Stadecker MJ, Asahi H, Finger E, Hernandez HJ, Rutitzky LI, Sun J. The immunobiology of Th1 polarization in high-pathology schistosomiasis. Immunol. Rev. 2004;201:168–179. doi: 10.1111/j.0105-2896.2004.00197.x. [DOI] [PubMed] [Google Scholar]
- 9.Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J. Clin. Invest. 1999;104:777–785. doi: 10.1172/JCI7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fallon PG, Richardson EJ, McKenzie GJ, McKenzie AN. Schistosome infection of transgenic mice defines distinct and contrasting pathogenic roles for IL-4 and IL-13: IL-13 is a profibrotic agent. J. Immunol. 2000;164:2585–2591. doi: 10.4049/jimmunol.164.5.2585. [DOI] [PubMed] [Google Scholar]
- 11.Rutitzky LI, Hernandez HJ, Stadecker MJ. Th1-polarizing immunization with egg antigens correlates with severe exacerbation of immunopathology and death in schistosome infection. Proc. Natl. Acad. Sci. USA. 2001;98:13243–13248. doi: 10.1073/pnas.231258498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rutitzky LI, Lopes da Rosa JR, Stadecker MJ. Severe CD4 T cell-mediated immunopathology in murine schistosomiasis is dependent on IL-12p40 and correlates with high levels of IL-17. J. Immunol. 2005;175:3920–3926. doi: 10.4049/jimmunol.175.6.3920. [DOI] [PubMed] [Google Scholar]
- 13.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 14.Chu CQ, Swart D, Alcorn D, Tocker J, Elkon KB. Interferon-gamma regulates susceptibility to collagen-induced arthritis through suppression of interleukin-17. Arthritis Rheum. 2007;56:1145–1151. doi: 10.1002/art.22453. [DOI] [PubMed] [Google Scholar]
- 15.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 16.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J. Biol. Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 17.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 18.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwakura Y, Nakae S, Saijo S, Ishigame H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol. Rev. 2008;226:57–79. doi: 10.1111/j.1600-065X.2008.00699.x. [DOI] [PubMed] [Google Scholar]
- 20.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Rutitzky LI, Bazzone L, Shainheit MG, Joyce-Shaikh B, Cua DJ, Stadecker MJ. IL-23 is required for the development of severe egg-induced immunopathology in schistosomiasis and for lesional expression of IL-17. J. Immunol. 2008;180:2486–2495. doi: 10.4049/jimmunol.180.4.2486. [DOI] [PubMed] [Google Scholar]
- 22.Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12-and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J. Exp. Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat. Med. 2008;14:337–342. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills KH. Induction, function and regulation of IL-17-producing T cells. Eur. J. Immunol. 2008;38:2636–2649. doi: 10.1002/eji.200838535. [DOI] [PubMed] [Google Scholar]
- 25.Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity. 2009;31:539–550. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Damsker JM, Hansen AM, Caspi RR. Th1 and Th17 cells: adversaries and collaborators. Ann. NY Acad. Sci. 2010;1183:211–221. doi: 10.1111/j.1749-6632.2009.05133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao JL, Sen AI, Kitaura M, Yoshie O, Rothenberg ME, Murphy PM, Luster AD. Identification of a mouse eosinophil receptor for the CC chemokine eotaxin. Biochem. Biophys. Res. Commun. 1996;223:679–684. doi: 10.1006/bbrc.1996.0955. [DOI] [PubMed] [Google Scholar]
- 28.Nakae S, Saijo S, Horai R, Sudo K, Mori S, Iwakura Y. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc. Natl. Acad. Sci. USA. 2003;100:5986–5990. doi: 10.1073/pnas.1035999100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shainheit MG, Smith PM, Bazzone LE, Wang AC, Rutitzky LI, Stadecker MJ. Dendritic cell IL-23 and IL-1 production in response to schistosome eggs induces Th17 cells in a mouse strain prone to severe immunopathology. J. Immunol. 2008;181:8559–8567. doi: 10.4049/jimmunol.181.12.8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, et al. Regulation of inflammatory responses by IL-17F. J. Exp. Med. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goerdt S, Orfanos CE. Other functions, other genes: alternative activation of antigen- presenting cells. Immunity. 1999;10:137–142. doi: 10.1016/s1074-7613(00)80014-x. [DOI] [PubMed] [Google Scholar]
- 32.Herbert DR, Holscher C, Mohrs M, Arendse B, Schwegmann A, Radwanska M, Leeto M, et al. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity. 2004;20:623–635. doi: 10.1016/s1074-7613(04)00107-4. [DOI] [PubMed] [Google Scholar]
- 33.Rutitzky LI, Smith PM, Stadecker MJ. T-bet protects against exacerbation of schistosome egg-induced immunopathology by regulating Th17-mediated inflammation. Eur. J. Immunol. 2009;39:2470–2481. doi: 10.1002/eji.200939325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 35.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 36.Ylikoski E, Lund R, Kylaniemi M, Filen S, Kilpelainen M, Savolainen J, Lahesmaa R. IL-12 up-regulates T-bet independently of IFN-gamma in human CD4+T cells. Eur. J. Immunol. 2005;35:3297–3306. doi: 10.1002/eji.200526101. [DOI] [PubMed] [Google Scholar]
- 37.Mohr E, Cunningham AF, Toellner KM, Bobat S, Coughlan RE, Bird RA, MacLennan IC, Serre K. IFN-gamma produced by CD8 T cells induces T-bet-dependent and -independent class switching in B cells in responses to alum-precipitated protein vaccine. Proc. Natl. Acad. Sci. USA. 2010;107:17292–17297. doi: 10.1073/pnas.1004879107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lazarevic V, Chen X, Shim JH, Hwang ES, Jang E, Bolm AN, Oukka M, et al. T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORgammat. Nat. Immunol. 2011;12:96–104. doi: 10.1038/ni.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 41.Irmler IM, Gajda M, Brauer R. Exacerbation of antigen-induced arthritis in IFN-gamma-deficient mice as a result of unrestricted IL-17 response. J. Immunol. 2007;179:6228–6236. doi: 10.4049/jimmunol.179.9.6228. [DOI] [PubMed] [Google Scholar]
- 42.Kim EY, Chi HH, Bouziane M, Gaur A, Moudgil KD. Regulation of autoimmune arthritis by the pro-inflammatory cytokine interferon-gamma. Clin. Immunol. 2008;127:98–106. doi: 10.1016/j.clim.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelchtermans H, Schurgers E, Geboes L, Mitera T, Van Damme J, Van Snick J, Uyttenhove C, Matthys P. Effector mechanisms of interleukin-17 in collagen-induced arthritis in the absence of interferon-gamma and counteraction by interferon-gamma. Arthritis Res. Ther. 2009;11:R122. doi: 10.1186/ar2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Doodes PD, Cao Y, Hamel KM, Wang Y, Rodeghero RL, Mikecz K, Glant TT, et al. IFN-gamma regulates the requirement for IL-17 in proteoglycan-induced arthritis. J. Immunol. 2010;184:1552–1559. doi: 10.4049/jimmunol.0902907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cihakova D, Rose NR. Pathogenesis of myocarditis and dilated cardiomyopathy. Adv. Immunol. 2008;99:95–114. doi: 10.1016/S0065-2776(08)00604-4. [DOI] [PubMed] [Google Scholar]
- 46.Valaperti A, Marty RR, Kania G, Germano D, Mauermann N, Dirnhofer S, Leimenstoll B, et al. CD11b+monocytes abrogate Th17 CD4+T cell-mediated experimental autoimmune myocarditis. J. Immunol. 2008;180:2686–2695. doi: 10.4049/jimmunol.180.4.2686. [DOI] [PubMed] [Google Scholar]
- 47.Afanasyeva M, Georgakopoulos D, Belardi DF, Bedja D, Fairweather D, Wang Y, Kaya Z, et al. Impaired up-regulation of CD25 on CD4+ T cells in IFN-gamma knockout mice is associated with progression of myocarditis to heart failure. Proc. Natl. Acad. Sci. USA. 2005;102:180–185. doi: 10.1073/pnas.0408241102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, Cua DJ, et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J. Exp. Med. 2006;203:2485–2494. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, Bowman EP, et al. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J. Exp. Med. 2008;205:799–810. doi: 10.1084/jem.20071258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Happel KI, Dubin PJ, Zheng M, Ghilardi N, Lockhart C, Quinton LJ, Odden AR, et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J. Exp. Med. 2005;202:761–769. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, Jelley-Gibbs DM, Ghilardi N, et al. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J. Immunol. 2005;175:788–795. doi: 10.4049/jimmunol.175.2.788. [DOI] [PubMed] [Google Scholar]
- 52.Higgins SC, Jarnicki AG, Lavelle EC, Mills KH. TLR4 mediates vaccine-induced protective cellular immunity to Bordetella pertussis: role of IL-17-producing T cells. J. Immunol. 2006;177:7980–7989. doi: 10.4049/jimmunol.177.11.7980. [DOI] [PubMed] [Google Scholar]
- 53.Masters SL, Mielke LA, Cornish AL, Sutton CE, O'Donnell J, Cengia LH, Roberts AW, et al. Regulation of interleukin-1beta by interferon-gamma is species specific, limited by suppressor of cytokine signalling 1 and influences interleukin-17 production. EMBO Rep. 2010;11:640–646. doi: 10.1038/embor.2010.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Flores-Villanueva PO, Zheng XX, Strom TB, Stadecker MJ. Recombinant IL-10 and IL-10/Fc treatment down-regulate egg antigen-specific delayed hypersensitivity reactions and egg granuloma formation in schistosomiasis. J. Immunol. 1996;156:3315–3320. [PubMed] [Google Scholar]
- 55.Bazzone LE, Smith PM, Rutitzky LI, Shainheit MG, Urban JF, Setiawan T, Blum AM, et al. Coinfection with the intestinal nematode Heligmosomoides polygyrus markedly reduces hepatic egg-induced immunopathology and proinflammatory cytokines in mouse models of severe schistosomiasis. Infect. Immun. 2008;76:5164–5172. doi: 10.1128/IAI.00673-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 57.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, et al. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 60.Boros D, Warren K. Delayed hypersensitivity-type granuloma formation and dermal reaction induced and elicited by a soluble factor isolated from Schistosoma mansoni soluble eggs. J. Exp. Med. 1970;132:488–507. doi: 10.1084/jem.132.3.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, Blumenschein W, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J. Clin. Invest. 2006;116:1317–1326. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rutitzky LI, Mirkin GA, Stadecker MJ. Apoptosis by neglect of CD4+Th cells in granulomas: a novel effector mechanism involved in the control of egg-induced immunopathology in murine schistosomiasis. J. Immunol. 2003;171:1859–1867. doi: 10.4049/jimmunol.171.4.1859. [DOI] [PubMed] [Google Scholar]