

Abstract
Sigma 1 receptor (S1R) is a eukaryotic membrane protein that functions as an inter-organelle signaling modulator and chaperone. Here we report an improved expression of S1R in Escherichia coli as a fusion to maltose binding protein (MBP) and a high-yield purification. Variants with linking amino acid sequences consisting of 0 to 5 alanine residues between MBP and S1R were created and tested in several E. coli expression strains in order to determine the best combination of construct and host for production of active MBP-S1R. Among the linker variations, the protein containing a 4-Ala linker exhibited superior expression characteristics (MBP-4A-S1R); this construct was most productively paired with Escherichia coli B834-pRARE2 and a chemically defined growth and expression medium. A 3-step purification was developed, including extraction from the E. coli membrane fraction using a mixture of Triton X-100 and n-dodecyl-beta-D-maltopyranoside identified by screening constrainted by retention of binding function, and purification by amylose affinity and gel filtration chromatographies. This procedure yields ~3.5 mg of purified fusion protein per L of bacterial culture medium. Purified MBP-4A-S1R showed a 175-fold purification from the starting cellular lysate with respect to specific ligand binding activity, and is stable during concentration and freeze-thaw cycling.
Keywords: sigma 1 receptor, maltose binding protein, fusion protein, Escherichia coli, selenomethionine labeling, integral membrane protein
Introduction
Sigma 1 receptor (S1R) is a 223 amino acid eukaryotic membrane protein found in the ER membrane of tissues of the endocrine, immune, and nervous systems. S1R interacts with progesterone and testosterone, and a diverse set of compounds including cocaine amphetamines, haloperidol, pentazocine, ditolylguanidine, hallucinogens and others [1–3]. S1R is implicated in inter-organelle signaling associated with neurological disorders and stroke, regulation of calcium homeostasis in mitochondria, sterol hormone synthesis and the etiology of addiction [4–10]. Knockout mice, which are otherwise viable, exhibit a variety of changes in psychological responses to stimuli, implicating S1R in pain response, learning, and psychoses [11–13]. S1R also interacts with other membrane proteins like acid-sensing channel and Nav1.5 voltage-gated Na+ channel, potentially providing modulator or chaperone-like properties [14,15].
S1R is a member of the ERG2_Sigma1 family (PFAM PF04622), which has a single domain architecture; greater than 100 homologous sequences have been identified in different eukaryotes. The family also includes fungal sterol binding proteins [16]. The primary sequence is highly conserved among different mammals, with >90% identity over 223 residues in human, chimpanzee, mouse, cow, rat, Mongolian gerbil and others [16]. Alternative splicing may produce transcript variants encoding distinct isoforms whose functions are not yet established. Although there are no three-dimensional structures known for this family, a membrane topology model for S1R has been assembled from biochemical and biophysical studies [17, 18]. S1R is thus predicted to be an α-helical membrane protein with two potential transmembrane domains [19].
S1R genes have been cloned from rodents and humans, and the protein has been expressed in Escherichia coli, Saccharomyces cerevisiae and CHO cells [2, 20–23]. The average yield of purified functional protein, whether from previous recombinant systems or from natural tissues, is low: e.g., 0.2 mg/L from E. coli culture and 0.2 mg from microsomes prepared from guinea pig liver.
Maltose binding protein (MBP) has been fused to membrane proteins in order to promote their expression, purification, and formation of crystals [24–30]. Moreover, periplasmic export of MBP has been shown to facilitate incorporation of appended membrane protein domains into the bacterial membrane [31–33]. Correspondingly, previous work showed that an MBP-S1R fusion containing a linker sequence for the Factor Xa protease recognition sequence could be expressed in E. coli BL21(DE3) and a functional form could be obtained, albeit in low yield, 0.6 mg of fusion protein per L of culture medium [2]. Our new results show that the E. coli strain used for expression and the linker region between MBP and S1R play important roles in production of the active form of the receptor. Arising from the improved expression, improved detergent extraction and improved purification protocols for MBP-S1R have been developed, and results from these are reported.
Materials and Methods
Reagents
All reagents were ACS grade unless otherwise specified. All buffers were prepared from deionized and distilled water (18MOhm) and filtered through a 0.8 μm filter.
Cloning
The guinea pig S1R gene is summarized as Uniprot Q60492. A plasmid encoding guinea pig S1R fused to periplasm-exportable maltose binding protein with a linker including a tobacco etch virus protease recognition site (MBP-TEV-S1R) was used as the template for PCR reactions. This plasmid was derived from MBP-Xa-S1R [1, 2]. The TEV protease linker between MBP and S1R was mutagenized to contain zero to five Ala residues by use of PIPE cloning [34, 35]. PCR was done using Pfu-UltraII polymerase (Stratagene, La Jolla, CA) and the primers listed in Table 1. All oligonucleotides were purchased from IDT (Coralville, IA). When the PCR reaction was completed, the template was destroyed by Dpn1 digestion [35]. The Dpn1-digested PCR product was purified using a kit (Qiagen, Valencia, CA) and the eluted DNA was used to transform E. coli 10G chemically competent cells (Lucigen, Middleton, WI). Plasmids were isolated from transformants and DNA sequencing was used to identify those containing the correct linker and coding region.
Table 1.
PCR primers used in this worka
| Primer name | Nucleotide sequence (5′-3′) | Length |
|---|---|---|
| gpS1R-F | ATGCAGTGGGCCGTGGGCCGGCGATG | 26 |
| gpS1R-A5R | GCCCACGGCCCACTGCATagctgcagctgcagcAGTCTGCGCGTCTTTCAGGGCTTC | 57 |
| gpS1R-A4R | GCCCACGGCCCACTGCATtgcagctgcagcAGTCTGCGCGTCTTTCAGGGCTTC | 54 |
| gpS1R-A3R | GCCCACGGCCCACTGCATagctgcagcAGTCTGCGCGTCTTTCAGGGCTTC | 51 |
| gpS1R-A2R | GCCCACGGCCCACTGCATtgcagcAGTCTGCGCGTCTTTCAGGGCTTC | 48 |
| gpS1R-A1R | GCCCACGGCCCACTGCATagcAGTCTGCGCGTCTTTCAGGGCTTC | 45 |
| gpS1R-NLR | GCCCACGGCCCACTGCATAGTCTGCGCGTCTTTCAGGGCTTC | 42 |
Each MBP-S1R variant was created using gpS1R-F and the appropriate reverse primer. Nucleotides shown in lower case encode the linker between MBP and S1R.
Expression strains
The progenitor MBP-TEV-S1R plasmid and new plasmids with variations in the linker region were transformed into the following Escherichia coli strains: BL21(DE3); B834-pRARE2; BL21(DE3)-RILP; C41(DE3)-pRARE2; and C43(DE3)-pRARE2. Chemically competent BL21(DE3) and B834 were from Novagen (Merck KGaA, Darmstadt, Germany), BL21(DE3)-RILP was from Stratagene, and C41(DE3) and C43(DE3) were from Lucigen. The rare codon supplementation plasmid pRARE2 was isolated from Rosetta 2 cells (Novagen) and then transformed into the appropriate strains [36]. All culture media were supplemented with 200 μg/mL of ampicillin; media used to grow strains transformed with the rare codon supplementation plasmids (pRARE2, RILP) were supplemented with 34 μg/mL of chloramphenicol.
Expression protocol
Starting inocula were grown in 3 mL of MDAG non-inducing medium at 37°C until the OD600 reached 0.2–0.3; the 3 mL culture was transferred to 100 mL of MDAG non-inducing medium [37]. The growth was continued at 25°C overnight with shaking. The scale-up of protein expression was carried out in selenomethionine-labeling medium 5SM having a modified sugar composition to permit IPTG induction (0.8% glycerol, 0.265% glucose, [37]). Polyethylene terephthalate 2-L soda bottles containing 480 mL of 5SM medium were inoculated with 20 mL of the overnight [38, 39]. The bottle cultures were grown at 37°C until the OD600 reached ~2. IPTG was then added to give a final concentration of 0.2 mM. The expression was continued at 25°C for ~20 h. Cells were harvested by centrifugation and stored at −80°C. A 0.1 g aliquot of cell paste was kept for expression analysis by denaturing electrophoresis.
Detergent solubilization screening
A 0.2 g aliquot of cell paste was re-suspended in 35 mL of 20 mM NaH2PO4, pH 7.5, containing 100 mM NaCl, 1 μM protease inhibitor E-64 (Sigma-Aldrich, Saint Louis, MO) and 0.3 mM (tris(2-carboxyethyl)phosphine). The cell suspension was sonicated on ice for 15 min using a Misonix 3000 sonicator (Farmingdale, NY) equipped with a micro tip horn (pulse on time of 2 s, pulse off time of 1 s) with output set to 7.0. The cell sonicate was centrifuged for 1 h at 75,000g between 4 and 8°C (Beckman-Coulter rotor JA- 25.50) and the resulting membrane fraction (pellet) was re-suspended in solubilization buffer containing 20 mM Tris-HCl, pH 8.0, 300 mM NaCl, and 1 mM 2-mercaptoethanol to give a final volume of 800 μL. The total protein content was measured by reducing agent compatible bichronic assay (BCA, ThermoFisher Scientific, Rockford, IL). The BCA assay was modified from the original protocol as follows. Instead of diluting with sample buffer, all standards and dilutions were prepared with deionized water. The values calculated for buffers were subtracted from the values obtained for sample dilutions.
A detergent screen was prepared to test the efficiency of combining Triton X-100 (5% or 10% w/v) with a second detergent for extraction of MBP-TEV-S1R. The following second detergents were included: n-decylphosphocholine (FC-10), n-dodecylphosphocholine (FC-12), lauryldimethylamine N-oxide (LDAO), 3-[(chloramidopropyl)-dimethylammonio]-1-propansulfonate (CHAPS), n-dodecyl-beta-D-maltopyranoside (DDM), sodium cholate, and n-octyl-beta-D-glucopyranoside (β-OG). Triton X-100 and sodium cholate were from Sigma, CHAPS was AppliChem GmbH (Darmstadt, Germany), and all other detergents from Affymetrix-Anatrace (Santa Clara, CA). Properties and applications of detergents used in this solubilization screen are discussed in detail in by Linke [40] and others [41,42,43]. A detergent master plate sufficient for 5 screening reactions was prepared in a 96 well plate. A 45 μL aliquot was transferred from the master plate to a corresponding well in an assay microplate; a control well was given buffer instead of detergent. A 5-μL aliquot of the re-suspended membrane fraction was then added to each well. The assay plate was covered and incubated at 4°C overnight or at room temperature for 3 h. After incubation, the assay microplate was centrifuged for 1 h at 5,500 rpm (Beckman-Coulter rotor JS-5.9) at 10°C. The supernatant (containing the solubilized protein) was transferred to a new microplate, while the pellet in the original plate was re-suspended in 50 μL of solubilization buffer. Aliquots (10 μL) of the solubilized (S) and non-solubilized fractions (P-pellet) were separated by SDS-PAGE. The solubilization efficiency was calculated by visually comparing the corresponding pellet (P) and solubilized (S) lanes. When greater than 50% of the protein was present in the S fraction, the detergent combination and concentration was considered acceptable for further investigation of specific ligand binding activity as described below. The final detergent composition, which optimizes extraction and purification efficiency and specific ligand binding activity, was set at 6.9 mg of Triton X-100 and 6.2 mg DDM per mg of protein in the E. coli membrane fraction. This ratio was successfully applied to all extractions of S1R linker constructs from E. coli membrane fraction reported herein.
Extraction from Escherichia coli membranes
Escherichia coli cell paste from a 1 L growth was suspended in 25 mL of 20 mM Tris-HCl, pH 8.0, containing 300 mM NaCl, 1 mM 2-mercaptoethanol, and 1 mM of EDTA per gram of cells. The cell suspension was supplemented to contain 1 μM of protease inhibitor E-64 (Sigma) and 0.25 mM phenylmethyl sulfonyl fluoride (Sigma). The cell suspension was placed on ice/water/KCl slurry to prevent overheating and sonicated in 75 mL batches using a Misonix 3000 sonicator equipped with a 1 cm probe for 15 min total processing time with a pulse sequence of 2 s on, 1 s off and output set to 7.0. The total cell lysate was centrifuged for 1 h at 75,000g between 4 and 8°C (Beckman-Coulter rotor JA- 25.50) to separate the membrane fraction from soluble proteins. The pelleted membrane fraction was re-suspended in 3 mL of the above buffer per initial gram of cell paste and then analyzed for total protein content using the modified reducing agent compatible BCA reaction. Membrane protein solubilization was initiated by directly adding 6.2 mg of DDM and 6.9 mg of Triton X-100 per mg of total protein in the re-suspended membrane fraction. The extraction was performed for 3 h with gentle stirring at 4°C and then the solution was centrifuged for 1 h at 75,000g (Beckman-Coulter rotor JA- 25.50). The supernatant, containing the solubilized protein, was collected and diluted with buffer to decrease the Triton X-100 concentration to 1.0% (w/v).
Purification
A 2.2 cm diameter column containing 10 mL of amylose resin (New England BioLabs, Ipswich, MA) was equilibrated in 20 mM Tris-HCl, pH 8.0, containing 300 mM NaCl, 1 mM 2-mercaptoethanol, 1 mM of EDTA and 1% (w/v) Triton X-100. The supernatant containing the fusion protein was loaded onto the column at a flow rate of 2 mL/min at 4°C. After loading, the column was washed with 10 column volumes of equilibration buffer and then with 3 column volumes of equilibration buffer lacking EDTA. The fusion protein was eluted with 10 column volumes of the EDTA-free buffer containing 10 mM maltose; 5 mL fractions were collected. The purity of fractions obtained from the amylose column was determined by 4–20% gradient SDS-PAGE (BioRad, Hercules, CA). Protein concentration was estimated using the SDS-PAGE based stain-free technology (BioRad) or the reducing agent compatible bichronic assay (ThermoFisher Scientific). Following analysis of purity and protein concentration, the appropriate fractions were pooled and Triton X-100 was added to a final concentration of 0.031% (w/v). The pooled fractions were concentrated using 50-kDa molecular weight cut off centrifugal concentrator to a protein concentration of ~5–10 mg/mL. Preparative gel filtration was performed using a 25 mL Superdex 200 10/300 GL column (GE Lifesciences, Pittsburgh, PA) and an AKTA purifier. The column was equilibrated in 10 mM HEPES, pH 7.2, containing 150 mM NaCl, 0.3 mM TCEP, and 0.018% (w/v) DDM. The concentrated protein sample was injected in 500 μL aliquots and fractions were collected. The collected fractions were analyzed for purity and protein concentration and appropriate fractions were combined and concentrated as described above using 100-kDa molecular weight cut off centrifugal concentrator.
Ligand binding assays
Ligand binding assays were performed in 20 mM Tris, pH 8.0, containing 0.1% (w/v) Triton X-100 according to published protocols with the following modifications [2, 44, 45]. Assays were performed in 100-μL total reaction volume in a 48-well block format. Each assay contained 80 ng of total protein and each reaction (total binding and non-specific binding) was carried out in triplicate. The final concentration of [H3]-(+)-pentazocine (specific activity 36 Ci/mmol, Perkin-Elmer, Waltham, MA) in both total and non-specific binding reactions was 100 nM. Haloperidol (Tocris, Bristol, UK) was used for masking in the non-specific binding reaction at final concentration of 10 μM. The incubation with ligands was performed for 90 min at 32°C, followed by filtration on a glass fiber filter (Whatman GF/B, Piscataway, NJ) in a Brandel cell harvester (Gaithersburg, MD). The glass filter was washed with 50 mM Tris, pH 8.0 and individual filters were transferred into vials containing 3 mL of scintillation cocktail (Ultima Gold, Perkin-Elmer). The level of radioactivity was measured the following day, employing a liquid scintillation counter (Packard model 1600 CA, Perkin Elmer). Raw count data were normalized to nmol of S1R in the assay and plotted as the percentage of specific binding activity of the original control sample, MBP-TEV-S1R. MBP purified in the same buffer and detergent conditions as the fusion protein was also assayed for ligand binding activity using the method described above. Less than 0.2% binding activity was detected.
Results
Expression construct design
We investigated whether the linker present in MBP-TEV-S1R (Table 2) could be replaced with shorter linkers in order to improve the expression and handling properties. Thus constructs with linkers consisting of one to five Ala residues and another construct that contained no additional amino acids between the MBP and S1R domains were produced by PIPE cloning [34, 35]. Because of the length of the TEV protease recognition site, and uncertainty regarding how this sequence would influence the secretion, we did not include this sequence in these constructs. The abbreviations for these constructs, their molecular weights, and the primary sequences of their linker regions are shown in Table 2.
Table 2.
Properties of MBP-S1R variants with a modified linker sequence
| Variant | Residues | MW (Da) | Sequence |
|---|---|---|---|
| MBP-TEV-S1R | 623 | 69240 | MBP-NSSSNNNNNNNNNNLGIENLYFQSGSAT-S1R |
| MBP-5A-S1Ra | 600 | 66598 | MBP-AAAAA-S1R |
| MBP-4A-S1R | 599 | 66527 | MBP-AAAA-S1R |
| MBP-3A-S1R | 598 | 66456 | MBP-AAA-S1R |
| MBP-2A-S1R | 597 | 66385 | MBP-AA-S1R |
| MBP-1A-S1R | 596 | 66313 | MBP-A-S1R |
| MBP-S1R | 595 | 66242 | MBP-S1R |
MBP-S1R variant having a 5 Ala residues in the linker between MBP and S1R. The other variants are named in a corresponding manner.
Expression of MBP-TEV-S1R
In preliminary tests, we found that use of modified selenomethionine-labeling medium coupled with induction using a low concentration of IPTG (0.2 mM) gave a higher yield of purified control protein MBP-TEV-S1R (~3 mg/L) than the previously published protocol starting with growth in Luria Bertani medium (2 mg/L) [37, 2]. Consequently, this medium and induction method was used in all subsequent experiments.
Extraction from E. coli membranes
Previous studies showed that Triton X-100 was useful for stabilizing the active form of S1R [2]. However, since less than 50% of the total MBP-S1R fusion was extracted from E. coli membranes using Triton X-100 alone, we examined mixtures of Triton X-100 and one additional detergent for their extraction capability. Fig. 1 shows the composition of the screen, which assessed the ability of combinations of Triton X-100 (5% or 10% w/v) and a second detergent (FC-10, FC-12, CHAPS, LDAO, β-OG, DDM, and sodium cholate) to solubilize MBP-TEV-S1R. Several combinations of detergents were successful in solubilizing MBP-TEV-S1R. Indeed, the yellow highlighted combinations of detergents gave ~50% solubilization efficiency, whereas the green highlighted combinations gave greater than 90% recovery. Moreover, the specific ligand binding activity was also satisfactory for MBP-TEV-S1R extracted with the Triton X-100/DDM and Triton X-100/FC-10 combinations. For subsequent experiments, DDM was preferred over FC-10 because of the lower critical micelle concentration (0.009% versus 0.35% w/v, respectively), the lower cost, and the potential for better behavior in crystallization trials. The optimization of the Triton X-100/DDM mixture provided by this screening procedure gave an ~4-fold decrease in the amount of detergent used relative to earlier purifications while improving the efficiency of extraction and also retaining the specific ligand binding activity.
Fig. 1.

Detergent mixtures used to extract MBP-S1R variants from Escherichia coli membranes. In A, 5% Triton X-100 (w/v) was mixed with various amounts of a second detergent; 10% Triton X-100 (w/v) was used in B. Bars with yellow color indicate at least 50% extraction; bars with green color indicate 90% or greater extraction; gray bars indicate less than 50% extraction.
Influence of linker
Fig. 2 compares the results of purification of short linker constructs after expression in E. coli strains BL21(DE3) and B834-pRARE2. On average, better yields of protein were obtained from B834-pRARE2. Fig. 2A shows that the MBP-3A-S1R and MBP-4A-S1R gave roughly double the yield after amylose affinity chromatography as compared to MBP-TEV-S1R (10 and 12 mg/L versus 5 mg/L, respectively). Moreover, Fig. 2B shows that the specific pentazocine binding activities of MBP-3A-S1R and MBP-4A-S1R expressed in B834-pRARE2 were higher than MBP-TEV-S1R expressed in BL21(DE3). While the specific binding of MBP-1A-S1R expressed in BL21(DE3) was comparable to (or slightly better) relative to other active constructs, the yield (6 mg/L) was significantly less than for either MBP-3A-S1R (10 mg/L) or MBP-4A-S1R (12 mg/L), so further studies of MBP-1A-S1R were not undertaken.
Fig. 2.

Protein yields and specific ligand binding activity determined for MBP-S1R variants with different linker regions. A, average protein yields from amylose affinity purification of MBP-SR1 variants expressed in E. coli strains BL21(DE3) (white bar) and B834-pRARE2 (black bar) compared to the yield of MBP-TEV-S1R expressed in BL21(DE3) (n = 3; error bars represent 1σ deviation). B, average specific binding activities (cpm/nmol) presented as a percentage of the specific ligand binding activity observed for MBP-TEV-S1R. MBP-4A-S1R and MBP-3A-S1R expressed in B834-pRARE2 gave the highest yield of the active protein, and so were used in subsequent studies.
MBP-3A-S1R and MBP-4A-S1R were also tested for expression in C41(DE3)-pRARE2 and C43(DE3)-pRARE2, two E. coli strains that were developed for overexpression of problematic proteins including integral membrane proteins [46, 47]. Fig. 3A shows that the yield from these two specialized strains was roughly equivalent to that obtained from B834-pRARE2. However, Fig. 3B shows that B834-pRARE2 gave the highest specific ligand binding activity of all tested strains.
Fig. 3.

Expression of MBP-3A-S1R and MBP-4A-S1R in E. coli strains BL21(DE3) (1), B834-pRARE2 (2), C41(DE3)-pRARE2 (3), BL21(DE3)-RILP (4) and C43(DE3)-pRARE2 (5). After expression and purification by amylose affinity, the average yields and ligand binding activity were determined. A, yields of the purified MBP-S1R variants (n = 3; error bars represent 1σ deviation). B, specific ligand binding activities of the purified MBP-S1R variants presented as a percentage of the binding activity of MBP-TEV-S1R.
Purification of MBP-S1R
Fig. 4 provides denaturing PAGE images of the purification of MBP-4A-S1R, while Table 3 summarizes a purification starting with 4 L of bacterial culture (~23 g of wet cell paste). Cell lysis, centrifugation and optimized two-detergent extraction from the bacterial membrane removed ~75% of the original cellular protein and also gave ~5-fold increase in the specific ligand binding activity. Amylose affinity chromatography gave another ~30-fold increase in specific ligand binding activity, and the protein obtained at this stage was greater than 90% pure based on visual inspection of the denaturing PAGE images. Gel filtration provided a further, modest increase in specific ligand binding activity. The protein-detergent complex was estimated to have molecular mass of ~470 kDa based on retention times observed during calibrated gel filtration.
Fig. 4.
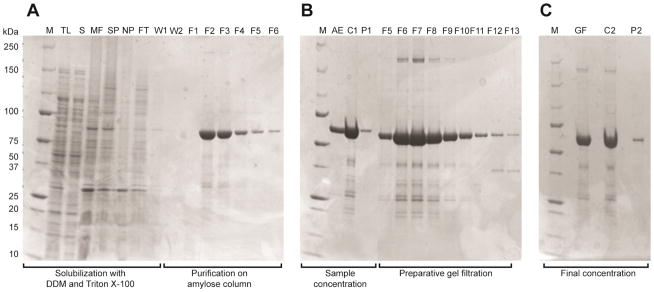
Denaturing PAGE analysis of the purification of MBP-4A-S1R. The lanes are annotated as follows: M, markers; TL, total lysate; S, soluble fraction; MF, membrane fraction; SP, solubilized membrane proteins; NP, non-solubilized membrane proteins; FT, flow-through of the amylose affinity column; W1, first column wash; W2, second column wash; F1–F6, fractions eluted from the amylose affinity column; AE, pooled fractions after elution from the amylose affinity column; C1, concentrated sample from amylose affinity chromatography; P1, precipitate formed during concentration; F5–F13, fractions eluted from the gel filtration column; GF, pooled fractions from gel filtration chromatography; C2, concentrated sample from gel filtration; P2, precipitate formed during concentration. A, extraction of MBP-4A-S1R from E. coli membranes and results of amylose affinity chromatography. Fractions F2–F5 were pooled and concentrated. B, Superdex 200 10/300 gel filtration chromatography. Fractions F5–F10 were pooled and concentrated. C, MBP-4A-S1R was purified to greater than 90% purity by visual inspection.
Table 3.
Summary of the purification of MBP-4A-S1R
| Volume | Protein concentration | Total protein | Total binding activity | Specific bindingb | Fold purification | |
|---|---|---|---|---|---|---|
| mL | mg/mL | mga | cpm x 109 | (cpm/mg) x 106 | ||
| Lysatea | 330 | 9.4 | 3102 | 4.4 | 1.4 | 1 |
| Membrane fraction | 75 | 9.7 | 728 | 5.6 | 7.7 | 5 |
| Amylose affinity | 43 | 0.6 | 26 | 5.8 | 225 | 158 |
| Gel filtration | 1 | 13.7 | 14 | 3.4 | 248 | 175c |
Prepared from 4 L of E. coli B834-pRARE2 culture, 23.3 g wet cells.
Normalized to mg of total protein instead of nmol of MBP-4A-S1R.
The 175-fold purification of MBP-4A-S1R was calculated based on increase in specific binding activity for the ligand [H3]-(+)-pentazocine.
The purified protein could be concentrated using centrifugation, and concentrations greater than 10 mg/mL could be routinely obtained in the identified buffer and detergent composition. Overall, MBP-4A-S1R was purified by 175-fold from the cell lysate. The purified protein was stable to freezing and storage at −80°C as indicated by minimal changes in specific ligand binding activity relative to original assay results. Furthermore, after 5 months of storage at 4°C, MBP-4A-S1R retained ~98% of the original specific ligand binding activity.
Discussion
Here we have presented an improved procedure for expressing and purifying MBP-S1R in a form that is suitable for additional research on biological function, biophysical characterizations, and potentially structure determination. Three key improvements in the methodology for MBP-S1R are summarized here.
Both the yield of fusion protein and the specific ligand binding activity were best in the rare codon supplemented strain E. coli B834-pRARE2. This strain has been successfully used in the Center for Eukaryotic Structural Genomics as part of the NIH-funded Protein Structure Initiative since 2003. We previously showed this strain can be effectively combined with a customized growth and expression medium that permits autoinduction if desired and high-level incorporation of selenomethionine for structure determination [38, 48]. This combination of host strain and medium was successfully applied to MBP-S1R. Rare codon supplementation in strain BL21(DE3)-RILP did not improve the average yields of active S1R fusion protein. Moreover, although the codon supplemented specialty strains C41(DE3)-pRARE2 and C43(DE3)-pRARE2 did give levels of protein expression roughly comparable to that observed with B834-pRARE2, these former strains gave significantly lower yield of active S1R fusion protein.
We found that shortening the linker between the MBP and S1R domains had a strong influence on the expression level and the specific ligand binding activity of the resulting fusion protein. The best case, based on the combination of high specific ligand binding activity and yield of purified protein, MBP-4A-S1R, had the two domains separated by only 4 Ala residues. Shorter linkers steadily decreased the yield of purified protein.
Detergent screening showed that active MBP-4A-S1R could be efficiently extracted from E. coli membranes by a combination of Triton X-100 and DDM. The detergent mixture decreased the total amount of detergent needed, and improved the extraction to near quantitative level while also retaining the specific ligand binding activity. The subsequent two-step chromatographic purification yielded purified protein that also retained high specific ligand binding activity. These improvements are of great advantage, as they may facilitate future studies on this enigmatic membrane protein.
Highlights.
Yield of active MBP-S1R depends on E. coli expression host and domain linker length
Escherichia coli B834-pRARE2 and a 4-Ala linker gave the highest yield of active protein
The fusion protein can be purified in high yield in a mixture of Triton-X100 and DDM
The purified fusion protein has high specific ligand binding activity
Acknowledgments
This work was supported by NIGMS PSI: Biology Network grant U54 GM094584 to B.G.F. The authors thank Dr. Arnold E. Ruoho, Dr. Uyen B. Chu and Dr. Subramaniam Ramachandran (University of Wisconsin-Madison) for providing the MBP-TEV-S1R plasmid used as the starting template in this work, for use of the scintillation counter used in this study, and their insightful discussions on S1R. The authors thank Dr. Emily Beebe (University of Wisconsin-Madison, USA) for generous assistance in protein analysis. We also thank Ms. Donna Troestler for assistance with manuscript preparation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chen Y, Hajipour AR, Sievert MK, Arbabian M, Ruoho AE. Characterization of the cocaine binding site on the sigma-1 receptor. Biochemistry. 2007;46:3532–3542. doi: 10.1021/bi061727o. [DOI] [PubMed] [Google Scholar]
- 2.Ramachandran S, Lu H, Prabhu U, Ruoho AE. Purification and characterization of the guinea pig sigma-1 receptor functionally expressed in Escherichia coli. Protein Expr Purif. 2007;51:283–292. doi: 10.1016/j.pep.2006.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fontanilla D, Johannessen M, Hajipour AR, Cozzi NV, Jackson MB, Ruoho AE. The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science. 2009;323:934–937. doi: 10.1126/science.1166127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furuse T, Hashimoto K. Sigma-1 receptor agonist fluvoxamine for delirium in patients with Alzheimer’s disease. Ann Gen Psychiatry. 2010;9:6. doi: 10.1186/1744-859X-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruscher K, Shamloo M, Rickhag M, Ladunga I, Soriano L, Gisselsson L, Toresson H, Ruslim-Litrus L, Oksenberg D, Urfer R, Johansson BB, Nikolich K, Wieloch T. The sigma-1 receptor enhances brain plasticity and functional recovery after experimental stroke. Brain. 2011;134:732–746. doi: 10.1093/brain/awq367. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 7.Shioda N, Ishikawa K, Tagashira H, Ishizuka T, Yawo H, Fukunaga K. Expression of a truncated form of the endoplasmic reticulum chaperone protein, sigma1 receptor, promotes mitochondrial energy depletion and apoptosis. J Biol Chem. 2012;287:23318–23331. doi: 10.1074/jbc.M112.349142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marriott KS, Prasad M, Thapliyal V, Bose HS. Sigma-1 receptor at the mitochondrial associated ER-membrane is responsible for mitochondrial metabolic regulation. J Pharmacol Exp Ther. 2012 doi: 10.1124/jpet.112.198168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maurice T, Su TP. The pharmacology of sigma-1 receptors. Pharmacol Ther. 2009;124:195–206. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen EC, McCracken KA, Liu Y, Pouw B, Matsumoto RR. Involvement of sigma (sigma) receptors in the acute actions of methamphetamine: receptor binding and behavioral studies. Neuropharmacology. 2005;49:638–645. doi: 10.1016/j.neuropharm.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 11.Su TP. Sigma receptors. Putative links between nervous, endocrine and immune systems. Eur J Biochem. 1991;200:633–642. doi: 10.1111/j.1432-1033.1991.tb16226.x. [DOI] [PubMed] [Google Scholar]
- 12.Su TP, Hayashi T. Understanding the molecular mechanism of sigma-1 receptors: towards a hypothesis that sigma-1 receptors are intracellular amplifiers for signal transduction. Curr Med Chem. 2003;10:2073–2080. doi: 10.2174/0929867033456783. [DOI] [PubMed] [Google Scholar]
- 13.Cendan CM, Pujalte JM, Portillo-Salido E, Montoliu L, Baeyens JM. Formalin-induced pain is reduced in sigma(1) receptor knockout mice. Eur J Pharmacol. 2005;511:73–74. doi: 10.1016/j.ejphar.2005.01.036. [DOI] [PubMed] [Google Scholar]
- 14.Balasuriya D, Stewart AP, Crottes D, Borgese F, Soriani O, Edwardson JM. The sigma-1 receptor binds to the Nav1.5 voltage-gated Na+ channel with four-fold symmetry. J Biol Chem. 2012 doi: 10.1074/jbc.M112.382077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carnally SM, Johannessen M, Henderson RM, Jackson MB, Edwardson JM. Demonstration of a direct interaction between sigma-1 receptors and acid-sensing ion channels. Biophys J. 2010;98:1182–1191. doi: 10.1016/j.bpj.2009.12.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jbilo O, Vidal H, Paul R, De Nys N, Bensaid M, Silve S, Carayon P, Davi D, Galiegue S, Bourrie B, Guillemot JC, Ferrara P, Loison G, Maffrand JP, Le Fur G, Casellas P. Purification and characterization of the human SR 31747A-binding protein. A nuclear membrane protein related to yeast sterol isomerase. J Biol Chem. 1997;272:27107–27115. doi: 10.1074/jbc.272.43.27107. [DOI] [PubMed] [Google Scholar]
- 17.Pal A, Chu UB, Ramachandran S, Grawoig D, Guo LW, Hajipour AR, Ruoho AE. Juxtaposition of the steroid binding domain-like I and II regions constitutes a ligand binding site in the sigma-1 receptor. J Biol Chem. 2008;283:19646–19656. doi: 10.1074/jbc.M802192200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fontanilla D, Hajipour AR, Pal A, Chu UB, Arbabian M, Ruoho AE. Probing the steroid binding domain-like I (SBDLI) of the sigma-1 receptor binding site using N-substituted photoaffinity labels. Biochemistry. 2008;47:7205–7217. doi: 10.1021/bi800564j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruoho AE, Chu UB, Ramachandran S, Fontanilla D, Mavlyutov T, Hajipour AR. The ligand binding region of the sigma-1 receptor: studies utilizing photoaffinity probes, sphingosine and N-alkylamines. Curr Pharm Des. 2012;18:920–929. doi: 10.2174/138161212799436584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanner M, Moebius FF, Flandorfer A, Knaus HG, Striessnig J, Kempner E, Glossmann H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci U S A. 1996;93:8072–8077. doi: 10.1073/pnas.93.15.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pan YX, Mei J, Xu J, Wan BL, Zuckerman A, Pasternak GW. Cloning and characterization of a mouse sigma1 receptor. J Neurochem. 1998;70:2279–2285. doi: 10.1046/j.1471-4159.1998.70062279.x. [DOI] [PubMed] [Google Scholar]
- 22.Mei J, Pasternak GW. Molecular cloning and pharmacological characterization of the rat sigma1 receptor. Biochem Pharmacol. 2001;62:349–355. doi: 10.1016/s0006-2952(01)00666-9. [DOI] [PubMed] [Google Scholar]
- 23.Kekuda R, Prasad PD, Fei YJ, Leibach FH, Ganapathy V. Cloning and functional expression of the human type 1 sigma receptor (hSigmaR1) Biochem Biophys Res Commun. 1996;229:553–558. doi: 10.1006/bbrc.1996.1842. [DOI] [PubMed] [Google Scholar]
- 24.Kapust RB, Waugh DS. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korepanova A, Moore JD, Nguyen HB, Hua Y, Cross TA, Gao F. Expression of membrane proteins from Mycobacterium tuberculosis in Escherichia coli as fusions with maltose binding protein. Protein Expr Purif. 2007;53:24–30. doi: 10.1016/j.pep.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sobrado P, Goren MA, James D, Amundson CK. A Protein Structure Initiative approach to expression, purification, and in situ delivery of human cytochrome b5 to membrane vesicles. Protein Expr Purif. 2008;58:229–241. doi: 10.1016/j.pep.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Center RJ, Kobe B, Wilson KA, Teh T, Howlett GJ, Kemp BE, Poumbourios P. Crystallization of a trimeric human T cell leukemia virus type 1 gp21 ectodomain fragment as a chimera with maltose-binding protein. Protein Sci. 1998;7:1612–1619. doi: 10.1002/pro.5560070715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ke A, Wolberger C. Insights into binding cooperativity of MATa1/MATalpha2 from the crystal structure of a MATa1 homeodomain-maltose binding protein chimera. Protein Sci. 2003;12:306–312. doi: 10.1110/ps.0219103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobe B, Center RJ, Kemp BE, Poumbourios P. Crystal structure of human T cell leukemia virus type 1 gp21 ectodomain crystallized as a maltose-binding protein chimera reveals structural evolution of retroviral transmembrane proteins. Proc Natl Acad Sci U S A. 1999;96:4319–4324. doi: 10.1073/pnas.96.8.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smyth DR, Mrozkiewicz MK, McGrath WJ, Listwan P, Kobe B. Crystal structures of fusion proteins with large-affinity tags. Protein Sci. 2003;12:1313–1322. doi: 10.1110/ps.0243403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bossé M, Handl CE, Lortie LA, Harel J, Dubreuil JD. Fusion of the genes encoding Escherichia coli heat-stable enterotoxin b (STb) and the maltose-binding protein to obtain mature STb enterotoxin. J Gen Microbiol. 1993;139:631–638. doi: 10.1099/00221287-139-3-631. [DOI] [PubMed] [Google Scholar]
- 32.Halfmann G, Brailly H, Bernadac A, Montero-Julian FA, Lazdunski C, Baty D. Targeting of interleukin-2 to the periplasm of Escherichia coli. J Gen Microbiol. 1993;139:2465–2473. doi: 10.1099/00221287-139-10-2465. [DOI] [PubMed] [Google Scholar]
- 33.Furukawa H, Haga T. Expression of functional M2 muscarinic acetylcholine receptor in Escherichia coli. J Biochem. 2000;127:151–161. doi: 10.1093/oxfordjournals.jbchem.a022577. [DOI] [PubMed] [Google Scholar]
- 34.Klock HE, Koesema EJ, Knuth MW, Lesley SA. Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins. 2008;71:982–994. doi: 10.1002/prot.21786. [DOI] [PubMed] [Google Scholar]
- 35.Klock HE, Lesley SA. The Polymerase Incomplete Primer Extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol Biol. 2009;498:91–103. doi: 10.1007/978-1-59745-196-3_6. [DOI] [PubMed] [Google Scholar]
- 36.Burgess-Brown NA, Sharma S, Sobott F, Loenarz C, Oppermann U, Gileadi O. Codon optimization can improve expression of human genes in Escherichia coli: A multi-gene study. Protein Expr Purif. 2008;59:94–102. doi: 10.1016/j.pep.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 37.Fox BG, Blommel PG. Autoinduction of protein expression. Curr Protoc Protein Sci Chapter. 2009;5(Unit 5):23. doi: 10.1002/0471140864.ps0523s56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blommel PG, Becker KJ, Duvnjak P, Fox BG. Enhanced bacterial protein expression during auto-induction obtained by alteration of lac repressor dosage and medium composition. Biotechnol Prog. 2007;23:585–598. doi: 10.1021/bp070011x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Millard CS, Stols L, Quartey P, Kim Y, Dementieva I, Donnelly MI. A less laborious approach to the high-throughput production of recombinant proteins in Escherichia coli using 2-liter plastic bottles. Protein Expr Purif. 2003;29:311–320. doi: 10.1016/s1046-5928(03)00063-9. [DOI] [PubMed] [Google Scholar]
- 40.Linke D. Chapter 34 Detergents: An overview. Methods Enzymol. 2009;463:603–617. doi: 10.1016/S0076-6879(09)63034-2. [DOI] [PubMed] [Google Scholar]
- 41.Lin SH, Guidotti G. Chapter 35 Purification of membrane proteins. Methods Enzymol. 2009;463:619–629. doi: 10.1016/S0076-6879(09)63035-4. [DOI] [PubMed] [Google Scholar]
- 42.Merkulova M, McKee M, Dip PV, Grüber G, Marshansky V. N-terminal domain of the V-ATPase a2-subunit displays integral membrane protein properties. Protein Sci. 2010;19:1850–1862. doi: 10.1002/pro.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arachea BT, Sun Z, Potente N, Malik R, Isailovic D, Viola RE. Detergent selection for enhanced extraction of membrane proteins. Protein Expr Purif. 2012;86:12–20. doi: 10.1016/j.pep.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 44.Ganapathy ME, Prasad PD, Huang W, Seth P, Leibach FH, Ganapathy V. Molecular and ligand-binding characterization of the sigma-receptor in the Jurkat human T lymphocyte cell line. J Pharmacol Exp Ther. 1999;289:251–260. [PubMed] [Google Scholar]
- 45.Torrence-Campbell C, Bowen WD. Differential solubilization of rat liver sigma 1 and sigma 2 receptors: retention of sigma 2 sites in particulate fractions. Eur J Pharmacol. 1996;304:201–210. doi: 10.1016/0014-2999(96)00109-4. [DOI] [PubMed] [Google Scholar]
- 46.Dumon-Seignovert L, Cariot G, Vuillard L. The toxicity of recombinant proteins in Escherichia coli: a comparison of overexpression in BL21(DE3), C41(DE3), and C43(DE3) Protein Expr Purif. 2004;37:203–206. doi: 10.1016/j.pep.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 47.Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 48.Markley JL, Aceti DJ, Bingman CA, Fox BG, Frederick RO, Makino S, Nichols KW, Phillips GN, Jr, Primm JG, Sahu SC, Vojtik FC, Volkman BF, Wrobel RL, Zolnai Z. The Center for Eukaryotic Structural Genomics. J Struct Funct Genomics. 2009;10:165–179. doi: 10.1007/s10969-008-9057-4. [DOI] [PMC free article] [PubMed] [Google Scholar]