

Abstract
Background
Elevated atmospheric CO2 (eCO2) has been shown to have significant effects on terrestrial ecosystems. However, little is known about its influence on the structure, composition, and functional potential of soil microbial communities, especially carbon (C) and nitrogen (N) cycling. A high-throughput functional gene array (GeoChip 3.0) was used to examine the composition, structure, and metabolic potential of soil microbial communities from a grassland field experiment after ten-year field exposure to ambient and elevated CO2 concentrations.
Results
Distinct microbial communities were established under eCO2. The abundance of three key C fixation genes encoding ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco), carbon monoxide dehydrogenase (CODH) and propionyl-CoA/acetyl-CoA carboxylase (PCC/ACC), significantly increased under eCO2, and so did some C degrading genes involved in starch, cellulose, and hemicellulose. Also, nifH and nirS involved in N cycling were significantly stimulated. In addition, based on variation partitioning analysis (VPA), the soil microbial community structure was largely shaped by direct and indirect eCO2-driven factors.
Conclusions
These findings suggest that the soil microbial community structure and their ecosystem functioning for C and N cycling were altered dramatically at eCO2. This study provides new insights into our understanding of the feedback response of soil microbial communities to elevated CO2 and global change.
Background
The concentrations of atmospheric CO2 have been increasing for the last 150 years and are predicted to increase to 550 ppm by the middle of this century [1]. This ongoing increase in atmospheric CO2 is due to the extensive use of fossil fuels and changes in land use patterns [2]. The rapid increase of CO2 in the atmosphere over the last century has led to an increase of global ecosystem carbon storage [3]. Terrestrial ecosystems are intimately connected to atmospheric CO2 levels and soil is the major organic C pool in all terrestrial biomes [4]. Studies of ecosystem responses to elevated CO2 have shown that eCO2 can have major effects on terrestrial ecosystems by enhancing plant photosynthetic CO2 fixation and primary productivity, and altered plant and soil characteristics [5-9]. However, the disparity between modeling and empirical studies suggests as yet incomplete understanding of the combined impacts of this global change factor on ecosystem functioning.
Since microorganisms mediate important biogeochemical processes such as soil C and N cycling, and are expected to influence future atmospheric CO2 concentrations, functional understanding of how eCO2 affects soil microbial community composition and structure will be necessary for robust prediction of atmospheric CO2 concentrations in the future. However, one of the major challenges for characterizing the functional diversity and their responses to the changes of atmospheric CO2 concentration is the extreme diversity and as-yet uncultivated status of many microorganisms. To date, most of the efforts to describe the effects of atmospheric CO2 concentration to soil microbial communities have been focused on phylogenetic composition [5,10,11]. Some studies [12,13] tried to examine the responses of soil microbial community to the changes of CO2 concentration. However, distinctly different results of the soil microbial diversity and activity under eCO2 have been obtained in different studies [11,14-17], and the possible relationships between the microbial community functional structure and the plant and soil parameters are still not clear.
Functional gene arrays (FGAs), such as GeoChip, which contain key genes encoding functional enzymes involved in biogeochemical cycling, have been successfully used for tracking and studying the biogeochemical processes in different ecosystems, including groundwater and aquatic ecosystems, soil, extreme environments, bioreactor systems, and oil-contaminated waters or soils [18,19]. Combined with multivariate statistical analyses [20], several systematic experimental evaluations have indicated that GeoChip can be used as a specific, sensitive tool for detecting the functional diversity, composition, structure, and metabolic potential of microbial communities, and correlating microbial communities to ecosystem processes and functioning [21-24].
We hypothesized that soil microbial community composition and structure would be altered directly or indirectly by eCO2, and that the functional gene groups involved in C and N cycling would be enhanced due to the increase of soil C input under eCO2[25]. To test those hypotheses, we conducted our experiments at the Cedar Creek Ecosystem Science Reserve in Minnesota (http://www.biocon.umn.edu/). A comprehensive functional gene array, GeoChip 3.0 [26], was used to analyze the function composition and structure of soil microbial communities under both ambient and elevated CO2 concentrations. Some key genes involved in C and N cycling were stimulated under CO2. This study provides new information for our understanding of the feedback response of soil microbial communities to eCO2.
Results
Overall responses of microbial C and N cycling genes under CO2
Based on the number of functional genes, Shannon diversity, evenness and dominance, no significant differences were detected in the overall microbial diversity (Additional file 1). Significant (p < 0.05) differences were observed in the abundance of C and N cycling genes between ambient CO2 (aCO2) and eCO2 microbial communities by detrended correspondence analysis (DCA) together with analysis of similarities (ANOSIM), non-parametric multivariate analysis of variance (Adonis) and Multi-Response Permutation Procedure (MRPP). The eCO2 samples were well separated from aCO2 ones by the first axis of DCA, which explained 10.4% and 10.1% for the genes involved in C cycling (Figure 1A) and N cycling (Figure 1B), respectively. These results suggest that most of the functional genes involved in C and N cycling were significantly stimulated, and that the functional composition and structure of soil microbial communities were also altered at eCO2. More details about individual key C and N cycling genes and their associated populations are described below.
Figure 1.
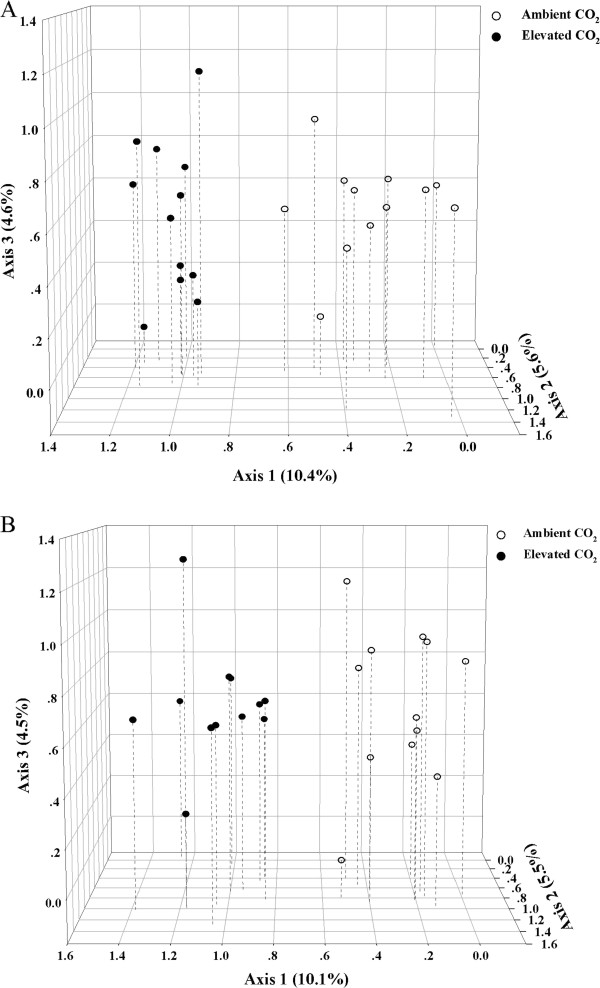
Detrended correspondence analysis (DCA) of the samples under ambient and elevated CO2 bsed on GeoChip 3.0 data of the genes involved in carbon (A) and nitrogen (B) cycling.
Responses of C cycling genes to eCO2
(i) Carbon fixation
Five pathways for autotrophic CO2 fixation have been identified [27]. Based on normalized signal intensities, 147 C fixation genes in four functional gene families were detected. Within this four functional gene families, two gene families encoding ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco) and carbon monoxide dehydrogenase (CODH) significantly increased (p < 0.05), and another one encoding propionyl-CoA/acetyl-CoA carboxylase (PCC/ACC) showed increase trend at p < 0.1 level under eCO2. Individual gene variants and dominant populations about those three gene families were examined to understand the potential of microbial CO2 fixation in soil at eCO2.
So far, Rubisco has been classified into four forms [28]. A total of 46 rbcL probes encoding the large subunit of Rubisco had positive signals with 27 shared by both CO2 conditions, 8 and 11 unique at aCO2 and eCO2, respectively. All four forms of Rubisco were detected, but more than 70% of the gene variants belonged to Form I, especially for those significantly changed and dominant variants mentioned above. Only two genes belonged to Form II with one (84181207 from Thiomicrospira pelophila) unique to eCO2 and the other (86748076 from Rhodopseudomonas palustris HaA2) exhibiting increased signal intensity at eCO2. One eCO2 unique gene (2648911 from Archaeoglobus fulgidus DSM 4304) belonged to Form III and one unchanged gene (149182238 from Bacillus sp. SG-1) belonged to Form IV (Figure 2). In addition, eight variants detected were clustered as the undefined Form. No significant change was observed in these rbcL genes detected, except two showed increase trends and two showed decrease at p < 0.1 level under eCO2 (Additional file 2). For the other two gene families, two and six significant increase genes were detected in CODH (Additional file 3) and PCC (Additional file 4), respectively. Details for these gene variants and dominant populations are described in the Additional file 5.
Figure 2.
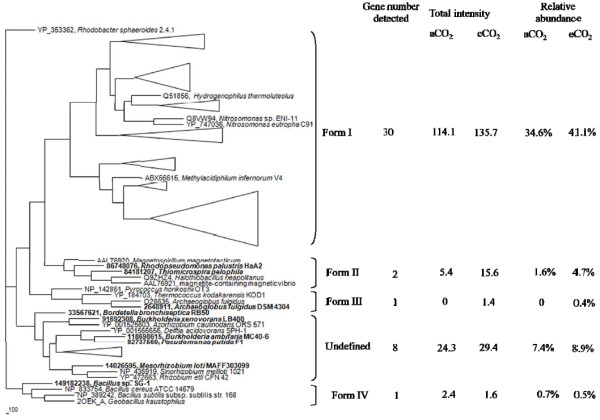
Maximum-likelihood phylogenetic tree of the deduced amino acid sequences of Rubisco large subunit genes obtained from GeoChip 3.0, showing the phylogenetic relationship among the five Rubisco clusters. The depth and width of each wedge is proportional to the branch lengths and number of Rubisco sequences, respectively. Some individual genes detected are shown in bold. The scale indicates the number of amino acid substitutions per site and the tree is outgroup rooted with YP_353362 (Rhodobacter sphaeroides 2.4.1).
(ii) Carbon degradation
GeoChip 3.0 targets many genes involved in labile C and recalcitrant C degradation. Overall, 429 C degradation genes in 24 functional gene families were detected and 26 genes showed significant (p < 0.05) changes with 15 increased and 11 decreased at eCO2 based on the signal intensity detected.
Based on the normalized average signal intensity of key gene families detected among 12 soil samples under aCO2 or eCO2, the genes involved in hydrolysis of starch and other labile polysaccharides such as α-amylases (EC 3.2.1.1), glucoamylases (EC 3.2.1.3) and pullulanases (EC 3.2.1.41) significantly (p < 0.05) increased at eCO2. Among 68 detected amyA probes, 44 were shared by both CO2 conditions. For those shared genes, six gene variants showed strongly increasing trends with four genes (84691156 from Parvularcula bermudensis HTCC2503, 113897923 from Herpetosiphon aurantiacus ATCC 23779, 72161237 from Thermobifida fusca YX, and 114197670 from Aspergillus terreus NIH2624) at p < 0.05 level and two genes (83643106 from Hahella chejuensis KCTC 2396 and 94984767 from Deinococcus geothermalis DSM 11300) at p < 0.10 level, and one gene variant (146337645 from Bradyrhizobium sp. ORS278) showed significant decrease at p < 0.05 level at eCO2 (Figure 3). Within the 24 unique amyA genes, 11 were detected at aCO2 and 13 were detected at eCO2, and they contributed approximately 8.6% (3.4% for aCO2 and 5.2% for eCO2) of the total amyA signal intensity. The significant increase genes, 84691156 (from Parvularcula bermudensis HTCC2503) and 113897923 (from Herpetosiphon aurantiacus ATCC 23779), also ranked as the first and second abundant amyA genes with 13.2% and 7.7% of the total amyA gene signal, respectively (Figure 3). These results suggested that starch degradation by microorganisms in soil may increase at eCO2. Similar trends about the gene variants and dominant populations were observed in glucoamylase (Additional file 6) and pullulanase (Additional file 7). Details for these two gene families are described in Additional file 5.
Figure 3.
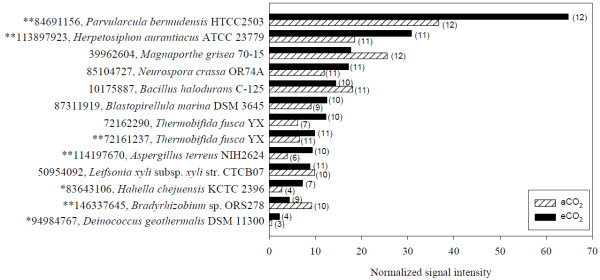
The top ten abundant and other significantly changed amyA genes. The number of the probes detected from eCO2 and aCO2 were presented following the bars in parentheses. The statistical significant results of response ratio were shown in front of the GenBank accession number of the probes (**p < 0.05, *p < 0.10).
Additionally, the abundance of key genes involved in the degradation of more complex C showed significantly increasing trends at eCO2, such as hemicellulose at p < 0.05 and cellulose at p < 0.1 level. For hemicellulose degradation, three gene families such as arabinofuranosidase (AFase, EC 3.2.1.55), cellobiase (EC 3.2.1.4) and xylanase (EC 3.2.1.8) were detected and the abundance of normalized signal intensity of AFase genes increased significantly (p < 0.05) in the normalized signal intensity under eCO2. The abundance of nine detected endoglucanase genes showed increases at p < 0.1 level under eCO2. Details regarding gene variants and dominant populations of endoglucanase (Additional file 8) and AFase (Additional file 9) genes are described in Additional file 5.
Finally, ten gene families encoding the enzymes for more complex or recalcitrant C degradation were detected with three for aromatic degradation (limonene-1,1-epoxide hydrolase, vanilate demethylase and vanillin dehydrogenase), three for chitin degradation (acetylglucosaminidase, endochitinase and exochitinase) and four for lignin degradation (glyoxal oxidase, lignin peroxidase, manganese peroxidase and phenol oxidase). However, based on the normalized signal intensity, only vanilate demethylase genes showed a significant increase (p < 0.05) under eCO2 (Additional file 10). The details about this gene are described in Additional file 5.
The above results clearly indicate that microbial CO2 fixation may increase, and that microbial degradation and utilization of labile C substrates (e.g., starch, cellulose) may also increase at eCO2, but the degradation of recalcitrant C (e.g., lignin) may not be stimulated by eCO2.
Responses of N cycling genes to eCO2
Sixteen enzymes/genes involved in different N cycling processes were selected in GeoChip 3.0 to target important N cycling processes, such as N2 fixation, nitrification, and denitrification. Based on the total signal intensity detected, significant changes were observed in nifH and nirS, but not other N cycling genes.
N2 fixation is exclusively performed by prokaryotes, and nifH encoding the iron protein of N synthase complex, nitrogenase, is the most widely used functional gene marker for N2 fixation [29] and also a phylogenetic marker for nifH-containing organisms [30]. A total of 147 nifH gene variants were detected with 92 shared by both aCO2 and eCO2 samples, 41 unique to eCO2, and 15 unique to aCO2 samples. The total normalized signal intensity of these detected nifH genes was significantly (p < 0.05) higher at eCO2 than that at aCO2. Ten gene variants were significantly (p < 0.05) increased, and five were significantly decreased at eCO2. More than 69% of the nifH genes detected were affiliated with uncultured or unidentified microorganisms, and five (44829093, 12001884, 780709, 89512880, and 3157614) had >3.0% of the total nifH gene signal intensity.
For 13 significantly increased nifH gene variants, ten were from the uncultured or unidentified bacteria, and three (116697525, 2897667, and 148568718) were derived from Syntrophobacter fumaroxidans MPOB, Paenibacillus macerans, and Roseiflexus sp. RS-1, respectively. Similarly, for five significantly decreased genes detected, three were from unidentified marine eubacterium and unidentified bacteria, and two (77463858 and 138897063) were derived from Rhodobacter sphaeroides 2.4.1 and Geobacillus thermodenitrificans NG80-2, respectively (Figure 4). It is also noted that nine of the top ten abundant genes were from uncultured or unidentified bacteria (Figure 4).
Figure 4.
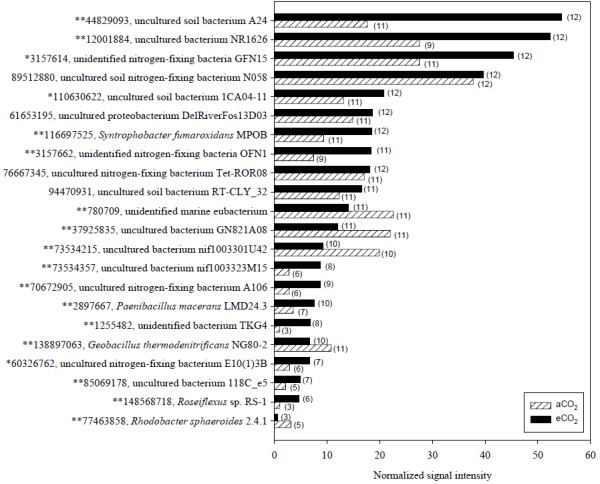
The top ten abundant and other significantly changed nifH genes. The number of the probes detected from eCO2 and aCO2 were presented following the bars in parentheses. The statistical significant results of response ratio were shown in front of the GenBank accession number of the probes (**p < 0.05, *p < 0.10).
NifH has been classified into five distinct evolutionary groups [31]. Based on the sequences of nifH probes, the detected nifH genes were clustered into Group I - IV. Within these four groups, Group III had 68 nifH genes detected, and Groups I, IV, and II had 24, 22, and 5 genes detected, respectively. There were 28 nifH genes for the undefined group (Figure 5). In the major group (Group III), 21.3% and 25.7% relative abundances were detected from aCO2 and eCO2 samples, respectively. Similar signal intensity distributions were observed in Group I, Group IV and the undefined Group with 7.2%, 8.3% and 7.0% relative abundances from the aCO2 samples and 11.8%, 9.3% and 8.9% from the eCO2 samples, respectively. Within five genes in Group II, the relative abundances from the two aCO2 genes and the three eCO2 were 0.2% and 0.3%, respectively. Among these five groups, significant increase in the total signal intensity under eCO2 was only observed in Group I, although higher total signal intensities at eCO2 were detected in all five groups (Figure 5).
Figure 5.
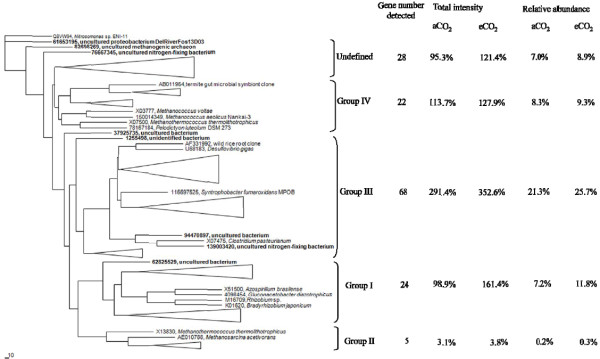
Maximum-likelihood phylogenetic tree of the deduced amino acid sequences of nifH sequences obtained from GeoChip 3.0, showing the phylogenetic relationship among the five nifH clusters. The depth and width of each wedge is proportional to the branch lengths and number of nifH sequences, respectively. Some individual genes detected are shown in bold. The scale indicates the number of amino acid substitutions per site and the tree is outgroup rooted with Q8VW94 (Nitrosomonas sp. ENI-11).
Among the 60 nirS genes detected, 31 were shared by both aCO2 and eCO2 samples (Additional file 11), whereas 23 and six were unique to eCO2 and aCO2, respectively (Additional file 12). Details for nirS gene are described in the Additional file 5. The above results indicate that N cycling may be significantly changed at eCO2, which was reflected in a significant increase in the abundance of detected nifH and nirS genes. Furthermore, the great nirS gene abundance would suggest the great N2O (a recognized greenhouse gas) emissions under eCO2 condition.
Relationships between the microbial community structure and environmental factors
The concentrations of atmospheric CO2 and nine environmental variables including four soil variables, soil N% at the depth of 0-10 cm (SN0-10) and 10–20 cm (SN10-20), soil C and N ratio at the depth of 10–20 cm (SCNR10-20), and soil pH (pH), and five plant variables, biomass of C4 plant species Andropogon gerardi (BAG) and Bouteloua gracilis (BBG), biomass of legume plant species Lupinus perennis (BLP), belowground plant C percentage (BPC), and the number of plant functional groups (PFG) were selected by forward selection based on variance inflation factor (VIF) with 999 Monte Carlo permutations. The VIF of these ten parameters were all less than 6.5. Although the rates of biogeochemical processes about nitrification, ammonification and net N mineralization were also detected, these three parameters were rejected by forward selection since their VIF were all higher than 100. The relationships between the functional structure of soil microbial communities and the ten parameters selected were analyzed by redundancy analysis (RDA) and the ordination plot (Figure 6) was very consistent with the DCA ordination patterns. The samples from aCO2 and eCO2 were well separated by the first axis of RDA with 19.4% explained by the first axis and a total of 47.6% explained with microbial communities (p = 0.047). Similar RDA results were obtained for subsets of functional genes, with 48.1% of the total variance explained for the C cycling genes (p = 0.037) and 48.2% of the total variance explained for the N cycling genes (p = 0.044). Within these variables, all detected functional genes and subsets of those genes were significantly different between CO2 treatments (p = 0.001).
Figure 6.
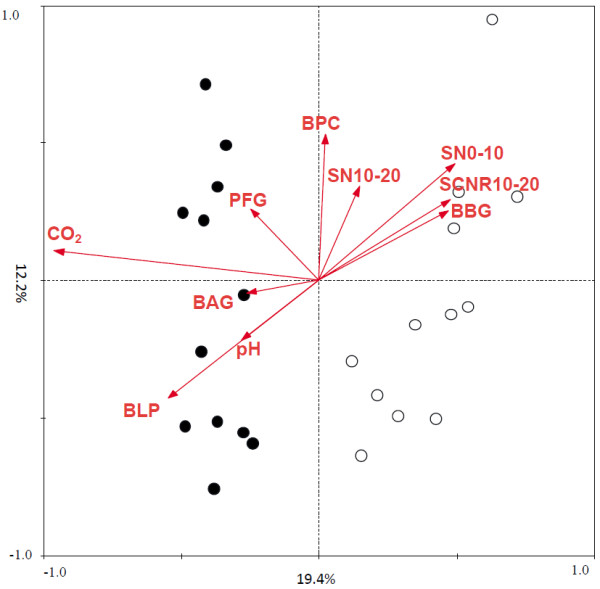
Biplot of redundancy analysis (RDA) of entire functional gene communities of soil samples from aCO2 and eCO2 conditions. Open circles represent samples collected from aCO2, whereas solid circles represent samples collected from eCO2. Four soil variables: soil N% at the depth of 0–10 ( SN0-10) and 10–20 cm (SN10-20), soil C and N ratio at the depth of 10–20 cm (SCNR10-20) and soil pH (pH), and five plant variables: biomass of C4 plant species Andropogon gerardi (BAG) and Bouteloua gracilis (BBG), biomass of legume plant species Lupinus perennis (BLP), below ground plant C percentage (BPC), and the number of plant functional groups (PFG), were selected by forward selection based variance inflation factor (VIF) with 999 Monte Carlo permutations.
To better understand the relationships between the functional structure of soil microbial communities and the plant and soil variables, variation partitioning analysis (VPA) was performed. After accounting for the effects of the CO2 treatment, the nine environmental variables could explain 42.2%, 42.8% and 42.8% of the total variation for all detected genes (p = 0.098), C cycling genes (p = 0.072), and N cycling genes (p = 0.087), respectively (Table 1). These five selected plant variables could significantly explain 24.7% (p = 0.010) of the variance for all detected genes, 24.6% (p = 0.022) for detected C cycling genes, and 25.1% (p = 0.014) for detected N cycling genes (Table 1). For the soil variables, these four selected variables also could explain 19.4% (p = 0.053) of the variance for all detected genes, 19.0% (p = 0.146) for detected C cycling genes, and 19.7% (p = 0.067) for detected N cycling genes (Table 1). Within these nine selected parameters, distinct differences were observed between the samples from aCO2 and eCO2 (p values ranged from 0.023 to 0.092), and the variance explained by four of the important variables, including pH (r = 0.411, p = 0.046), BLP (r = 0.378, p = 0.069), BPC (r = −0.345, p = 0.098), and PFG (r = 0.385, p = 0.063).
Table 1.
The relationships of microbial community functional structure to plant and soil characteristics by RDA and VPAa
| All genes detected | C cycling genes | N cycling genes | ||
|---|---|---|---|---|
| With nine selected variables |
First axis explanation (%) |
19.1 |
20.3 |
19.6 |
| |
Total explanation (%) |
42.2 |
42.8 |
42.8 |
| |
F |
1.138 |
1.167 |
1.163 |
| |
p |
0.098 |
0.072 |
0.087 |
| Explanations of the selected plant variables (%) |
Total |
24.7 |
24.6 |
25.1 |
| |
The number of plant functional groups (PFG) |
5.9 |
4.5 |
5.1 |
| |
Belowground plant C percentage (BPC) |
4.4 |
4.5 |
4.5 |
| |
Biomass of C4 plant species Andropogon gerardi (BAG) |
4.4 |
3.7 |
4.5 |
| |
Biomass of C4 plant species Bouteloua gracilis (BBG) |
3.7 |
4.5 |
3.8 |
| |
Biomass of legume plant species Lupinus perennis (BLP) |
6.0 |
6.0 |
6.4 |
| Explanations of the selected soil variables (%) |
Total |
19.4 |
19.0 |
19.7 |
| |
Soil N% at the depth of 0-10 cm (SN0-10) |
5.7 |
5.2 |
4.5 |
| |
Soil N% at the depth of 10-20 cm (SN10-20) |
4.4 |
4.5 |
5.1 |
| |
Soil C and N ratio at the depth of 10–20 cm (SCNR10-20) |
4.4 |
4.5 |
3.8 |
| pH | 4.4 | 5.2 | 5.1 |
a The covariables for plant and soil variables were close zero.
Discussion
It is hypothesized that eCO2 may affect soil microbial C and N cycling due to the stimulation of plant photosynthesis, growth, and C allocation belowground [25,32,33] . Previous studies from the BioCON experiment showed that eCO2 led to changes in soil microbial biomass, community structure, functional activities [13,34,35], soil properties, such as pH and moisture [36], and microbial interactions [37]. Also, another study with Mojave Desert soils indicated that eCO2 increased microbial use of C substrates [17]. Consistently, our GeoChip data showed that the composition and structure of functional genes involved in C cycling dramatically shifted with a general increase in abundance at eCO2. First, this is reflected in an increase of abundances of microbial C fixation genes. Three key C fixation genes increased significantly at eCO2, including Rubisco for the Calvin–Benson–Bassham (CBB) cycle [38], CODH for the reductive acetyl-CoA pathway [39], and PCC/ACC for the 3-hydroxypropionate/malyl-CoA cycle [40]. It is expected that Form II Rubiscos would be favored at high CO2 and low O2 based on the kinetic properties [28]. Indeed, two Form II Rubiscos genes from Thiomicrospira pelophila (γ-Proteobacteria) and Rhodopseudomonas palustris HaA2 (α-Proteobacteria) were unique or increased at eCO2, respectively. For Thiomicrospira, the Form II Rubiscos are presumably expressed in the more anaerobic environments at high CO2[28], while R. palustris has extremely flexible metabolic characteristics including CO2 and N2 fixation under anaerobic and phototrophic conditions [41]. The second most abundant CODH gene was also detected from R. palustris and increased significantly at eCO2, and its dominant populations were found to be acetogenic bacteria, which may function for converting CO2 to biomass under anaerobic conditions. Since the knowledge of microbial C fixation processes in soil is still limited, mechanisms of the response of microbial C fixation genes to eCO2 need further study. Second, significant increases were observed in the abundance of genes involved in degradation of labile C substrates, such as the genes encoding α-amylase, glucoamylase and pullulanase for starch degradation, arabinofuranosidase for hemicellulose degradation, and endoglucanase for cellulose degradation. However, no significant change was shown in the abundance of genes involved in recalcitrant C (e.g., lignin) degradation. Therefore, our results indicate that eCO2 significantly affected metabolic potentials for C fixation and degradation. However, it appears that such changes have little effect on soil C storage [25], probably due to accelerated degradation of increased C inputs, which is consistent with increased soil CO2 flux over the course of the experiment.
Another important question is whether eCO2 affects soil N cycling processes and/or soil N dynamics. Our previous study has showed that soil N supply is probably an important constraint on global terrestrial productivity in response to eCO2[32]. When N is limiting, decomposers may respond to increased C inputs by decomposing soil organic matter to gain access to N and constrain the plant biomass accumulation at eCO2[42,43]. In this study, our GeoChip analysis showed that the abundance of nifH genes significantly increased at eCO2. Presumably, an increase in N2 fixation under eCO2 may lead to enhanced CO2 fertilization of plant biomass production by alleviating some of the N constraints on plant response to eCO2. In the plots examined in the present study, no N fertilizer was supplemented, but significant increases were observed in the total plant biomass and aboveground plant biomass, especially the biomass of legume plant species Lupinus perennis, which may be associated with significant increases of N2 fixers in soil under eCO2 measured by the abundance of nifH genes in this study. At eCO2, if the increased nifH abundance is interpreted as potential increase of soil microbial N2 fixation, such increase could supplement N nutrients for the plant growth to eliminate the N limitation constraint. In addition, the abundance of nirS genes significantly increased at eCO2 while all others genes involved in denitrification remained unaffected. The results suggest that eCO2 could significantly impact microbial N2 fixation and denitrification, and probably enhance the production of the greenhouse gas N2O. However, it appears that no significant changes were observed in soil N dynamics under eCO2, which may be largely due to the large N pool size in soil.
It is largely unknown whether or how eCO2 and eCO2-induced effects, such as increased C inputs into soil and changes in soil properties, shape soil microbial community structure. The direct effects of elevated atmospheric CO2 concentration on soil microbial communities were expected to be negligible compared to potential indirect effects such as increased plant C inputs to soil, since the CO2 concentrations in the pore space of soil generally is much higher than those in the atmosphere even under ambient CO2 concentrations [5]. However, this has not been well studied. Based on our GeoChip data, VPA showed the CO2 treatment could significantly explain 8.9% of the total variation of microbial community structure, 9.6% of detected functional genes involved in C cycling, and 9.4% of detected functional genes in N cycling in this study. After accounting for the effects of the CO2 treatment, the selected variables from plant and soil could significantly explain more than 42% of the total variances of microbial community structure. Our previous studies have demonstrated that increased C inputs at eCO2 stimulate microbial activity and regulate their composition [13,25]. Consistently, our statistical analysis suggests that the biomass of N2-fixing legume species (BLP) and the number of plant functional groups (PFG) have significantly positive correlations with the atmospheric CO2 level. These strong correlations could arise because increased plant-derived substrates at eCO2 could fuel heterotrophic metabolism in soil [44]. Such a strong correlation with the biomass of N2-fixing legume species (BLP) may result in an increased amount of N derived from the atmosphere. Therefore, significant increases in plant biomass were associated with the significant increase in the abundance of nifH genes, but little effect was seen in soil N dynamics.
Soil microbial community structure may be shaped by soil properties, such as pH and moisture [45]. For example, soil pH and moisture changed at eCO2 in the BioCON study [6,46], and a significant correlation between the soil microbial community compositions and soil pH was observed with a survey of 88 soils across North and South America [47]. In this study, soil N% at the depth of 0-10 cm (SN0-10) and 10–20 cm (SN10-20), soil C and N ratio at the depth of 10–20 cm (SCNR10-20), and soil pH (pH) were identified as the most important soil factors shaping microbial community structures. In addition, significant correlations were also observed between the plant and soil factors, such as positive correlations between pH and BBG, pH and PFG, SCNR10-20 and BBG, and negative correlations between SCNR10-20 and BLP. These results suggested that, in addition to direct effects of atmospheric CO2 on soil microbial C and N cycling, such as CO2 fixation, eCO2-induced indirect effects on plant and soil properties significantly impact the soil microbial community structure and modify their ecosystem functioning. The simultaneous enhances in the processes involved in CO2 fixation, C degradation, N fixations and partial denitrification could be the reason that no significant difference was detected in total soil C and N.
Conclusions
GeoChip was successfully used to illuminate the response of soil microbial communities to eCO2. The results showed that microbial C and N cycling were altered dramatically at eCO2, and the eCO2-induced effects, such as increased plant biomass and altered soil pH, may largely shape the soil microbial community structure and regulate their ecosystem functioning. However, the impact of these changes on soil C and N dynamics need to be further investigated. This study provides important insights into our understanding of the feedback response of soil microbial communities to elevated CO2 and global change.
Methods
Site, sampling and environmental variable analysis
This study was conducted within the BioCON experiment site [6] located at the Cedar Creek Ecosystem Science Reserve, MN, USA. The main BioCON field experiment has 296 plots (2 by 2 m) in six 20-meter-diameter rings, three for an aCO2 concentration of 368 μmol/mol and three for an elevated CO2 concentration of 560 μmol/mol using a FACE system as described by Reich et al. [6]. In this study, soil samples without plant root from 24 plots (12 biological replicates from ambient CO2 and 12 biological replicates from elevated CO2. All with 16 native plant species including four C4 grasses, four C3 grasses, four N-fixing legumes and four non-N-fixing herbaceous species, and no additional N supply) were collected in July 2007. The aboveground and belowground biomass, plant C and N concentrations, soil parameters, and in situ net N mineralization and net nitrification were measured as previously described [6,32]. More detailed information about sampling is provided in Additional file 13.
GeoChip analysis
DNA extraction, amplification and labeling, as well as the purification of labeled DNA, were carried out according the methods described by Xu et al. [23]. GeoChip 3.0 [26] was used to analyze the functional structure of the soil microbial communities. Details for GeoChip hybridization, image processing and data pre-processing are described in Additional file 13.
Statistical analysis
Pre-processed GeoChip data were further analyzed with different statistical methods: (i) detrended correspondence analysis (DCA) [48], combined with analysis of similarities (ANOSIM), non-parametric multivariate analysis of variance (Adonis) and Multi-Response Permutation Procedure (MRPP), for determining the overall functional changes in the microbial communities; (ii) microbial diversity index, Significant Pearson’s linear correlation (r) analysis, analyses of variance (ANOVA) and response ratio (RR) [3]; (iii) redundancy analysis (RDA) for revealing the individual or set of environmental variables that significantly explained the variation in functional microbial communities; (iv) variation partitioning for RDA were used to select the minimum number of environmental variables explaining the largest amount of variation in the model [20,49]. More details about the data analysis are described in Additional file 13.
Competing interests
The authors have declared that no competing interests exist.
Authors’ contributions
Conceived and designed the experiments: MX, ZH, SEH, PBR and JZ. MX, LW, JDN performed the experiments. MX, ZH and DY analyzed the data. MX, ZH and JZ interpreted the data. MX and ZH drafted the manuscript. SEH, PBR and JZ were involved in editing and revising the manuscript critically in preparation for submission. All authors read and approved the final manuscript.
Supplementary Material
A table listing the overall microbial community diversity detected by GeoChip under ambient CO2 (aCO2) and elevated CO2 (eCO2).
A figure about the normalized signal intensities of rbcL gene detected.
A figure about the normalized signal intensities of CODH gene detected.
A figure about the significantly changed and other top ten abundant pcc genes.
The supplemental results about the responses of carbon and nitrogen cycling genes to eCO2.
A figure about the normalized signal intensities of glucoamylase encoding gene detected.
A figure about the normalized signal intensities of pulA gene detected.
A figure about the normalized signal intensities of endoglucanase gene detected.
A figure about the normalized signal intensities of ara gene detected.
A figure about the normalized signal intensities of vanA gene detected.
A figure about the normalized signal intensities of shared nirS gene detected.
A table listing the nirS genes only detected at aCO2 or eCO2.
The supplemental descriptions for materials and methods.
Contributor Information
Meiying Xu, Email: xumy@gdim.cn.
Zhili He, Email: zhili.he@ou.edu.
Ye Deng, Email: dengye@ou.edu.
Liyou Wu, Email: liyou.wu-1@ou.edu.
Joy D van Nostrand, Email: joy.vannostrand@ou.edu.
Sarah E Hobbie, Email: shobbie@umn.edu.
Peter B Reich, Email: preich@umn.edu.
Jizhong Zhou, Email: jzhou@ou.edu.
Acknowledgements
This work is supported by the United States Department of Agriculture (Project 2007-35319-18305) through NSF-USDA Microbial Observatories Program; by US Department of Energy (contract DE-SC0004601), by the National Science Foundation under Grant Numbers DEB-0716587 and DEB-0620652 as well as DEB-0322057, DEB-0080382, DEB-0218039 DEB-0219104, DEB-0217631, DEB-0716587 (BioComplexity, Cedar Creek LTER and LTREB projects); the DOE Program for Ecosystem Research; the Minnesota Environment and Natural Resources Trust Fund; and the Team Project of the Natural Science Foundation of Guangdong Province, China (9351007002000001).
References
- IPCC. Intergovernmental Panel on Climate Change. Climate Change 2007: The Physical Science Basis: Fourth Assessment Report of the Intergovernmental Panel on Climate. Change. Cambridge: Cambridge University Press; 2007. [Google Scholar]
- Houghton JT, Ding Y, Griggs DJ, Noguer M, Linden PJ, Xiaosu D. Climate Change 2001: The Scientific Basis: Contributions of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge: Cambridge University Press; 2001. p. 881. [Google Scholar]
- Luo Y, Hui D, Zhang D. Elevated CO2 stimulates net accumulations of carbon and nitrogen in land ecosystems: a meta-analysis. Ecology. 2006;87(1):53–63. doi: 10.1890/04-1724. [DOI] [PubMed] [Google Scholar]
- Heimann M, Reichstein M. Terrestrial ecosystem carbon dynamics and climate feedbacks. Nature. 2008;451(7176):289–292. doi: 10.1038/nature06591. [DOI] [PubMed] [Google Scholar]
- Drigo B, Kowalchuk G, Van Veen J. Climate change goes underground: effects of elevated atmospheric CO2 on microbial community structure and activities in the rhizosphere. Biol Fertil Soils. 2008;44(5):667–679. doi: 10.1007/s00374-008-0277-3. [DOI] [Google Scholar]
- Reich PB, Knops J, Tilman D, Craine J, Ellsworth D, Tjoelker M, Lee T, Wedin D, Naeem S, Bahauddin D. et al. Plant diversity enhances ecosystem responses to elevated CO2 and nitrogen deposition. Nature. 2001;410(6830):809–812. doi: 10.1038/35071062. [DOI] [PubMed] [Google Scholar]
- Ainsworth EA, Long SP. What have we learned from 15 years of free-air CO2 enrichment (FACE)? A meta-analytic review of the responses of photosynthesis, canopy properties and plant production to rising CO2. New Phytol. 2005;165:351–372. doi: 10.1111/j.1469-8137.2004.01224.x. [DOI] [PubMed] [Google Scholar]
- Zak DR, Pregitzer KS, King JS, Holmes WE. Elevated atmospheric CO2, fine roots and the response of soil microorganisms: a review and hypothesis. New Phytol. 2000;147(1):201–222. doi: 10.1046/j.1469-8137.2000.00687.x. [DOI] [Google Scholar]
- Souza L, Belote RT, Kardol P, Weltzin JF, Norby RJ. CO2 enrichment accelerates successional development of an understory plant community. Journal of Plant Ecology-Uk. 2010;3(1):33–39. doi: 10.1093/jpe/rtp032. [DOI] [Google Scholar]
- Balser TC, Firestone MK. Linking microbial community composition and soil processes in a California annual grassland and mixed-conifer forest. Biogeochemistry. 2005;73(2):395–415. doi: 10.1007/s10533-004-0372-y. [DOI] [Google Scholar]
- Lesaulnier C, Papamichail D, McCorkle S, Ollivier B, Skiena S, Taghavi S, Zak D, van der Lelie D. Elevated atmospheric CO2 affects soil microbial diversity associated with trembling aspen. Environ Microbiol. 2008;10(4):926–941. doi: 10.1111/j.1462-2920.2007.01512.x. [DOI] [PubMed] [Google Scholar]
- Finzi AC, Sinsabaugh RL, Long TM, Osgood MP. Micorbial community responses to atmospheric carbon dioxide enrichment in a warm-temperate forest. Ecosystems. 2006;9:215–226. doi: 10.1007/s10021-005-0078-6. [DOI] [Google Scholar]
- Chung H, Zak DR, Reich PB, Ellsworth DS. Plant species richness, elevated CO2, and atmospheric nitrogen deposition alter soil microbial community composition and function. Glob Chang Biol. 2007;13:980–989. doi: 10.1111/j.1365-2486.2007.01313.x. [DOI] [Google Scholar]
- Jossi M, Fromin N, Tarnawski S, Kohler F, Gillet F, Aragno M, Hamelin J. How elevated CO2 modifies total and metabolically active bacterial communities in the rhizosphere of two perennial grasses grown under field conditions. FEMS Microbiol Ecol. 2006;55:339–350. doi: 10.1111/j.1574-6941.2005.00040.x. [DOI] [PubMed] [Google Scholar]
- Carney MC, Hungate BA, Drake BG, Megonigal JP. Altered soil microbial community at elevated CO2 leads to loss of soil carbon. Proc Natl Acad Sci USA. 2007;104(12):4990–4995. doi: 10.1073/pnas.0610045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin EE, Castro HF, Sides KE, Schadt CW, Classen AT. Assessment of 10 years of CO2 fumigation on soil microbial communities and function in a sweetgum plantation. Soil Biol Biochem. 2009;41(3):514–520. doi: 10.1016/j.soilbio.2008.12.010. [DOI] [Google Scholar]
- Jin VL, Evans RD. Elevated CO2 increases microbial carbon substrate use and nitrogen cycling in Mojave Desert soils. Glob Chang Biol. 2007;13(2):452–465. doi: 10.1111/j.1365-2486.2006.01308.x. [DOI] [Google Scholar]
- He Z, Deng Y, Zhou J. Development of functional gene microarrays for microbial community analysis. Curr Opin Biotechnol. 2012;23(1):49–55. doi: 10.1016/j.copbio.2011.11.001. [DOI] [PubMed] [Google Scholar]
- He Z, Van Nostrand JD, Zhou J. Applications of functional gene microarrays for profiling microbial communities. Curr Opin Biotechnol. 2012;23(3):460–466. doi: 10.1016/j.copbio.2011.12.021. [DOI] [PubMed] [Google Scholar]
- Ramette A, Tiedje JM. Multiscale responses of microbial life to spatial distance and environmental heterogeneity in a patchy ecosystem. Proc Natl Acad Sci. 2007;104(8):2761–2766. doi: 10.1073/pnas.0610671104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Kang S, Schadt CW, Garten CT Jr. Spatial scaling of functional gene diversity across various microbial taxa. Proc Natl Acad Sci USA. 2008;105(22):7768–7773. doi: 10.1073/pnas.0709016105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Zhou H, Meng J, Peng X, Jiang L, Sun P, Zhang C, Van Nostrand JD, Deng Y, He Z. et al. GeoChip-based analysis of metabolic diversity of microbial communities at the Juan de Fuca Ridge hydrothermal vent. Proc Natl Acad Sci USA. 2009;106(12):4840–4845. doi: 10.1073/pnas.0810418106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Wu W-M, Wu L, He Z, Van Nostrand JD, Deng Y, Luo J, Carley J, Ginder-Vogel M, Gentry TJ. et al. Responses of microbial community functional structures to pilot-scale uranium in situ bioremediation. ISME J. 2010;4(8):1060–1070. doi: 10.1038/ismej.2010.31. [DOI] [PubMed] [Google Scholar]
- Naeem S, Duffy JE, Zavaleta E. The functions of biological diversity in an age of extinction. Science. 2012;336(6087):1401–1406. doi: 10.1126/science.1215855. [DOI] [PubMed] [Google Scholar]
- Adair EC, Peter BR, Sarah EH, Johannes MHK. Interactive effects of time, CO2, N, and diversity on total belowground carbon allocation and ecosystem carbon storage in a grassland community. Ecosystems. 2009;12(6):1037–1052. doi: 10.1007/s10021-009-9278-9. [DOI] [Google Scholar]
- He Z, Deng Y, Van Nostrand JD, Tu Q, Xu M, Hemme CL, Li X, Wu L, Gentry TJ, Yin Y. et al. GeoChip 3.0 as a high-throughput tool for analyzing microbial community composition, structure and functional activity. ISME J. 2010;4(9):1167–1179. doi: 10.1038/ismej.2010.46. [DOI] [PubMed] [Google Scholar]
- Berg IA, Kockelkorn D, Buckel W, Fuchs G. A 3-hydroxypropionate/ 4-hydroxybutyrate autotrophic carbon dioxide assimilation pathway in archaea. Science. 2007;318(5857):1782–1786. doi: 10.1126/science.1149976. [DOI] [PubMed] [Google Scholar]
- Badger MR, Bek EJ. Multiple rubisco forms in proteobacteria: their functional significance in relation to CO2 acquisition by the CBB cycle. J Exp Bot. 2008;59(7):1525–1541. doi: 10.1093/jxb/erm297. [DOI] [PubMed] [Google Scholar]
- Hageman RV, Burris RH. Nitrogenase and nitrogenase reductase associate and dissociate with each catalytic cycle. Proc Natl Acad Sci USA. 1978;75(6):2699–2702. doi: 10.1073/pnas.75.6.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehr JP, Jenkins BD, Short SM, Steward GF. Nitrogenase gene diversity and microbial community structure: a cross-system comparison. Environ Microbiol. 2003;5(7):539–554. doi: 10.1046/j.1462-2920.2003.00451.x. [DOI] [PubMed] [Google Scholar]
- Raymond J, Siefert JL, Staples CR, Blankenship RE. The natural history of nitrogen fixation. Mol Biol Evol. 2004;21(3):541–554. doi: 10.1093/molbev/msh047. [DOI] [PubMed] [Google Scholar]
- Reich PB, Hobbie SE, Lee T, Ellsworth DS, West JB, Tilman D, Knops JMH, Naeem S, Trost J. Nitrogen limitation constrains sustainability of ecosystem response to CO2. Nature. 2006;440(7086):922–925. doi: 10.1038/nature04486. [DOI] [PubMed] [Google Scholar]
- Lee TD, Barrott SH, Reich PB. Photosynthetic responses of 13 grassland species across 11 years of free-air CO2 enrichment is modest, consistent and independent of N supply. Glob Chang Biol. 2011;17(9):2893–2904. doi: 10.1111/j.1365-2486.2011.02435.x. [DOI] [Google Scholar]
- Dijkstra FA, Hobbie SE, Reich PB, Knops JMH. Divergent effects of elevated CO2, N fertilization, and plant diversity on soil C and N dynamics in a grassland field experiment. Plant Soil. 2005;272(1):41–52. [Google Scholar]
- Deng Y, He Z, Xu M, Qin Y, Van Nostrand JD, Wu L, Roe BA, Wiley G, Hobbie SE, Reich PB. et al. Elevated carbon dioxide alters the structure of soil microbial communities. Appl Environ Microbiol. 2012;78(8):2991–2995. doi: 10.1128/AEM.06924-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich PB. Elevated CO2 reduces losses of plant diversity caused by nitrogen deposition. Science. 2009;326(5958):1399–1402. doi: 10.1126/science.1178820. [DOI] [PubMed] [Google Scholar]
- Zhou J, Deng Y, Luo F, He Z, Tu Q, Zhi X. Functional molecular ecological networks. mBio. 2010;1(4):e00169-10. doi: 10.1128/mBio.00169-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvin M. Nobel prize for chemistry. Nature. 1961;192:799. [Google Scholar]
- Evans MC, Buchanan BB, Arnon DI. A new ferredoxin-dependent carbon reduction cycle in a photosynthetic bacterium. Proc Natl Acad Sci U S A. 1966;55:928–934. doi: 10.1073/pnas.55.4.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herter S, Fuchs G, Bacher A, Eisenreich W. A bicyclic autotrophic CO2 fixation pathway in chloroflexus aurantiacus. J Biol Chem. 2002;277(23):20277–20283. doi: 10.1074/jbc.M201030200. [DOI] [PubMed] [Google Scholar]
- Larimer FW, Chain P, Hauser L, Lamerdin J, Malfatti S, Do L, Land ML, Pelletier DA, Beatty JT, Lang AS. et al. Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nat Biotech. 2004;22(1):55–61. doi: 10.1038/nbt923. [DOI] [PubMed] [Google Scholar]
- Langley JA, McKinley DC, Wolf AA, Hungate BA, Drake BG, Megonigal JP. Priming depletes soil carbon and releases nitrogen in a scrub-oak ecosystem exposed to elevated CO2. Soil Biol Biochem. 2009;41(1):54–60. doi: 10.1016/j.soilbio.2008.09.016. [DOI] [Google Scholar]
- Billings SA, Lichter J, Ziegler SE, Hungate BA, Richter DB. A call to investigate drivers of soil organic matter retention vs. mineralization in a high CO2 world. Soil Biol Biochem. 2010;42(4):665–668. doi: 10.1016/j.soilbio.2010.01.002. [DOI] [Google Scholar]
- Zak DR, Tilman D, Parmenter RR, Rice CW, Fisher FM, Vose J, Milchunas D, Martin CW. Plant production and soil microorganisms in late-successional ecosystems: A continental-scale study. Ecology. 1994;75(8):2333–2347. doi: 10.2307/1940888. [DOI] [Google Scholar]
- Marschner P, Yang CH, Lieberei R, Crowley DE. Soil and plant specific effects on bacterial community composition in the rhizosphere. Soil Biol Biochem. 2001;33(11):1437–1445. doi: 10.1016/S0038-0717(01)00052-9. [DOI] [Google Scholar]
- He Z, Xu M, Deng Y, Kang S, Kellogg L, Wu L, Van Nostrand JD, Hobbie SE, Reich PB, Zhou J. Metagenomic analysis reveals a marked divergence in the structure of belowground microbial communities at elevated CO2. Ecol Lett. 2010;13(5):564–575. doi: 10.1111/j.1461-0248.2010.01453.x. [DOI] [PubMed] [Google Scholar]
- Lauber CL, Hamady M, Knight R, Fierer N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol. 2009;75(15):5111–5120. doi: 10.1128/AEM.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MO, Gauch HG. Deterended correspondence analysis, an improved ordination technique. Vegetatio. 1980;42:47–58. doi: 10.1007/BF00048870. [DOI] [Google Scholar]
- Ramette A. Multivariate analyses in microbial ecology. FEMS Microbiol Ecol. 2007;62(2):142–160. doi: 10.1111/j.1574-6941.2007.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A table listing the overall microbial community diversity detected by GeoChip under ambient CO2 (aCO2) and elevated CO2 (eCO2).
A figure about the normalized signal intensities of rbcL gene detected.
A figure about the normalized signal intensities of CODH gene detected.
A figure about the significantly changed and other top ten abundant pcc genes.
The supplemental results about the responses of carbon and nitrogen cycling genes to eCO2.
A figure about the normalized signal intensities of glucoamylase encoding gene detected.
A figure about the normalized signal intensities of pulA gene detected.
A figure about the normalized signal intensities of endoglucanase gene detected.
A figure about the normalized signal intensities of ara gene detected.
A figure about the normalized signal intensities of vanA gene detected.
A figure about the normalized signal intensities of shared nirS gene detected.
A table listing the nirS genes only detected at aCO2 or eCO2.
The supplemental descriptions for materials and methods.