

Abstract
We present herein characteristics of a conjugate in which dL5, a fluorogen-activating protein (FAP) and AEAEAKAK, an amphiphilic peptide are combined to form a solid-phase fluorescence-detection platform. The FAP dL5 is a covalently linked dimer of two identical light chain variable fragments which activates the fluorescence of the fluorogen malachite green (MG). The amphiphilic peptide of sequence AEAEAKAK is a building block of stimuli-responsive materials that undergoes sol-gel phase transition at high ionic strengths. We hypothesize that the novel bi-functional protein containing both the FAP and the amphiphile, termed dL5_EAK, co-assembles with the self-assembling peptide [AEAEAKAK]2 (EAK16-II) to form an insoluble membrane composite whereby the fluorescence enhancement function of the FAP domain remains intact. Denaturing polyacrylamide electrophoresis indicated that greater than 78% of dL5_EAK incorporates into the EAK16-II membrane. Conversely, less than 32% of dL5 without the EAK sequence associates with the insoluble fraction of EAK16-II in buffers. Membranes containing dL5_EAK and EAK16-II exhibited at least 4-fold higher fluorescence intensity compared to mixtures containing dL5 and EAK16-II. Scanning electron microscopy revealed the presence of particulates, presumably FAPs, scattering on the membrane fibrils. The evidence suggests a system of materials that can be developed into in situ-forming local sensors by immobilizing dL5 into coacervate, on which MG can be detected. It is envisioned that dL5 membranes can be established in diseased locales to monitor infiltration and migration of inflammatory cells marked with antibodies conjugated to MG.
Introduction
Molecular mechanisms in mammalian systems have been studied by genetically coupling fluorescent proteins with the polypeptides of interest.1 Fluorogen activating proteins (FAPs) represent a new class of fluorescence-based molecular reporters.2–8 The underpinning mechanism entails intense fluorescence enhancement of small organic dyes, or fluorogens, upon binding to FAP. The enhancement results from FAP constraining the rapid rotation around a single bond within the fluorogen chromophore. FAPs were discovered from screening combinatorial libraries of human single-chain variable fragments (scFv) that bind to pegylated forms of the fluorogenic dyes malachite green (MG) and thiazole orange (TO).2
Among the FAPs discovered was the scFv λ light chain L5, which enhances the fluorescence of the cell-impermeant derivatives of MG, “MG-2p” and “MG-11p” (Fig 1.) by 4,100-fold.2 This dramatic amplification can be attributed to the high affinity of L5 for MG (Kd= 320 nM) and extremely low background fluorescence. Affinity maturation of L5 on yeast cells has yielded several variants with even higher affinities for MG, particularly the double mutant E52D and L91S (L5 NP138)4, dimers of which, linked via a (G4S)4 linker, bind MG with Kd in the picomolar range (manuscript in preparation by CSG). These FAP-fluorogen pairs have been validated in a kinetic analysis of beta 2-adrenergic receptor (β2AR) internalization3 and other applications currently in development.6
Figure 1.

Structures of the malachite green fluorogens used in these studies. Details of their synthesis and NMR analysis have been described previously. 2
We hypothesize that the switchable “off-on” functionality of FAP-fluorogen modules can be exploited in developing novel biosensors for other biomedical applications. For example, leukocytes emigrating from transplants may be tracked by pre-treating the organ with MG-conjugated antibodies against graft surface markers. Cells migrating through concentrated FAP membranes would be detected with fluorescence. (Fig. 2) The very tight binding of fluorogen to linked dimers of L5 NP 138 suggests that fluorogen would stay bound, and would remain fluorescent, even at relatively low fluorogen concentrations. On the other hand, detection of fluorescence in model organisms would require concentration of FAP/fluorogen pairs to a localized area, rather than in a diffuse solution. The method of protein immobilization is critical in optimizing the efficiency of biosensors for use in animal models. Random adsorption to materials may damage native protein conformations and alter the sensitivity and specificity of detection.9 Covalent conjugation of protein sensors might result in overloading and reduced functions due to steric hindrance. Herein we report a system by which the sensing component (FAP) co-assembles into an immobilizing material.
Figure 2.

Conceptual representation of an application envisioned for dL5_EAK membranes in vivo. In this scenario, membranes composed of dL5_EAK and EAK16-II would be used to intercept trafficking of inflammation-associated leukocytes to lymph nodes. Antibodies conjugated with MG would be introduced at the site of inflammation to label trafficking leukocytes. Association of cells with dL5_EAK could be detected by MG fluorescence generated by binding of MG and the FAP domain.
Stimuli-responsive materials have been tested in animal models as injectable scaffolds for tissue regeneration and as drug delivery depots.10–14 EAK16-II (amino acids sequence AEAEAKAKAEAEAKAK) and related peptides are building blocks of materials responsive to elevated ionic strengths. 13,14 These low molecular weight peptides (< 5 kDa), with periodic repeats of alternating polar and nonpolar residues, adopt β-strand conformations in which the polar and non-polar side chains orient toward opposite sides. The peptides undergo solution-gel phase transitions upon exposure to high concentrations of salts. Consequently, such peptides dissolved in deionized water can be injected to form gels or membranes in a physiological environment (animal tissues) in which the salt concentration is relatively high (e.g. Na+ >100 mM). We have previously reported a design by which IgG molecules can be concentrated and oriented on a co-assembly of amphiphilic peptides EAK16-II and EAKH6 (EAK16-II appended with six histidines at the C-terminus).15 Upon injecting into phosphate buffers, EAK16-II and EAKH6 co-assemble to form membranes on which His-tags are detectable for days afterward. The ability of EAK16-II to accommodate proteins containing complementary amphiphilic sequences can be exploited in developing novel FAP-based sensors. While direct conjugation of FAP to EAK16-II negates the self-assembling propensity, introducing amphiphilic domains into L5 would incorporate a fluorescence signal into injectable gel membranes to track and quantify biomarkers in animal systems for a variety of purposes. Conjugation of MG to antibodies or protein ligands could have a wide variety of uses as capture or detection of disease-associated cells or molecules by allowing detection by fluorescence.
In the current study, we report evidence supporting functionalization of the EAK16-II membrane with dimerized L5 NP138 (dL5), which has a higher affinity for MG compared to the monomer. This was accomplished by engineering a construct containing both the dL5 and EAK16-II peptide sequences (referred hereafter as dL5_EAK). Data are presented demonstrating the co-assembly of dL5_EAK and EAK16-II into β-sheet fibril membranes. The functionalized membranes retain the ability to amplify MG fluorescence after addition of MG.
Experimental Procedures
FAP plasmid construction
The plasmid pET21 GST HRV dL5 was constructed by PCR amplification of L5 (E52D L91S, referred to as L5 in this work) monomer and ligation into the bacterial expression vector pET21 GST HRV. An annealed DNA insert encoding the amino acid repeat (G4S)4 was ligated downstream from the L5 subunit, followed by the ligation of PCR amplified DNA encoding an additional L5 subunit. The plasmid pET21 GST HRV dL5_EAK was constructed by ligation of double stranded DNA encoding 3 additional repeats of (G4S) followed by 3 repeats of the amino acids AEAEAKAK downstream of the dL5 sequence of pET21 GST HRV dL5. Plasmid sequences were verified by DNA sequencing. Details on oligonucleotide primer and insert sequences and restriction enzymes used to construct plasmids can be found in the supporting information section.
Protein expression and purification
Expression and purification of dL5 and dL5_EAKwas carried out by transforming pET21 GST- HRV-dL5 and pET21 GST-HRV-dL5_EAK into calcium competent Rosetta-Gami 2(DE3) E. coli. (Novagen, Madison, WI). Transformed Rosetta-Gami bacteria were grown in 500 ml volumes and induced for expression of protein using 500 μM IPTG and 0.4% glucose overnight at 20°C. Cells were pelleted, lysed and clarified. FAP protein was purified using Ni-NTA agarose (Qiagen, Valencia, CA) with elution following cleavage using HRV3C protease. Purified protein was dialyzed into PBS after elution and quantified by 280 nm absorption measurements. Details on protein expression and purification are described in the supporting information section.
Fluorogen and peptide synthesis
Malachite Green with 11 ethyleneoxide units (MG-11p) and 2 ethyleneoxide units (MG-2p) were provided by the Technology Center for Networks and Pathways in the Molecular Imaging and Biosensor Center at Carnegie Mellon University. Synthesis and NMR characterization of MG-2p and MG-11p have been described previously.2 N-acetylated and C-amidated EAK16-II was custom synthesized by EZBiolab (Carmel, IN) at greater than 90% purity. Sequences were confirmed by mass spectroscopy. Aliquots of EAK16-II peptides were reconstituted in sterile deionized water (18.2 MΩ at 25°C) at 5 mg/ml.
Fluorescence spectroscopic measurements of soluble dL5 and dL5_EAK
Absorbance and emission spectra of MG-2p bound to dL5 and dL5_EAK were taken using a TECAN Safire 2 fluorescence plate reader with Costar 3596 96-well plate (Corning Inc., Corning, NY). dL5 or dL5_EAK (1.25 μM ) in PBS and MG-2p (125 μM ) were mixed in the 96 well plate, followed by a 10 minute incubation at room temp. Excitation scans (400–800 nm) were collected in 5 nm increments measuring emission at 680 nm with an instrument gain of 86 and Z position of 9,020 μm. Emission scans (510–800 nm) were performed (excitation = 480 nm) in 1 nm increments with an instrument gain of 101 and Z position of 8,020 μm. All instrument Z positions were auto-optimized by the TECAN Safire 2.
dL5 and dL5_EAK membrane EAK16-II membrane preparation and fluorescence quantification
Insoluble protein/peptide membranes were made by mixing dL5 or dL5_EAK with EAK16-II at ratios of 1:50 and 1:100 in PBS + (Phosphate buffered saline with 0.1% Pluronic F-127 (Invitrogen, Carlsbad, CA)) with 0.0002% Congo Red (Thermo Fisher, Waltham, MA) followed by incubation at room temperature on a rotator. Samples were centrifuged, supernatants removed and re-suspended in PBS+ three times. Insoluble pellets were re-suspended in PBS+. 125 μM fluorogen was added for excitation and emission spectra while 1 μM fluorogen was added for single point measurement of protein to peptide fluorescence values. Detailed descriptions of protein and peptide concentrations and settings for the TECAN Safire 2 fluorescence plate reader are given in the supporting information section.
Microscopic analyses
Bright field imaging was conducted as follows. 8 μl of 5 mg/ml EAK 16-II was mixed with 4.4 μl of 58 μM dL5_EAK to give a molar ratio of 100:1. This mixture (5 μl ) was added to 5 μl of 0.01% Congo red in PBS and spotted onto a glass slide (Fisher Scientific, Waltham, MA). Control samples of 5 mg/ml EAK16-II alone and 58 μM dL5_EAK alone were mixed similarly with Congo Red and spotted onto microscope slides. Images were captured immediately after spotting using a Vista Vision™ inverted microscope (VWR, Radnor, PA) equipped with a ProgRes C3 cooled CCD camera (Jenoptik AG, Jena, Germany).
Fluorescence microscopy was performed using a Zeiss Axioplan 2 imaging microscope equipped with a mercury lamp with samples prepared on PET track-etched membrane 24 well format cell culture insert filters. dL5 and dL5_EAK were mixed at a 1:100 ratio with EAK16-II and Congo red was added to visualize membrane formation on the filter. Details of sample preparation are described in the supporting information section. Congo red fluorescence was visualized using a 605/55 filter (Chroma, Lititz, PA) while MG/dL5 fluorescence was visualized using a 710/75 filter (Chroma, Lititz, PA). Images were collected using a Zeiss Axio cam MRm camera.
Sample membranes for scanning electron microscopy (SEM) were prepared by mixing dL5 or dL5_EAK with EAK16II at a ratio of 1:100 with no fluorogen or Congo red present. Minor differences in the dL5_EAK/EAK16II sample are detailed in the supporting information section. Samples were subsequently fixed in 10% formalin, rinsed with deionized water three times, then lyophilized before being mounted on double-sided carbon tape affixed to the aluminum specimen holder of the SEM instrument. A Hitachi S-3400N scanning electron microscope was used to examine the samples. Images were taken at a working distance between 5–7 mm with a 5.0 kV accelerating voltage under vacuum (<1 Pa). The average size of particles in each image was evaluated using an interactive measurement program in Axiovision 4.6.3. By selecting particles in the image field, the bound height and width of each particle was reported and these values were then summed and divided by two in order to represent an individual particle size. Measurements consisted of selecting all of the particles that could be individually seen of three representative images, from three different areas, which resulted in a total of 42 particles in 450 μm2 imaged.
Sodium dodecyl sulfate polyacrylamide electrophoresis (SDS-PAGE)
200 μl solutions containing EAK16-II (620 μM) with dL5 (6.2 μM) or dL5_EAK (6.2 μM) were incubated in PBS at 37°C overnight and spun at 6,000 rcf in an Eppendorf 5414 R model centrifuge (Eppendorf, Happauge, NY) for 2 minutes with the supernatants analyzed using denaturing Novex® NuPAGE 4–12% Bis-Tris gels (catalog # NPO322, Life Technologies, Carlsbad, CA). Samples were prepared by mixing 5 μl of supernatant with 5 μl of of NuPAGE LDS sample buffer (Life Technologies, Carlsbad, CA) containing 10% beta-mercaptoethanol and 10 μl PBS and heated at 70°C for 10 minutes with the entire 20 μl loaded into the gel well. Gels were run at 200 V for 35 min in 1X NuPAGE MES SDS running buffer (Life Technologies, Carlsbad, CA). Proteins were visualized using the SilverQuest™ staining kit (Life Technologies, Carlsbad, CA) with a reported detection limit of 0.3 ng and imaged using the Kodak 440 Image Station (Kodak, Rochester, NY). Band intensities were quantified using ImageJ (version 1.45s) with percent of dL5 or dL5_EAK protein incorporated into insoluble EAK16-II membranes quantified by subtracting the intensity of the protein band with EAK116-II present after centrifugation from the intensity of the samples without EAK16-II present.
Results
dL5_EAK was engineered by introducing the nucleotide sequences encoding both domains into a modified pET21 plasmid expression vector. A glycine-serine linker (G4S)3 separating the FAP and amphiphilic [AEAEAKAK]3 domains was inserted to provide conformational flexibility necessary for efficient binding of the fluorogen and incorporation into β-sheet fibrils (Fig. 3). In the presence of fluorogen, dL5 and dL5_EAK exhibited excitation and emission maxima at 637 nm and 664 nm, respectively (Fig. 4). A characteristic smaller excitation peak was observed at 480 nm. These mirrored the excitation (639 nm) and emission (666 nm) of fluorogen bound in dL5.2
Figure 3.

Schematic representation of the bi-functional FAP-amphiphile conjugate, dL5_EAK. (a) Primary structure domains in dL5_EAK separated via glycine-serine linkers. (b) Schematic depiction of dL5_EAK binding fluorogen and incorporation of the amphiphilic sequence into the EAK16-II fibril network. (Molecular schematics are not drawn to scale.)
Figure 4.
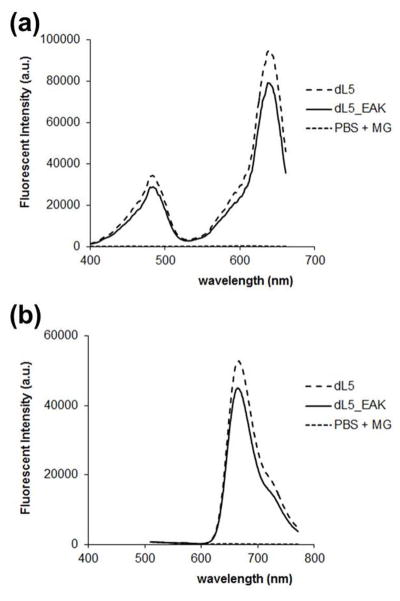
Fluorescence spectra of parent and modified FAP with malachite green. Aqueous solutions of dL5 and dL5_EAK (both at 1.25 μM) were incubated with MG-2p (12.5 μM); (a) excitation scan with emission monitored at 680 nm +/−10 nm and (b) emission scan using 480 +/−10 nm excitation. The 480 nm excitation was used to render a wider range of emission due to the peak excitation maximum (640 nm) being very close to the emission maximum (680).
The excitation and emission spectra (Fig. 5, b and c) of dL5_EAK integrated into membranes (1:100) resembled that of free dL5_EAK (Fig. 4). Low fluorescence was observed in membranes prepared by mixing dL5 and EAK16-II. The propensity for dL5 with or without the amphiphilic domain to integrate into the EAK16-II membrane was examined at protein to EAK16-II molar ratios of 1:50 and 1:100 (Fig. 5d). Fluorescence was higher in membranes containing dL5_EAK compared to those formed by mixing EAK16-II with dL5. The difference in fluorescence intensity was found to be more than 4-fold, indicating low non-specific adsorption of dL5 to the EAK16-II fibrils. Taken together, these data indicate a preferential incorporation of dL5_EAK into EAK16-II fibrils, and that the FAP domain remained functional when incorporated into the membrane. Additional analysis of dL5_EAK at 1:100 molar ratio indicated that the membranes were stable for up to five days at room temperature with no loss of fluorescence over that time period (data not shown). Subsequent studies employed dL5_EAK to EAK16-II at a molar ratio of 1:100.
Figure 5.

Differential fluorescence enhancement of membranes prepared with EAK16-II mixed with dL5 or dL5_EAK. (a) Samples were prepared by mixing the respective FAP proteins with EAK16-II with MG in PBS+ containing Congo red. (b) Excitation spectra (emission = 680 nm +/− 10 nm) and (c) emission spectra (excitation = 480 nm +/− 10 nm) of dL5_EAK membrane and dL5 mixed with EAK16-II. (d) Average fluorescence intensity (excitation = 635 nm; emission = 680 nm) of re-suspended membrane pellets of EAK 16-II prepared with dL5_EAK or dL5 at the indicated molar ratios (FAP to EAK16-II). No fluorescence above unbound fluorogen background was detected for MG with EAK16-II alone (data not shown).
The morphological features of dL5_EAK membranes were examined using optical and scanning electron microscopy (Fig. 6). Bright field micrographs show that similar to EAK16-II membranes, mixtures of dL5_EAK and EAK16-II in buffers exhibited an orange color in the presence of Congo red (Fig. 6a), which binds to hydrophobic cavities in the cross-linked fibrils.16 Addition of dL5_EAK to EAK16-II resulted in retracted rather than smooth membranes. It was evident that collapsing of EAK16-II β-sheet fibrils could take place in the presence of dL5_EAK. No membranes were visible (in the presence of Congo red) in dL5_EAK without EAK16-II at room temperature or 37°C in PBS (Fig. 6a “dL5_EAK”). SEM revealed that membranes formed by dL5_EAK/EAK16-II contained entangled but less dense fibrils compared to the membranes formed by EAK16-II alone (Fig. 6b). The average width of dL5_EAK/EAK16-II fibrils was estimated to be 11 nm (±4.9), close to those in pure EAK16-II (9.3 nm ±2.1). Particulates were found on dL5_EAK/EAK16-II but not on EAK16-II membranes (Fig. 6b). The size range (2.5–25nm) suggests dL5_EAK might localize on the membrane as monomers and oligomers. These microscopic studies confirmed that dL5_EAK and EAK16-II co-assembled into a fibrous composite.
Figure 6.
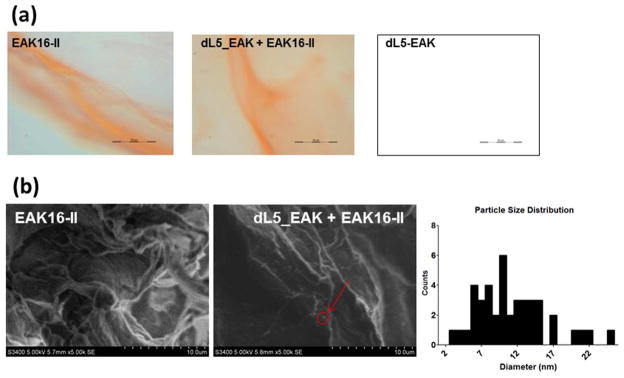
Morphological features of EAK16-II and dL5_EAK/EAK16-II membranes as revealed in (a) Optical images (40X; scale bar=50 μm) captured using a cooled CCD camera immediately after solutions containing 5 μl of EAK16-II (left panel), 5 μl of a 1:100 ratio of dL5_EAK to EAK16-II (middle panel) and 5 μl of 58 μM dL5_EAK alone (right panel) were pipetted into an equal volume of PBS with 0.01% Congo red and spotted on glass slides. β sheath structures formed by self-assembly of EAK16-II domains are visible by Congo red incorporation. (b) SEM images (5000X magnification) of fixed and lyophilized membrane samples. Size distribution of the particles in the dL5_EAK/EAK16-II image was evaluated using an interactive measurement program in Axiovision 4.6.3.
Direct evidence for the functionalization of EAK16-II membranes was demonstrated using fluorescence microscopy (Fig. 7). Intense MG fluorescence (Fig. 7e) was observed concentrating on the dL5_EAK membrane co-localizing with Congo red fluorescence (Fig. 7c). Conversely, low MG fluorescence (Fig. 7f) appeared on membranes (Fig. 7d) formed with EAK16-II and dL5. These results are consistent with fluorimetric studies showing that the dL5 mixed with EAK16-II exhibited approximately one-fourth fluorescence of that detected with the dL5_EAK membrane (Fig 5b). The fluorescence in the dL5_EAK membranes appeared in patches rather than uniformly distributed, suggesting possible aggregation of the dL5_EAK polypeptides prior to integration into EAK16-II β-sheet fibrils. Taken together, these data provide proof that dL5_EAK/EAK16-II membranes were capable of enhancing MG fluorescence.
Figure 7.

Fluorescence micrographs of EAK16-II membranes prepared with dL5 or dL5_EAK. Bright field images of (a) dL5_EAK mixed with EAK16-II [exposure time (et) = 79 milliseconds (ms)] and (b) dL5 mixed with EAK16-II (et = 56 ms). Congo red fluorescence through a 605/55 filter of membranes prepared with (c) dL5_EAK and EAK16-II (et = 538 ms) and (d) dL5 and EAK16-II (et = 763 ms). Malachite green fluorescence measured through a 710/75 filter in (e) dL5_EAK mixed with EAK16-II (et = 957 ms) and (f) dL5 and EAK16-II (et = 1.34 seconds).
The fraction of dL5_EAK incorporated into EAK16-II membranes was determined using SDS-PAGE (Fig. 8). Insoluble membranes were formed with EAK16-II containing either dL5_EAK or dL5 and the samples were allowed to settle in buffers (Fig 5a). Proteins that remained in solution were subjected to electrophoresis in reducing conditions. The results indicate that greater than 78% of dL5_EAK were associated with EAK16-II in the insoluble fraction (Fig. 8). Conversely, less than 32% of control dL5 remained associated with EAK16-II. No EAK16-II was detected in the soluble fraction by gel electrophoresis. dL5 migrated through the matrix to the expected size (26 kDa), while the dL5_EAK band migrated with an apparent MW closer to 40 kDa, higher than the theoretical mass of 28.4 kDa. This might due to incomplete denaturation of the protein conformation or addition of localized positive charge in lysine residues on the EAK repeat sequence on the protein. MALDI-TOF mass spectroscopy experiments were performed on dL5 and dL5_EAK to verify molecular mass and showed that dL5 is 25.6 kDa while dL5_EAK is 28.4 kDa. (Figure S1, supporting material) Taken together, these data provided evidence that dL5_EAK efficiently incorporated into EAK16-II fibrils.
Figure 8.
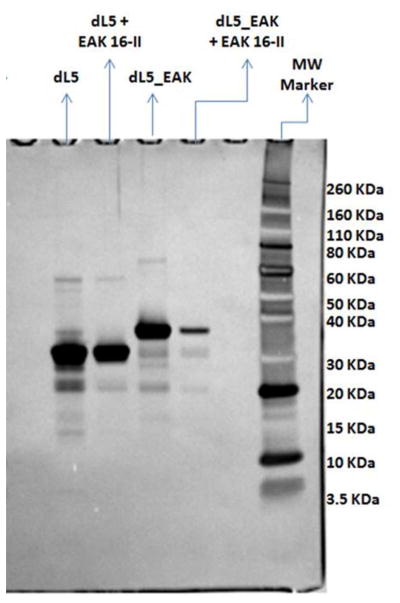
Analysis of protein incorporated into membranes using SDS-PAGE and detected by SilverQuest staining. Protein/peptide samples were set up at 1:100 ratios and incubated overnight along with protein only samples, centrifuged for 2 minutes with supernatant loaded onto the gel. Electrophoresis was conducted under reducing conditions. Band intensities were quantified using Image J. Percent incorporation of dL5 and dL5_EAK into EAK16-II membranes was determined based on the amount of proteins added to the system without EAK16-II (lanes labeled “dL5_EAK” and “dL5”). Bands other than the major protein bands are most likely due to impurities in protein preparations or degradation products brought to attention by the sensitivity of SilverQuest staining. dL5_EAK migrates at a higher Mw than expected, however, MALDI-TOF experiments have shown the mass of dL5_EAK is in fact 28.4 kDA (MALDI-TOF results in supplementary materials).
Discussion
The current study demonstrated the adaptation of a FAP-fluorogen module into a self-gelling material system. Various biosensing platforms have been explored for analyzing samples ex vivo.17 Real-time measurements of metabolic biomarkers or pharmaceuticals in vivo typically require surgical implantation. An unmet need is the option to render in situ sensing mechanisms without the need for surgery. The dL5_EAK/EAK16-II composite may be injected using conventional syringes to establish local sensing mechanisms.15 Traditional biosensors utilize silicon, metal, and glass as the solid support, which may complicate deployment in vivo due to potential inflammatory responses.18 Peptide-based materials generally have better biocompatibility; testing of EAK16-II and related sequences in rodents so far have shown no signs of acute inflammation.19 While MG itself is sometimes considered toxic, primarily due to its accumulation in mitochondria and nuclei,20 addition of a polyethylene glycol (PEG) tail prevents its diffusion through the cellular and mitochondrial membranes.6 This cell-impermanent MG derivative should mitigate the concerns for toxicities.
The PEG tail may potentially be conjugated to other molecules of interest such as antibodies or receptor ligands. MG-11p has been conjugated to biotin in initial FAP screening as previously described2, as well as to other fluorescent dyes to create FRET based fluorogens4. We envision a sensing design in which the dL5_EAK/EAK16-II membrane is established locally in vivo, capturing MG conjugated with a ligand, antibody or other biomolecular conjugate as a way to track or capture biomolecules or cells of interest. Work in the Meng group has shown that EAK16-II materials incorporating protein domains are stable for several days in animal models21. The MG fluorogen conjugated to biomolecules of interest could be used for tracking cell migration in vivo (Fig. 2). Given the diversity of FAPs discovered with their range of affinities for various fluorogens, the immobilization strategy described herein could form the basis for a variety of biomedical applications. We would like to point out that the proposed use of antibody conjugated fluorogen depicted in Fig. 2 would result in background fluorescence owing to unbound fluorogen conjugated IgG interacting with immobilized FAP. However, we expect unbound antibodies would diffuse from the site of injection therefore should render only weak fluorescence. Conversely, migrating cells bound with multiple antibodies would generate concentrated and stronger fluorescence on FAP membranes. Studies are ongoing to validate such a system in vitro and in vivo.
The present study centers on using an innovative way to immobilize sensor proteins whereby the bi-functional construct dL5_EAK forms β-strand alignment with the fibrillar EAK16-II. The data presented herein show that dL5 containing the amphiphilic sequence (AEAEAKAK)3 assembles into peptide β-sheet fibrils and the co-assembly retained the ability to enhance MG fluorescence. Incorporation of dL5_EAK was highly efficient, with close to 80% of the protein incorporated into the EAK16-II membrane. Spectroscopic analyses show that dL5_EAK enhanced MG fluorescence identical to that of dL5, suggesting that the FAP domain in the conjugate remained in its native conformation and was solvent accessible. EAK16-II membranes prepared with dL5_EAK exhibited strong fluorescence upon addition of MG, indicating the fluorescence enhancement function was retained after the immobilization.
In summary, a system of materials whereby a sensing mechanism is efficiently immobilized is described. Appending an amphiphilic domain into protein sensors may be a general strategy for establishing local fluorescence detection mechanisms. The components could be maintained in solution (low ionic strength buffer) and be delivered in vivo using conventional syringes.15 This system may be tailored to different applications by adjusting the density of the FAP, or by simultaneous immobilization of FAPs with different binding affinities.6 Conjugation of MG fluorogen to biological molecules of interest and introduction of both dL5_EAK in EAK16-II membranes with conjugated fluorogen could be used as a mechanism to capture and detect undesired or rare cell types. This will be the focus of future work using these materials.
Supplementary Material
Acknowledgments
This work was supported in part by a C.U.R.E. award from the Pennsylvania Department of Health and the Hunkele Dreaded Disease Fund (both to W.S.M) and NIH TCNP Grant U54RR022241 (A.W.). Fluorogens were synthesized by Dr. Brigitte Schmidt (Carnegie Mellon MBIC). The HRV 3C protease and modified pET21 plasmid used for protein expression were generous gifts of Dr. Joseph Franke (Carnegie Mellon). Zachery Drennen, of Washington and Jefferson College (PA), was a trainee with the Duquesne University Undergraduate Research Program supported in part by NIH R25DA032519 and NSF CHE-1126465. We thank Dr. Jennifer Aitken (Duquesne University) for assistance with scanning electron microscopy and Dr. Lauren Ernst (Carnegie Mellon) for assistance with fluorescent microscopy and manuscript proofreading. MALDI-TOF mass spectroscopy was performed by Dr. Mark Bier at the Carnegie Mellon University Center for Molecular Analysis. The Hitachi S-3000N microscope was supported in part by NSF CHE-0923183 awarded to Duquesne University.
References
- 1.Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–24. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 2.Szent-Gyorgyi C, Schmidt BF, Creeger Y, Fisher GW, Zakel KL, Adler S, Fitzpatrick JA, Woolford CA, Yan Q, Vasilev KV, Berget PB, Bruchez MP, Jarvik JW, Waggoner A. Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat Biotechnol. 2008;26:235–40. doi: 10.1038/nbt1368. [DOI] [PubMed] [Google Scholar]
- 3.Fisher GW, Adler SA, Fuhrman MH, Waggoner AS, Bruchez MP, Jarvik JW. Detection and quantification of beta2AR internalization in living cells using FAP-based biosensor technology. J Biomol Screen. 2010;15:703–9. doi: 10.1177/1087057110370892. [DOI] [PubMed] [Google Scholar]
- 4.Szent-Gyorgyi C, Schmidt BF, Fitzpatrick JA, Bruchez MP. Fluorogenic dendrons with multiple donor chromophores as bright genetically targeted and activated probes. J Am Chem Soc. 2010;132:11103–9. doi: 10.1021/ja9099328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Senutovitch N, Stanfield RL, Bhattacharyya S, Rule GS, Wilson IA, Armitage BA, Waggoner AS, Berget PB. A variable light domain fluorogen activating protein homodimerizes to activate dimethylindole red. Biochemistry. 2012;51:2471–85. doi: 10.1021/bi201422g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saunders MJ, Szent-Gyorgyi C, Fisher GW, Jarvik JW, Bruchez MP, Waggoner AS. Fluorogen activating proteins in flow cytometry for the study of surface molecules and receptors. Methods. 2012;57:308–17. doi: 10.1016/j.ymeth.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zanotti KJ, Silva GL, Creeger Y, Robertson KL, Waggoner AS, Berget PB, Armitage BA. Blue fluorescent dye-protein complexes based on fluorogenic cyanine dyes and single chain antibody fragments. Org Biomol Chem. 2011;9:1012–20. doi: 10.1039/c0ob00444h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ozhalici-Unal H, Pow CL, Marks SA, Jesper LD, Silva GL, Shank NI, Jones EW, Burnette JM, 3rd, Berget PB, Armitage BA. A rainbow of fluoromodules: a promiscuous scFv protein binds to and activates a diverse set of fluorogenic cyanine dyes. J Am Chem Soc. 2008;130:12620–1. doi: 10.1021/ja805042p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leckband DE. The influence of protein and interfacial structure on the self-assembly of oriented protein arrays. Adv Biophys. 1997;34:173–90. doi: 10.1016/s0065-227x(97)89639-6. [DOI] [PubMed] [Google Scholar]
- 10.Hoffman AS, Stayton PS. Conjugates of stimuli-responsive polymers and proteins. Progress in Polymer Science (Oxford) 2007;32:922–932. [Google Scholar]
- 11.Williams DF. On the nature of biomaterials. Biomaterials. 2009;30:5897–5909. doi: 10.1016/j.biomaterials.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 12.Zhang S, Marini DM, Hwang W, Santoso S. Design of nanostructured biological materials through self-assembly of peptides and proteins. Curr Opin Chem Biol. 2002;6:865–71. doi: 10.1016/s1367-5931(02)00391-5. [DOI] [PubMed] [Google Scholar]
- 13.Zhang S, Lockshin C, Cook R, Rich A. Unusually stable beta-sheet formation in an ionic self-complementary oligopeptide. Biopolymers. 1994;34:663–72. doi: 10.1002/bip.360340508. [DOI] [PubMed] [Google Scholar]
- 14.Zhang S, Holmes T, Lockshin C, Rich A. Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane. Proc Natl Acad Sci U S A. 1993;90:3334–8. doi: 10.1073/pnas.90.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng Y, Wen Y, George AM, Steinbach AM, Phillips BE, Giannoukakis N, Gawalt ES, Meng WS. A peptide-based material platform for displaying antibodies to engage T cells. Biomaterials. 2011;32:249–57. doi: 10.1016/j.biomaterials.2010.08.083. [DOI] [PubMed] [Google Scholar]
- 16.Klunk WE, Pettegrew J, Abraham DJ. Quantitative evaluation of congo red binding to amyloid-like proteins with a beta-pleated sheet conformation. Journal of Histochemistry & Cytochemistry. 1989;37:1273. doi: 10.1177/37.8.2666510. [DOI] [PubMed] [Google Scholar]
- 17.Myszka DG. Improving biosensor analysis. Journal of molecular recognition. 1999;12:279–284. doi: 10.1002/(SICI)1099-1352(199909/10)12:5<279::AID-JMR473>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 18.Zahn T, Braunbeck T. Cytotoxic Effects of Sublethal Concentrations of Malachite Green in Isolated Hepatocytes from Rainbow-Trout (Oncorhynchus-Mykiss) Toxicol in Vitro. 1995;9:729–741. doi: 10.1016/0887-2333(95)00056-e. [DOI] [PubMed] [Google Scholar]
- 19.Holmes TC, de Lacalle S, Su X, Liu G, Rich A, Zhang S. Extensive neurite outgrowth and active synapse formation on self-assembling peptide scaffolds. Proc Natl Acad Sci U S A. 2000;97:6728–33. doi: 10.1073/pnas.97.12.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zahn T, Braunbeck T. Cytotoxic effects of sublethal concentrations of malachite green in isolated hepatocytes from rainbow trout (< i> Oncorhynchus mykiss</i>) Toxicology in vitro. 1995;9:729–741. doi: 10.1016/0887-2333(95)00056-e. [DOI] [PubMed] [Google Scholar]
- 21.Wen Y, Kolonich HR, Kruszewski KM, Giannoukakis N, Gawalt ES, Meng WS. Retaining Antibodies in Tumors with a Self-Assembling Injectable System. Mol Pharmaceutics. 2013;10:1035–1044. doi: 10.1021/mp300504z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.