

Abstract
Background
The activity of transposable elements can be regulated by different means. DNA CpG methylation is known to decrease or inhibit transpositional activity of diverse transposons. However, very surprisingly, it was previously shown that CpG methylation of the Sleeping Beauty (SB) transposon significantly enhanced transposition in mouse embryonic stem cells.
Results
In order to investigate the unexpected response of SB transposition to CpG methylation, related transposons from the Tc1/mariner superfamily, that is, Tc1, Himar1, Hsmar1, Frog Prince (FP) and Minos were tested to see how transposition was affected by CpG methylation. A significant increase of >20-fold in transposition of SB, FP and Minos was seen, whereas Tc1, Himar1 and Hsmar1 showed no difference in transposition upon CpG-methylation. The terminal inverted repeats (TIRs) of the SB, FP and Minos elements share a common structure, in which each TIR contains two functionally important binding sites for the transposase (termed the IR/DR structure). The group of IR/DR elements showed increased excision after CpG methylation compared to untreated transposon donor plasmids. We found that de novo CpG methylation is not required for transposition. A mutated FP donor plasmid with depleted CpG sites in both TIRs was as efficient in transposition as the wild-type transposon, indicating that CpG sites inside the TIRs are not responsible for altered binding of factors potentially modulating transposition. By using an in vivo one-hybrid DNA-binding assay in cultured human cells we found that CpG methylation had no appreciable effect on the affinity of SB transposase to its binding sites. However, chromatin immunoprecipitation indicated that CpG-methylated transposon donor plasmids are associated with a condensed chromatin structure characterized by trimethylated histone H3K9. Finally, DNA compaction by protamine was found to enhance SB transposition.
Conclusions
We have shown that DNA CpG methylation upregulates transposition of IR/DR elements in the Tc1/mariner superfamily. CpG methylation provokes the formation of a tight chromatin structure at the transposon DNA, likely aiding the formation of a catalytically active complex by facilitating synapsis of sites bound by the transposase.
Background
Co-evolution of transposable elements (TEs) with their host species gave rise to several mechanisms that regulate the transposition reaction [1], for example, cell or tissue type [2,3], cell-cycle timing [4], transcriptional regulation [5-7], posttranscriptional regulation by small RNAs [8-10], regulation of the transposase protein [11], interactions with DNA repair [12], and target site selectivity. The activity of TEs can be regulated by chromatin at different stages of the transpositional reaction. For example, the formation of a catalytically active synaptic complex requires expression of the transposase and a DNA topology that makes the element accessible for the protein machinery required for catalysis. The eukaryotic genome is typically organized into either of two types of chromatin: euchromatin, a relatively relaxed chromatin structure, in which the DNA is packed less tightly and heterochromatin, a more inaccessible and highly condensed fraction of the genome. Heterochromatic regions carry characteristic features, which distinguish them from euchromatic DNA, such as dense cytosine-methylation (5-Me-C) of CpG sites, hypo-acetylation of lysine residues in the N-terminal tails of histones H3 and H4 and methylation of specific lysine residues such as lysine 9 in histone H3. In contrast to euchromatin, which is largely composed of unique (protein coding) sequences, the DNA sequence of heterochromatin is usually repetitive and gene poor [13]. One class of repetitive sequences found in heterochromatic regions of different genomes are TEs, and therefore it is believed that the accumulation of transposable DNA sequences in heterochromatic regions provides a safe place, where the deleterious potential of these elements can be kept on a leash [14]. Indeed, there is a strong correlation between chromatin structure and the activity of TEs. For example, recruiting transposable DNAs into heterochromatic regions may provide efficient silencing of transcription of element-encoded proteins, and thus provides genome stability. In addition to its repressive function on transcription, heterochromatin also exerts a repressive influence on recombination [15,16]; hence, the containment of repeated sequences in heterochromatic regions may prevent irregular recombination and genome instability.
CpG methylation is known to decrease or inhibit transpositional activity of diverse transposons. However, very surprisingly, Yusa et al. showed that CpG methylation of the Sleeping Beauty (SB) transposon and to a smaller extent the Tc3 element of Caenorhabditis elegans produced elevated transpositional activity in mouse embryonic stem (ES) cells [17]. Chromatin immunoprecipitation experiments revealed that hyperactive genomic donor sites have the characteristics of a heterochromatic structure. The SB transposase was found to co-localize with heterochromatin protein 1 (HP1), a well-established marker for heterochromatin, suggesting that the transposase preferentially associates with heterochromatic DNA [18]. Based on these results, it was postulated that heterochromatin formation at the transposon donor site can upregulate SB transposition [17].
We addressed the question whether transposition of other Tc1/mariner elements is also enhanced by CpG methylation. The elements of the Tc1/mariner superfamily can be subdivided into two groups based on the size and structure of their terminal inverted repeats (TIRs) (Figure 1) [19]. The first group is characterized by short TIRs (approximately 25 bp to 60 bp) containing a single transposase binding site (TBS) per TIR, whereas the second group has longer TIRs (>200 bp) harboring two TBSs in the same orientation, which are thus referred to as direct repeats (DRs). This arrangement is called the IR/DR structure [19]. Here we extend the original observations on SB transposition by examining the effects of CpG methylation on transposition of other Tc1/mariner elements. Transposition of six different transposons: SB, Frog Prince (FP) [20], Minos[21], Tc1[22], Himar1[23] and Hsmar1[24] was analyzed. We found that the enhancing effect of CpG methylation is not restricted to SB, but does not universally apply within the Tc1/mariner family either. A shared feature of SB, FP and Minos transposon TIRs is the IR/DR structure. We propose a model where formation of a condensed chromatin structure at transposon sequences is responsible for the observed increase in transposition.
Figure 1.

Structure of Tc1/mariner transposons. The transposons can be grouped by their different TIR structure. IR/DR TEs have long TIRs with two transposase binding sites (TBSs) per TIR in the same orientation (direct repeats). Simple-structured TEs have short TIRs with only one TBS. Frog Prince has 26 bp-long DRs, but the TBSs are only 21 bp. DR: direct repeat; ssTIR: simple-structured TIR; TBS: transposase binding site; TE: transposable element; TIR: terminal inverted repeat; IR/DR: inverted repeat/direct repeat.
Results
Transpositional activities of different Tc1/mariner elements upon CpG methylation
It was previously found that SB transposition is enhanced by CpG methylation [17]. In order to extend these observations to other Tc1/mariner transposons, the effect of CpG methylation on Tc1 and the Tc1-like elements FP and Minos, and the mariner-like elements Himar1 and Hsmar1 was investigated. We generated transposon donor plasmids containing the respective TIRs flanking a neomycin-resistance gene trap cassette cloned into identical plasmid backbones. Because CpG methylation would silence a transgene promoter (thereby compromising the selection of antibiotic-resistant cell colonies upon transposition), we utilized a splice acceptor site upstream of the neomycin-resistance gene, so that expression of the selectable marker is dependent on transposition into an endogenous, expressed gene. The donor plasmids were CpG-methylated in vitro, and methylated or untreated donor plasmids were co-transfected with the respective transposase-expressing helper plasmids into cultured HeLa cells and selected with G418. Confirming previous findings [17], transposition of SB was enhanced >tenfold upon CpG methylation (Figure 2). Varying degrees of enhancement of transposition were also seen for the FP and Minos elements; the greatest increase in transposition, >30-fold, was seen for the FP element (Figure 2). In contrast, transposition of Tc1, Himar1 and Hsmar1 was not or only weakly affected by CpG methylation (Figure 2). These data indicate that Tc1 family transposons of the IR/DR structure group are responsive to CpG methylation.
Figure 2.

Transposition activity of Tc1/mariner elements upon CpG methylation. CpG-methylated (black bars) or untreated (grey) Tc1-, Himar1-, Hsmar1-, Minos-, SB-, FP-gene trap donor plasmids were co-transfected with/without transposase into HeLa cells and selected with G418. The transposition ratio was calculated by dividing the numbers of G418-resistant colonies obtained in the presence of transposase by the number of colonies obtained in the absence of transposase. Error bars are the standard deviation; the asterisk indicates that the error bar cannot be displayed for FP (±38.6). FP: Frog Prince; SB: Sleeping Beauty.
CpG methylation does not affect binding of the Sleeping Beauty transposase to transposon inverted repeats in vivo
Transposition is a multistep process consisting of: (i) binding the transposase to the TIRs, (ii) synaptic complex formation, (iii) transposon excision, (iv) target capture and (v) transposon integration. Each of the studied IR/DR TEs contains at least one CpG site in the TIRs (SB: 2, FP: 1, Minos: 11). In order to address whether CpG methylation affects binding of the SB transposase to its binding sites in the TIRs, we applied a one-hybrid assay [25] to cultured cells. The reporter plasmid contained a luciferase gene behind a minimal promoter and four SB transposase binding sites upstream of the promoter. Untreated or CpG-methylated reporter plasmids were co-transfected with (i) the transcriptional activator plasmid expressing N123-AD containing the N-terminal DNA-binding domain of the SB transposase fused to a VP16-transactivation domain that induces luciferase expression upon TBS binding and (ii) a plasmid expressing full-length SB transposase that binds TBS, but does not transactivate luciferase expression, and thus acts as a competitor of N123-AD. The activator induced luciferase expression >350-fold, and co-expression of the competitor reduced luciferase expression up to tenfold for both untreated and CpG-methylated reporters (Additional file 1: Table S1). CpG methylation of the reporter plasmid reduced luciferase expression, presumably by promoter silencing (Figure 3). However, the extent of reduction was always similar in the presence or absence of the activator and the competitor (Figure 3). We conclude that the SB transposase does not have either reduced or enhanced affinity for CpG-methylated DNA.
Figure 3.
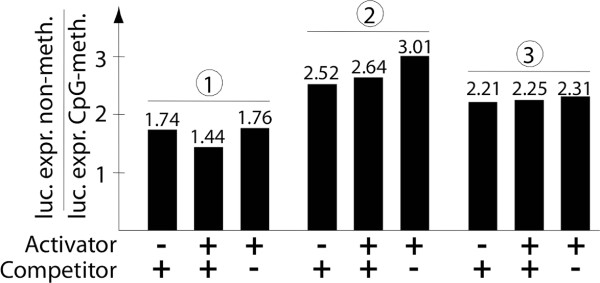
In vivo binding of SB transposase. Untreated or CpG-methylated p5SB-luc reporter plasmids were co-transfected with/without the activator plasmid pc-SB(123)-AD and with/without a full-length SB transposase competitor into HeLa cells. After 48 h luciferase expression was determined. To compare the effect of CpG methylation, ratios were calculated by dividing the value for luciferase expression with untreated donor plasmids by the value for luciferase expression with CpG-methylated donors. Three independent experiments are shown. No statistically significant differences were obtained.
Methylation of CpG sites in transposon inverted repeats is not required for enhanced transposition
Methylation of CpG sites in the transposon TIRs may affect the affinity of DNA-binding proteins [26], for example, it might increase binding of an enhancer or reduce binding of an inhibitor of transposition, thereby enhancing transposition. Both SB and Minos contain CpG sites in their TBSs and, therefore, mutagenesis of the TBS sequence is expected to drastically affect or abolish transposase binding. However, FP has only one CpG site in its TIRs outside the TBSs (plus 435 additional CpG sites distributed all across the rest of the plasmid). We mutated the CpG site at both TIRs of FP to a CpA sequence that cannot be methylated, and compared the transpositional activity of the mutant transposon to the wild-type transposon in the absence and presence of CpG methylation. No differences in transposition were detected either for untreated or for CpG-methylated donor plasmids (Figure 4). We conclude that it is not a particular CpG site inside the TIRs, but rather the global CpG content of the transposon plasmid that is responsible for the observed enhancing effect of CpG methylation.
Figure 4.
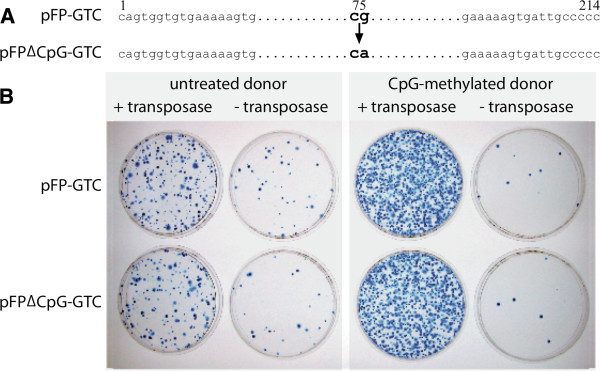
Transposition of CpG-free FP TIRs. (A) CpG sites inside the FP TIRs were mutated to obtain pFPΔCpG-GTC. (B) Transposition assay of CpG-methylated or untreated pFPΔCpG-GTC and pFP-GTC was performed in HeLa cells in the presence or absence of transposase. FP: Frog Prince; TIR: terminal inverted repeat.
CpG methylation enhances Sleeping Beauty transposon excision in vivo
In order to identify the step(s) at which CpG methylation influences transposition, we subjected the six elements to a PCR-based excision assay. CpG-methylated or untreated donor plasmids were co-transfected with the respective helper plasmids into cultured HeLa cells and were re-isolated 48 h post-transfection. Amplification of transposon excision sites of the donor plasmids via nested PCR produces a 308-bp-long amplicon, whose band intensity in gel electrophoresis was used as measure of transposon excision. Consistent with the effect on transposition, excision of IR/DR-elements SB, FP and Minos was enhanced by CpG methylation (Figure 5A). By serial dilution of PCR input DNA, we quantified the increase of excision efficiency for FP, which is enhanced at least 16-fold by CpG methylation. This is in accordance with the 20-fold increase in overall transposition upon CpG methylation obtained in the colony-based assay. Again, the group of simple-structured TIR (ssTIR) elements showed no increase in excision activity; in contrast, excision was significantly lower or even undetectable following CpG methylation. Thus, the enhancing effect of CpG methylation on transposon excision is limited to the elements of the IR/DR group.
Figure 5.
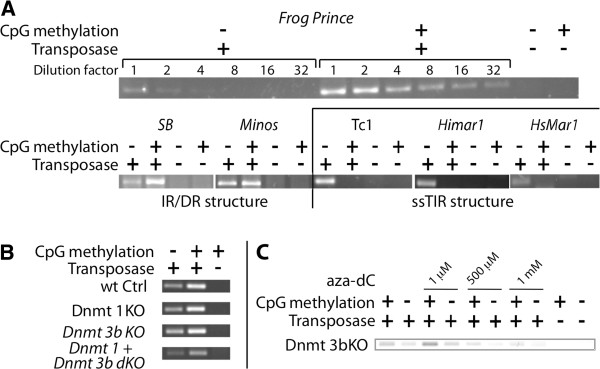
Excision activity upon CpG methylation. CpG-methylated or untreated donor plasmids were co-transfected with/without a helper plasmid; 48 h post-transfection plasmids were extracted, purified and DNA amounts were normalized. After two rounds of nested PCR, the product band intensity represents the relative efficiency of transposon excision. (A) Excision activity of different TEs in HeLa cells. Upper panel: Dilutions of PCR input DNA for quantification of FP excision. (B) Excision of SB in different HCT116 knockout cell lines. (C) SB excision in HCT116 Dnmt3b knockout cells. Increasing amounts of aza-deoxycytidine (aza-dC) block de novo CpG methylation. aza-dC: aza-deoxycytidine; FP: Frog Prince; SB: Sleeping Beauty; PCR: polymerase chain reaction; TE: transposable element.
Transiently transfected plasmids can become CpG-methylated in the cell de novo[27]. CpG methylation in mammals is catalyzed by three DNA-methyltransferases (Dnmts): Dnmt1 is responsible for the maintenance of CpG methylation patterns, and Dnmt3a and Dnmt3b perform de novo CpG methylation. We tested the excision activity of untreated and CpG-methylated SB transposon donors in HCT116 knockout/knockdown cell lines lacking either Dnmt1 or Dnmt3b or both [28,29] by PCR-based excision assay (Figure 5B). All cell lines supported transposon excision. However, in these cells Dnmt3a is still active and could provide de novo CpG methylation of transfected donor plasmids. Thus, to inhibit the activity of Dnmt3a, we used the chemical aza-deoxycytidine, which is a known inhibitor of cellular de novo CpG methylation. Aza-deoxycytidine was added to cultured, HCT116-derived Dnmt3b knockout cells in a range of 1 μM to 1 mM two days prior to co-transfection of the transposon plasmids. The PCR-based excision assay (Figure 5C) showed that transposon excision is still detectable at high concentrations of aza-deoxycytidine. Even though PCR band intensity became weaker at increasing aza-deoxycytidine concentrations, this is likely due to the cytotoxic effects of aza-deoxycytidine, which is accompanied by reduced cell survival and cell growth. The enhancing effect of in vitro CpG methylation was not affected by aza-deoxycytidine treatment. We conclude that host-mediated de novo CpG methylation is not required for transposition.
CpG-methylated transposon plasmids are associated with condensed chromatin
As CpG methylation did not seem to have a direct influence on transposition, we hypothesized that it affects the transposition process indirectly, by inducing heterochromatin formation and persistence. Indeed, genomic, CpG-methylated transposons were shown to be associated with heterochromatin [17]. We utilized a chromatin immunoprecipitation (ChIP) assay to investigate chromatin structure on transposon donor plasmids. CpG-methylated and non-methylated pFP-GTC donor plasmids were transfected into HeLa cells, cross-linked and immunoprecipitated using antibodies against acetylated histone H3 (a marker for open chromatin) or against trimethylated H3K9 (a marker for condensed heterochromatin). The co-precipitated DNA was purified and analyzed by (i) semi-quantitative PCR and (ii) transformation of recovered donor plasmids into Escherichia coli and subsequent colony counting (Figure 6).
Figure 6.

CpG-methylated transposon plasmids are associated with condensed chromatin. CpG-methylated or untreated pFP-GTC donor plasmids were transfected into HeLa cells, cross-linked, and precipitated with anti-acetylated histone H3 (anti-AcH3) antibodies indicating euchromatin or anti-trimethylated histone H3 lysine 9 (anti-H3triMeK9) antibodies indicating heterochromatin. DNA was purified and quantified by E. coli transformation (A) or by semi-quantitative PCR after serial dilutions of input DNA (B). anti-AcH3: anti-acetylated histone H3; anti-H3triMeK9: anti-trimethylated histone H3 lysine 9; PCR: polymerase chain reaction.
As quantified by bacterial transformation, anti-acetylated histone H3 (anti-AcH3) antibodies precipitated threefold less CpG-methylated transposon plasmids than non-methylated transposon plasmids, suggesting an enrichment of non-methylated plasmids in open chromatin (Figure 6A). Conversely, immunoprecipitation with anti-trimethylated histone H3 lysine 9 (anti-H3triMeK9) antibodies resulted in reduced recovery of non-methylated plasmids compared to CpG-methylated plasmids, implying an enrichment of condensed chromatin status for CpG-methylated plasmids. PCR analyses of the immunoprecipitated DNA samples showed, in accordance with the results obtained by bacterial transformation, a relative enrichment of non-methylated plasmids in the euchromatic fraction (precipitation with anti-AcH3 antibody) and a relative enrichment of methylated plasmids in the heterochromatic fraction (precipitation with anti-H3triMeK9 antibody) (Figure 6B). We conclude that CpG-methylated plasmids are associated with condensed chromatin.
Precomplexing transposon DNA with protamine enhances Sleeping Beauty transposition
To test if DNA condensation is an underlying structural determinant of enhanced transposition in our experiments, we sought to examine the effect of condensation introduced into transposon DNA by means other than CpG methylation. We opted to package and condense transposon donor plasmids using protamine. Protamines are small, basic, arginine-rich peptides that largely replace histones in sperm chromatin, and package DNA into the most condensed eukaryotic DNA known [30].
Antibiotic-resistance-gene-containing SB transposon donor plasmids were pre-incubated with protamine, and transfected into human HeLa cells expressing SB transposase, followed by antibiotic selection and counting of resistant cell colonies. Pre-incubation of SB transposons with protamine resulted in an approximately threefold enhancement of transposition compared to uncondensed plasmids (Figure 7). The enhancing effect of protamine on stable gene transfer was only manifested with transposition-competent vectors; treatment of transpositionally incompetent vectors with protamine apparently had no effect on colony numbers (Figure 7). We conclude that condensation of transposon DNA has an enhancing effect on SB transposition.
Figure 7.

Protamine enhances SB transposition. Transpositionally competent as well as incompetent (negative control) SB transposon plasmids were precomplexed with protamine in vitro, followed by transfection into transgenic HeLa cells stably expressing the SB transposase. The transfected cells have undergone antibiotic selection to allow a quantitative measurement of transposition efficiencies represented by antibiotic-resistant colony numbers. SB: Sleeping Beauty.
Discussion
The original finding that CpG methylation enhances Sleeping Beauty transposition [17] was very surprising, as CpG methylation has been known to play a crucial role in the cellular defense against TEs. We now extend these observations to other members of the Tc1/mariner superfamily, and show that this phenomenon is not restricted to SB, but seems to be an intrinsic feature associated with the characteristic IR/DR structure of the SB, Frog Prince and Minos elements (Figure 2).
We tested several hypotheses that provided possible explanations for the enhancing effect of CpG methylation on transposition. It is formally possible that CpG methylation is a prerequisite for transposition of SB, FP and Minos. In this case donor plasmids that are CpG-methylated in vitro would have a head start against non-methylated donors, which would need to be methylated by cellular factors following transfection. We tested transposition of non-methylated donor plasmids in Dnmt1 and/or Dnmt3b knockout/knockdown cell lines and in the presence of aza-deoxycytidine, which blocks de novo CpG methylation. Transposition was as efficient under these conditions as in wild-type or untreated cells (Figure 5), thus we conclude that CpG methylation is not required for transposition.
An alternative hypothesis predicts that one (or several) particular CpG sites inside the TIRs might affect CpG methylation. An enhancer of transposition might possibly be attracted by methylated CpG sites or, alternatively, a potential repressor might be distracted. However, a mutated FP donor plasmid with depleted CpG sites in both TIRs was as efficient in transposition as the wild-type transposon (Figure 4). Thus, CpG sites inside the TIRs do not seem to be responsible for the binding of any transposition-enhancing factor or the blocking of any transposition repressor.
Binding of the SB, FP and Minos transposases might directly be supported by CpG sites inside the TBSs, the TIRs or close to them. Alternatively, CpG methylation could increase transposition indirectly; for example by the formation of heterochromatin following binding of methylated CpG sites by the HP1 or MeCP2 (a protein that binds specifically to methylated DNA) proteins [31,32]. Yusa et al.[17] tested SB transposase binding to TBSs in vitro and did not detect any influence from CpG methylation. In line with these previous observations, the expression of luciferase as a reporter for TBS binding in an in vivo one-hybrid assay was affected by CpG methylation to the same extent in all background controls, binding experiments and under competition conditions (Figure 3). We thus conclude that CpG methylation had neither a direct nor an indirect effect on SB transposase binding.
Our findings suggest an indirect effect of CpG methylation on transposition rather than a direct influence. Because (i) enhancement of CpG methylation is detectable at the transposon excision step (Figure 5), (ii) CpG methylation was found to induce the formation of a condensed chromatin structure (heterochromatin) (Figure 6), and (iii) DNA compaction by protamine was found to enhance transposition (Figure 7), we propose a model, in which CpG methylation and subsequent chromatin condensation aids synaptic complex formation. Indeed, transposition critically depends on the formation of a synaptic complex that is built up of the transposon TIRs, transposase proteins and additional, host-encoded proteins. It is formally possible that CpG methylation-induced chromatin condensation brings the TIRs closer together, thereby promoting synaptic complex assembly. However, our studies on several, closely related Tc1/mariner-elements revealed the selective importance of TIR structure for the observed sensitivity for CpG methylation. Namely, only the SB, FP and Minos elements were responsive to CpG methylation; these IR/DR elements share a common structure of two TBSs per TIR (Figure 1). Heterochromatin formation results in tight packaging of DNA and histones. As a result, DNA sites that are usually far away from each other; for example, the two TBSs inside one TIR, might be brought closer together (Figure 8). The physical proximity of the inner and outer TBSs might assist the formation of transposase dimers as soon as they bind, thereby facilitating the formation of a catalytically active synaptic complex.
Figure 8.

A model of a molecular mechanism of enhanced synaptic complex assembly by condensed DNA packed into heterochromatin. CpG methylation induces heterochromatin condensation whereby the TBSs inside the TIRs will come into close proximity. Hence, the formation of transposase dimers is improved, which subsequently enhances the formation of synaptic complexes. TBS: transposase binding site; TIR: terminal inverted repeat.
Conclusions
CpG methylation/heterochromatin is one of the regulatory mechanisms that silence and inhibit TE activity. The potential of TEs to escape a regulatory mechanism imposed by the host is a strong evolutionary advantage (at least for the transposon). Assuming that the transposase source is provided by a transcriptionally active element located in euchromatin, host-cell induced CpG methylation/heterochromatin-based silencing of TEs can be offset by higher transposition efficiency out of condensed chromatin, thereby constituting a potential mechanism for SB and other, similar-structured transposons to escape CpG methylation-mediated silencing.
Methods
Plasmid construction
A 200 bp-minivariant of Himar1 (GenBank #U11644) was amplified out of pMM2611 [33] via PCR using the primer Himar1/IR (TAACAGGTTGGCTGATAAGTCCCC). A 1.6 kb fragment of Tc1 (GenBank #X01005) was amplified out of pTc1_Ex/01 with primer Tc1/IR (TACAGTGCTGGCCAAAAAGATATCC). The PCR products were purified, phosphorylated and ligated into a SmaI digested, dephosphorylated vector pUC19. After transformation in E. coli DH10B cells, plasmids were isolated, test digested and sequenced. pUC19-Himar1 was SmaI digested, pUC19-Tc1 was StyI digested; both were dephosphorylated and ligated with the Klenow-treated gene trap cassette (GTC) (HindIII-NotI-XmnI digest of plasmid Δ170_CMV_zeo#1) [20]. After transformation in E. coli DH10B, plasmids were prepared and control digested to verify the expected plasmid layout. For Minos (GenBank #X61695) the plasmid pMiLRneo[34] was HindIII-NotI digested and ligated with the formerly described HindIII-NotI fragment of the GTC. The TIRs harboring the GTC were amplified out using PCR primer Minos/IR (TACGAGCCCCAACCACTATTAATTC). The PCR product was purified and ligated into SmaI digested pUC19. The product was checked by test digestion and sequencing. pSB-GTC (= GT/neo_CMV/zeo #2), derived from pT/neo [35], pFP-GTC (= pFP/GT-neo) [20] and pHsMar1-GTC and pHsmar1-GTCrev [24] were provided by C Miskey. The plasmids containing the transposases have been previously published: Tc1: pCMV/Tc1 [36], Himar1: pCMV/Himar3x [36], pSB10: [35], Minos: pJGD/ILMi [37], FP: pFV-FP [20], Hsmar1: pHsmar1[24]. pFPΔCpG-GTC: FP TIRs on plasmid pFP-GTC were mutated using the Stratagene QuikChange Multi Site Directed Mutagenesis Kit following the manufacturer’s instructions with phosphorylated primers FP/IR_CpG_delete_syn (P-TGTTTGTCACACTTAA-GTGTTTCAGAACATCAAA-CCAATTTAAACAATAG) and FP/IR_CpG_delete_anti (P-CTATTGTTTAAATTGGTTTGATGTTCTGAAACA-CTTAAGTGTGACAAACA). The mutated plasmid pFPΔCpG-GTC was transformed in QuikChange XL1-Blue Supercompetent Cells (Stratagene), purified and sequenced.
In vitro CpG methylation
Donor plasmids were CpG-methylated by SssI CpG methylase (NEB), and purified with the Qiagen PCR purification kit. Complete methylation was tested by control digests using methylation-sensitive restriction enzymes NotI (pMiLRgeo) or SalI (all others), and compared to a control digest of the respective untreated donor plasmids.
Cell culture, transfections and DNA condensation by protamine
HeLa cells were cultured in DMEM with 4.5 g/L glucose and 110 mg/L pyruvate supplemented with 10% fetal calf serum (FCS) and an antibiotic/antimycotic cocktail (DMEM+/+) at 37°C. Cells were passaged via trypsinization with 1:5 dilution of 0.5% trypsin in 5.3 mM ethylenediaminetetraacetic acid (EDTA). Transfections were done at 60% to 80% cell confluency in DMEM without antibiotics (DMEM+/−) with the Fugene6 transfection reagent (Roche). For transposition assays, 6 × 105 cells were transfected with CpG-methylated or untreated donor plasmids (500 ng) together with transposase expression plasmids (50 ng). Complexing with protamine sulfate was done as described previously [38] by pre-incubating 500 ng plasmid DNA with 1 μg protamine sulfate (Sigma-Aldrich) for 15 min at room temperature, followed by the addition of Fugene6 as described above.
Excision PCR
Cells (6 × 105) were transfected with equal amounts of CpG-methylated or untreated donor plasmids plus equal amounts of transposase-carrying helper or control plasmids. Typically 250 ng or 500 ng of donor plasmid was used and 50 ng of helper plasmid. Two days after transfection, the cells were harvested, plasmid DNA was prepared with the Qiagen Miniprep kit in a volume of 50 μL. A 1:10 dilution of 1 μL of the extracted DNA served as template for input normalization PCR utilizing primers Amp-For (TGCACGAGTGGGTTACATCGAACT) and Amp-Rev (TTGTTGCCATTGCTACAGGCATCG). Normalized DNA amounts served as templates for nested PCR utilizing pUC2 (GCGAAAGGGGGATGTGCTGCAAGG) and pUC5 (TCTTTCCTGCGTTATCCCCTGATTC) in the first, and pUC19-3F (GTTTTCCCAGTCACGACGTT) and pUC19-3R (TGTGGAATTGTGAGCGGATA) in the second PCR. PCR protocol: 95°C 3 min, 30 cycles of 95°C 30 s, 58°C 20 s, 64°C 10 s, and 72°C 2 min. The PCR products were subjected to gel electrophoresis.
Chemical block of CpG methylation
Two days prior to transfection cells were treated with different amounts of 5-aza-2′-deoxycytidine (aza-dC), ranging from 1 μM to 1 mM. Aza-dC addition was repeated daily after previous medium exchange. Then, 6 h before transfection, aza-dC was removed from the cells and added again 6 h post-transfection.
One-hybrid assay
HeLa cells (6 × 105) were transfected with 300 ng CpG-methylated or untreated p5SB-luc reporter plasmid, 90 ng pc-SB(123)-AD activator plasmid [25] and 1.1 μg pCMV-SB10 competitor plasmid (or filled with adequate control plasmids). Two days later, transfected cells were washed, treated with CCLR buffer (5× buffer: 125 mM Tris*H3PO4 pH 7.8, 10 mM dithiothreitol (DTT), 10 mM 1,2 cyclohexane-diaminetetra-acetic acid (CDTA), 50% glycerol, 5% Triton X-100), incubated for 15 min on ice and vortexed for 15 s. Cell debris was pelleted for 2 min with 12,000g at 4°C. 30 μL of the supernatant was combined with 100 μL luciferase buffer (20 mM tricine-NaOH pH7.8, 1.07 mM Mg(CO3)4 Mg(OH)2*5H2O, 2.67 mM MgSO4, 0.1 mM EDTA, 470 μM luciferin, 530 μM ATP, 33.3 mM DTT), vortexed briefly and measured in the luminometer.
Chromatin immunoprecipitation
Cells (4×106) were transfected in either four 5-cm or one 10-cm culture dish with approximately 3 μg CpG-methylated or non-methylated pFP-GTC donor plasmid. 24 h after incubation, 37% formaldehyde was added to the medium to an end concentration of 1%. The culture dishes were put into a plastic bag, sealed and incubated for 10 min at 37°C. After removal of the medium and two washing steps with ice-cold PBS containing protease inhibitors (1 mM PMSF, 1 μg/mL aprotinin, 1 μg/mL pepstatin A), the cells were scraped, transferred into 1.5 mL Eppendorf tubes and centrifuged for 4 min at 4°C at 2,000g. The pellet was resuspended in 400 μL lysis buffer [1% sodium dodecyl sulfate (SDS), 10 mM EDTA, 50 mM Tris–HCl pH 8.1, protease inhibitors]. Each sample was split into 2× 200 μL samples, incubated for 10 min on ice and centrifuged for 10 min at 4°C at 13,000 rpm. Supernatants were transferred to fresh 2 ml Eppendorf tubes and 1,800 μL ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris–HCl pH 8.1, 167 mM NaCl, protease inhibitors) and 80 μL salmon sperm DNA/Protein A agarose-50% slurry (0.2 mg/mL sonicated salmon sperm DNA, 0.5 mg/mL BSA, approximately 1.5 mg/mL recombinant Protein A, 0.05% sodium azide) were added. The Eppendorf tubes were incubated for 30 min on a rotating plate at 4°C. The agarose was pelleted using brief centrifugation and the supernatants were transferred to new tubes. Polyclonal antibodies anti-acetylated histone H3 (anti-AcH3) (provided with the ChIP Kit, Upstate #06-599, Lot 27610) or anti-trimethylated histone H3 lysine 9 (anti-H3triMeK9) (Abcam #ab8898) were added in varying amounts (4 μL to 10 μL) and incubated overnight at 4°C on a rotating plate. The next day, 60 μL of salmon sperm DNA/Protein A agarose-50% slurry was added and incubated for 1 h at 4°C with rotation. The agarose was pelleted at 900 rpm for 1 min at 4°C and washed with low salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 8.1, 150 mM NaCl), high salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 8.1, 500 mM NaCl), LiCl immune complex wash buffer [0.25 M LiCl, 1% Nonidet P-40 (NP40), 1% deoxycholate, 1 mM EDTA, pH 8.0] and twice with TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8.0). The chromatin was eluted by the repeated addition of 250 μL elution buffer (1% SDS, 0.1 M NaHCO3) to the pellet, vortexing and 15 min incubation followed by pelleting the agarose and saving the supernatant in a fresh 1.5 mL tube. To the combined (500 μL) eluates, 20 μL 5 M NaCl was added and heated for 4 h at 65°C to reverse cross-linking (samples were stored at this point overnight at −20°C). Using 10 μL 0.5 M EDTA, 20 μL 1 M Tris–HCl pH 6.5 and 2 μL proteinase K, histones and other DNA-bound proteins were digested for 1 h at 45°C. The remaining residues were extracted twice with phenol/chloroform and the DNA was precipitated with isopropanol (standard protocol). The DNA was taken up in H2O and re-purified by dialysis. For the quantification of plasmids, the extracted DNA was either transformed into electrocompetent E. coli DH10B or used in a semi-quantitative PCR (primer Amp-For and Amp-Rev). Transformed cells were plated on ampicillin/zeocin/LB (Luria-Bertani) agar and incubated overnight at 37°C. Colonies were counted the next morning.
Abbreviations
anti-AcH3: Anti-acetylated histone H3; anti-H3triMeK9: Anti-trimethylated histone H3 lysine 9; aza-dC: aza-deoxycytidine (5-aza-2′-deoxycytidine); BSA: Bovine serum albumin; CDTA: 1,2 cyclohexane-diaminetetra-acetic acid; ChIP: Chromatin immunoprecipitation; DTT: Dithiothreitol; DMEM: Dulbecco’s modified Eagle’s medium; Dnmt: DNA-methyltransferase; EDTA: Ethylenediaminetetraacetic acid; ES: Embryonic stem; FCS: Fetal calf serum; FP: Frog Prince; GTC: Gene trap cassette; HP1: Heterochromatin protein 1; LB agar: Luria-Bertani agar; NP40: Nonidet P-40; PCR: Polymerase chain reaction; SB: Sleeping Beauty; SDS: Sodium dodecyl sulfate; ssTIR: Simple-structured TIR; TBS: Transposase binding site; TE: Transposable element; TIR: Terminal inverted repeat.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TJ designed and performed all of the experiments in the laboratory of ZIv. ZIv designed and coordinated the study. ZIz participated in the design and coordination of the study. CM participated in both design and execution of some of the experiments. TJ and ZIv wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
One-hybrid assay reads. Reporter, activator and competitor plasmids were co‒transfected into HeLa cells. Cells were harvested 48 h post‒transfection, and protein extracts were used in luciferase assays. Three repetitions are shown.
Contributor Information
Tobias Jursch, Email: tobias.jursch@gmx.de.
Csaba Miskey, Email: csaba.miskey@pei.de.
Zsuzsanna Izsvák, Email: zizsvak@mdc-berlin.de.
Zoltán Ivics, Email: zoltan.ivics@pei.de.
Acknowledgements
We thank B Savakis for kindly providing Minos transposon vectors. The luciferase-based Sleeping Beauty reporter and activator plasmids were obtained from S Yant. TJ was funded by the International MDC/HU PhD Program.
References
- Walisko O, Jursch T, Izsvak Z, Ivics Z. In: Transposons and the Dynamic Genome. Lankenau D-H, editor. Berlin/Heidelberg: Springer; 2009. Transposon–host cell interactions in the regulation of Sleeping Beauty transposition; pp. 109–132. [Google Scholar]
- Dupressoir A, Heidmann T. Germ line-specific expression of intracisternal A-particle retrotransposons in transgenic mice. Mol Cell Biol. 1996;16:4495–4503. doi: 10.1128/mcb.16.8.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergun S, Buschmann C, Heukeshoven J, Dammann K, Schnieders F, Lauke H, Chalajour F, Kilic N, Stratling WH, Schumann GG. Cell type-specific expression of LINE-1 open reading frames 1 and 2 in fetal and adult human tissues. J Biol Chem. 2004;279:27753–27763. doi: 10.1074/jbc.M312985200. [DOI] [PubMed] [Google Scholar]
- Walisko O, Izsvak Z, Szabo K, Kaufman CD, Herold S, Ivics Z. Sleeping Beauty transposase modulates cell-cycle progression through interaction with Miz-1. Proc Natl Acad Sci USA. 2006;103:4062–4067. doi: 10.1073/pnas.0507683103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohe AR, Hartl DL. Autoregulation of mariner transposase activity by overproduction and dominant-negative complementation. Mol Biol Evol. 1996;13:549–555. doi: 10.1093/oxfordjournals.molbev.a025615. [DOI] [PubMed] [Google Scholar]
- Lippman Z, Martienssen R. The role of RNA interference in heterochromatic silencing. Nature. 2004;431:364–370. doi: 10.1038/nature02875. [DOI] [PubMed] [Google Scholar]
- Walisko O, Schorn A, Rolfs F, Devaraj A, Miskey C, Izsvak Z, Ivics Z. Transcriptional activities of the Sleeping Beauty transposon and shielding its genetic cargo with insulators. Mol Ther. 2008;16:359–369. doi: 10.1038/sj.mt.6300366. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Hannon GJ, Brennecke J. The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science. 2007;318:761–764. doi: 10.1126/science.1146484. [DOI] [PubMed] [Google Scholar]
- Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007;8:272–285. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- Sijen T, Plasterk RH. Transposon silencing in the Caenorhabditis elegans germ line by natural RNAi. Nature. 2003;426:310–314. doi: 10.1038/nature02107. [DOI] [PubMed] [Google Scholar]
- Sinzelle L, Kapitonov VV, Grzela DP, Jursch T, Jurka J, Izsvak Z, Ivics Z. Transposition of a reconstructed Harbinger element in human cells and functional homology with two transposon-derived cellular genes. Proc Natl Acad Sci USA. 2008;105:4715–4720. doi: 10.1073/pnas.0707746105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izsvak Z, Stuwe EE, Fiedler D, Katzer A, Jeggo PA, Ivics Z. Healing the wounds inflicted by Sleeping Beauty transposition by double-strand break repair in mammalian somatic cells. Mol Cell. 2004;13:279–290. doi: 10.1016/S1097-2765(03)00524-0. [DOI] [PubMed] [Google Scholar]
- Grewal SI, Jia S. Heterochromatin revisited. Nat Rev Genet. 2007;8:35–46. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- Bender J. Chromatin-based silencing mechanisms. Curr Opin Plant Biol. 2004;7:521–526. doi: 10.1016/j.pbi.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Clarke L, Amstutz H, Fishel B, Carbon J. Analysis of centromeric DNA in the fission yeast Schizosaccharomyces pombe. Proc Natl Acad Sci USA. 1986;83:8253–8257. doi: 10.1073/pnas.83.21.8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaseko Y, Adachi Y, Funahashi S, Niwa O, Yanagida M. Chromosome walking shows a highly homologous repetitive sequence present in all the centromere regions of fission yeast. EMBO J. 1986;5:1011–1021. doi: 10.1002/j.1460-2075.1986.tb04316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusa K, Takeda J, Horie K. Enhancement of Sleeping Beauty transposition by CpG methylation: possible role of heterochromatin formation. Mol Cell Biol. 2004;24:4004–4018. doi: 10.1128/MCB.24.9.4004-4018.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda R, Kokubu C, Yusa K, Keng VW, Horie K, Takeda J. Sleeping Beauty transposase has an affinity for heterochromatin conformation. Mol Cell Biol. 2007;27:1665–1676. doi: 10.1128/MCB.01500-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izsvak Z, Ivics Z, Hackett PB. Characterization of a Tc1-like transposable element in zebrafish (Danio rerio) Mol Gen Genet. 1995;247:312–322. doi: 10.1007/BF00293199. [DOI] [PubMed] [Google Scholar]
- Miskey C, Izsvak Z, Plasterk RH, Ivics Z. The Frog Prince: a reconstructed transposon from Rana pipiens with high transpositional activity in vertebrate cells. Nucleic Acids Res. 2003;31:6873–6881. doi: 10.1093/nar/gkg910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz G, Savakis C. Minos, a new transposable element from Drosophila hydei, is a member of the Tc1-like family of transposons. Nucleic Acids Res. 1991;19:6646. doi: 10.1093/nar/19.23.6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmons SW, Yesner L, Ruan KS, Katzenberg D. Evidence for a transposon in Caenorhabditis elegans. Cell. 1983;32:55–65. doi: 10.1016/0092-8674(83)90496-8. [DOI] [PubMed] [Google Scholar]
- Jacobson JW, Medhora MM, Hartl DL. Molecular structure of a somatically unstable transposable element in Drosophila. Proc Natl Acad Sci USA. 1986;83:8684–8688. doi: 10.1073/pnas.83.22.8684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miskey C, Papp B, Mates L, Sinzelle L, Keller H, Izsvak Z, Ivics Z. The ancient mariner sails again: transposition of the human Hsmar1 element by a reconstructed transposase and activities of the SETMAR protein on transposon ends. Mol Cell Biol. 2007;27:4589–4600. doi: 10.1128/MCB.02027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant SR, Huang Y, Akache B, Kay MA. Site-directed transposon integration in human cells. Nucleic Acids Res. 2007;35:e50. doi: 10.1093/nar/gkm089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Meehan RR, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993;21:4886–4892. doi: 10.1093/nar/21.21.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto G, Schafer A, Marhold J, Stach D, Swaminathan SK, Handa V, Doderlein G, Maltry N, Wu W, Lyko F, Niehrs C. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445:671–675. doi: 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW. et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552–556. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- Egger G, Jeong S, Escobar SG, Cortez CC, Li TW, Saito Y, Yoo CB, Jones PA, Liang G. Identification of DNMT1 (DNA methyltransferase 1) hypomorphs in somatic knockouts suggests an essential role for DNMT1 in cell survival. Proc Natl Acad Sci USA. 2006;103:14080–14085. doi: 10.1073/pnas.0604602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes-Mascorro G, Serrano H, Rosado A. Sperm chromatin. Arch Androl. 2000;45:215–225. doi: 10.1080/01485010050193995. [DOI] [PubMed] [Google Scholar]
- Bird AP, Wolffe AP. Methylation-induced repression – belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/S0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Pelicic V, Morelle S, Lampe D, Nassif X. Mutagenesis of Neisseria meningitidis by in vitro transposition of Himar1 mariner. J Bacteriol. 2000;182:5391–5398. doi: 10.1128/JB.182.19.5391-5398.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinakis AG, Zagoraiou L, Vassilatis DK, Savakis C. Genome-wide insertional mutagenesis in human cells by the Drosophila mobile element Minos. EMBO Rep. 2000;1:416–421. doi: 10.1093/embo-reports/kvd089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–510. doi: 10.1016/S0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- Fischer SE, Wienholds E, Plasterk RH. Regulated transposition of a fish transposon in the mouse germ line. Proc Natl Acad Sci USA. 2001;98:6759–6764. doi: 10.1073/pnas.121569298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagoraiou L, Drabek D, Alexaki S, Guy JA, Klinakis AG, Langeveld A, Skavdis G, Mamalaki C, Grosveld F, Savakis C. In vivo transposition of Minos, a Drosophila mobile element, in mammalian tissues. Proc Natl Acad Sci USA. 2001;98:11474–11478. doi: 10.1073/pnas.201392398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorgi FL, Bhattacharya S, Huang L. Protamine sulfate enhances lipid-mediated gene transfer. Gene Ther. 1997;4:961–968. doi: 10.1038/sj.gt.3300484. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
One-hybrid assay reads. Reporter, activator and competitor plasmids were co‒transfected into HeLa cells. Cells were harvested 48 h post‒transfection, and protein extracts were used in luciferase assays. Three repetitions are shown.