

Abstract
Background
Malaria is a major health and socio-economical problem in tropical and sub-tropical areas of the world. Several methodologies have been used to assess parasite viability during the adaption of field strains to culture or the assessment of drug potential, but these are in general not able to provide an accurate real-time assessment of whether parasites are alive or dead.
Methods
Different commercial dyes and kits were assessed for their potential to allow for the real-time detection of whether a blood stage malaria parasite is dead or alive.
Results
Here, a methodology is presented based on the potential-sensitive mitochondrial probe JC-1, which allows for the real-time visualization of live (red staining) and/or dead (absence of red staining) blood stage parasites in vitro and ex vivo. This method is applicable across malaria parasite species and strains and allows to visualize all parasite blood stages including gametocytes. Further, this methodology has been assessed also for use in drug sensitivity testing.
Conclusions
The JC-1 staining approach is a versatile methodology that can be used to assess parasite viability during the adaptation of field samples to culture and during drug treatment. It was found to hold promise in the assessment of drugs expected to lead to delayed death phenotypes and it currently being evaluated as a method for the assessment of parasite viability during the adaptation of patient-derived Plasmodium vivax to long-term in vitro culture.
Background
Malaria, a disease caused by apicomplexan parasites of the genus Plasmodium, with Plasmodium falciparum causing 250–500 million clinical cases and up to 1.2 million deaths every year, is a major health- and socio-economical problem in the tropical and sub-tropical areas of the world [1]. Today, malaria occurs in 90 countries: an estimated 1.26 billion people (29% of the world’s population) live in areas where malaria is currently on the rise, while 400 million people live with endemic malaria unchanged by current control measures [2]. While vector and transmission control measures (e.g. bed nets) are in place in most of the risk countries [3,4] and the world’s scientific community is looking for effective vaccines [5], drugs are the weapon of choice both for malaria prophylaxis [6,7] and treatment [8,9].
As the focus shifted towards malaria eradication, there is a need to adapt new field strains and additional parasite species. For example, Plasmodium vivax, the second most important human malaria, has been largely neglected due to absence of in vitro culture systems and there is an increasing need to adapt it to long-term in vitro culture for research purposes and develop new therapies. In addition, there are challenges derived from the rapidly spreading resistance to existing anti-malarial drugs, mainly in P. falciparum and from the need to tackle emerging zoonoses (e.g. Plasmodium knowlesi[10,11]). Thus, the search for potent anti-malarials with innovative mechanism of action remains a priority.
The challenges inherent to the adaptation of field strains to culture, to the development of currently lacking, innovative, continuous parasite culture methods for specific malaria parasite species (e.g. P. vivax or the closely related primate malaria Plasmodium cynomolgi and to in vivo, ex vivo and in vitro drug potency studies would benefit from the availability of a sensitive and specific live-dead staining of parasites that would make it possible to asses real time parasite viability.
A number of ex vivo and in vitro reference methods have been established for the screening of anti-malarial activity, whereby drug potency is usually expressed as the concentration required to inhibit parasite growth by 50% (IC50). The Mark III WHO Giemsa thick blood smear method [12] is the gold standard in the field, while a variety of medium- to high-throughput drug screening methods based on either nucleic acid incorporation of radio-labelled hypoxanthine [13], SYBR green I dye [14,15], the detection of parasite specific enzymes (HRP-2 [16,17], LDH [18-20]) or the use of transgenic fluorescent parasites [21] have been introduced for laboratory use. Other screening methods are pathway specific and have been used to determine whether a compound can interfere with a certain specific parasite metabolic pathway (e.g. haemozoin formation [20,22]). Drawbacks of these methods include, but are not limited to, the subjective nature of the measurement, its reproducibility, the use of radioactive material, and the need for dedicated spaces, specialized equipment and specific disposal procedures.
Here, a methodology is presented based on the potential-sensitive mitochondrial probe JC-1, which allows for the real-time visualization of live (red staining) and/or dead (absence of red staining) parasites in vitro and ex vivo. This method is applicable across parasite species and strains (P. falciparum FCR3 and NF54, P. knowlesi H, P. cynomolgi M) and allows visualizing all parasite blood stages including gametocytes. This methodology has been assessed also for use in drug sensitivity testing and it is currently being evaluated as a method for the assessment of parasite viability during the adaptation of patient-derived P. vivax to long-term in vitro culture.
Methods
Parasites
Plasmodium falciparum NF54, FCR3 strains and P. knowlesi H strain were cultured in vitro in human A + red blood cells (RBC) (P. falciparum) (Sanquin Blood Bank, The Netherlands) or in rhesus monkey RBC (P. knowlesi) in complete medium composed of RPMI-1640 (with 25 mM HEPES, Invitrogen) supplemented with gentamycin and 10%-20% human serum (A+) in standard gassed (5% O2, 5% CO2, 90% N2) culture flasks and kept under shaking in an incubator at 37°C [23-25]. Parasites for live/dead staining were obtained at a parasitaemia between 3-10%, stained with JC-1 as reported below and immediately evaluated with a fluorescence microscope (Nikon Microphot-FXA equipped with green filter XF22, blue filter UV, red/green XF53, red XF40 and a Nikon FX35 PX camera).
Plasmodium cynomolgi ex vivo samples were obtained from infected rhesus monkeys from unrelated experiments after ethical permission from the ethical committee as required under Dutch law. Blood containing P. cynomolgi M strain trophozoite and gametocyte stages at around 2% parasitaemia was collected, immediately stained with JC-1 (using the protocol reported below) and the staining was evaluated as described above.
Parasite staining with JC-1 and drug assays using LDH or JC-1 staining as read-out
Drug assays were performed in culture flasks or 96-well plates by adding serial drug dilutions to synchronized P. falciparum or P. knowlesi ring-stage cultures as previously described [13,26,27]. Briefly, the concentration range in each of the JC-1 assays was based on the IC50 values, previously obtained using pLDH and hypoxanthine radioactive testing for each drug on the specific parasite strain. From stock solutions of each drug, 10-fold serial dilutions were made and plated in triplicate. As a read out of drug activity either the established pLDH enzymatic assay [18,19] or JC-1 staining were used.
For JC-1 staining for fluorescent microscope-based counting and parasite visualization, a 200 μl sample was taken from a standard culture or ex vivo experiment, spun down at 930 × g for 10 min at RT and medium was removed. When drug testing was carried out in plates, the plate was spun down and the medium removed. Pellets containing parasite infected erythrocytes were washed five times with sterile PBS at RT and re-suspend at 5% haematocrit and 1% parasitaemia in 500 μl (plate: 100 μl) Ringer’s solution either containing 4 nM DAPI (for manual counting of slides, Nikon microphot-FXA microscope) or 10 μl/ml Hoechst, Bisbenzimide H 33258 (Sigma, for counting with a high-content imager, the Operetta®, Perkin Elmer). Five μl JC-1 dye (Molecular Probes, Invitrogen) was added to the sample (final concentration: 2 μM), which was then incubated for 30 minutes at 37°C in the dark with continuous agitation. After the incubation period, cells were washed again 5 times with PBS to remove excess dye. Samples were either used to make wet slides (manual counting) or resuspended in 1 mL PBS and aliquoted as specified below for Operetta based counting.
In infected RBC containing both living and dead parasite, the JC-1 staining is characterized by a diffused green fluorescence (JC-1 monomer) across the RBC and parasite’s cytoplasm. Living parasites are further characterized by strong red mitochondrial fluorescence as the JC-1 monomer aggregates in presence of the mitochondrial membrane potential characteristic of a living parasite. This red signal is absent in dead parasites, which have lost their mitochondrial functionality and thus also their mitochondrial membrane potential.
Manual counting was performed using the fluorescence microscope. Up to 50 parasites/slide were counted. If fewer than 50 parasites were seen in slides from drug-treated samples, we assumed the remainder had died and lysed, and they were, therefore, noted as dead.
Operetta® based counting was performed as follows: after plated parasites were JC-1 stained and re-suspended in 1 ml PBS as described above, 50 μl of the suspension was transferred to a 96-well plate and an additional 50 μl PBS was added. The positive control containing untreated parasites was plated twice, each in triplicate, once at the start of the plate and once at the end to ensure consistency. RBCs were allowed to settle for about 5 min and a snapshot of the wells was taken using the bright-field filter to check for the cell density in each well. If the cells could be viewed individually the density was considered appropriate for fluorescence measurement, otherwise the RBC suspension was adjusted appropriately. In order to achieve accurate counting, pictures of 25 fields per well were taken on which all red (JC-1 aggregate) and green (JC-1 monomer only) parasites were afterwards counted manually. Manual counting was preferred, as Operetta counts of non-adherent cells would require development and validation of new algorithms, which were beyond the scope of our study. Operetta settings used for making the pictures were selected as follows: 40× long Working Distance objective, excitation 50%, transmission 50%. Visualization of the different dyes was performed using the standard Operetta® filters for DAPI, Texas Red and JC-1 pH8 monomer (green). Both gamma and contrast settings were optimized to achieve optimal pictures for assessing the status of the parasites. The intensity values used to name a parasite alive, dying or dead are <350 units dead, >350 < 450 units dying and >450 units alive. These units were validated by conducting side-by-side experiments using the red channel of a fluorescent microscope to evaluate the staining’s correlation with parasite viability: signal absent (parasite dead), faint red signal at times difficult to detect (parasite dying), signal bright red (parasite alive).
All parasites in each well were counted and their state (living, dead, dying) assessed. The untreated controls were used to calculate the final percent inhibition using the formula below. If there were fewer parasites present in the treated well as compared to the mean untreated control, the missing parasites were assessed as dead.
Results and discussion
JC1 is a viability stain distinguishing living from dead malaria parasites
This study aimed at developing a fluorescent parasite staining protocol that would allow to distinguish live from dead parasites during the adaptation to long-term in vitro culture of Plasmodium species, and would also be useful in drug-efficacy assays. For this, we evaluated dyes with differential membrane permeability and/or specifically labelling parasite organelles key to life. Nuclear stains tested in P. falciparum and P. knowlesi included SYTO-16, a combination of SYTO-10 and ethidium homodimer-2 and combinations of potential insensitive anionic and cationic dyes (acridine orange with ethidium bromide or propidium iodide), while mitochondrial stains tested in P. falciparum and P. knowlesi included MitoTrack, TMRE and the potential-sensitive Mitoprobe JC-1.
Nuclear dyes (e.g. SYTO-16) used to distinguish live from apoptotic/necrotic eukaryotic cells have different permeability characteristics (neutral or ionic) and staining thus depends on the integrity of the plasma membrane. However, when these dyes are applied to P. falciparum- and P. knowlesi-infected RBCs, mature schizonts are systematically stained as dead, due to the increased membrane permeability of the late Plasmodium blood life-cycle stages. Although staining selectivity was observed with dyes of the SYTO family in the case of rings, trophozoites and free merozoites (Additional file 1 and Additional file 2), these dyes were not considered useful for our applications.
In 1998, Borel et al. [28,29] described a new methodology employing a combination of an anionic and a cationic dye (acridine orange/ethidium bromide or bisbenzimide (Hoechst 33258)/propidium iodide) to distinguish living from dead Toxoplasma gondii. While acridine orange has been evaluated in the field as an alternative to Giemsa for malaria diagnosis [29-31] and found to be promising, the double dye method did not yield consistent results when P. falciparum or P. knowlesi drug-treated blood stage cultures were examined for the number of living and dead parasites. This may be due to the fact that this method is also partially based on differential membrane permeability, as the Hoechst, Bisbenzimide H 33258 (Sigma) dye is able to reach the nucleus when the cell membrane is intact by passive diffusion, while propidium iodide only reaches the nucleus when the membrane is compromised. This yields the same problem for mature schizonts as discussed above. Further, some mitochondrial stains (TMRE, MitoTrack) were evaluated on P. falciparum- and P. knowlesi-infected blood cultures, which gave a bright red labelling of living parasites, but gave at times residual vacuolar staining in dead parasites. Another mitochondrial stain, the potential-sensitive Mitoprobe JC-1, responds to the difference in mitochondrial membrane potential in living and dead cells. When the mitochondrial potential is normal (living parasite) the dye aggregates in the mitochondrion, labelling the mitochondrion red; when the potential is disrupted less of the dye aggregates resulting in a faint red signal (dying parasite), which may at times be difficult to detect, while when the potential is absent (dead parasite) the dye is only present in its monomeric form in the cytoplasm, thus labelling the parasite cytoplasm green. The fluorescent JC-1 staining of in vitro cultured living P. falciparum parasites growing in human RBC was evaluated in different parasite stages across the blood stage life cycle (rings, trophozoites, schizonts, merozoites and gametocytes, Figure 1). This demonstrated that all blood stage parasites could be clearly distinguished. Importantly, also mature schizont stages are stained as live parasites, unlike the other staining procedures mentioned above as JC-1 staining is not dependent on parasite membrane permeability, but only on the mitochondrial membrane potential, which is not altered in schizonts. In living P. falciparum parasites, the JC-1 red staining pattern corresponds to a previously reported pattern in which the malarial mitochondrion was labelled with Discosoma red fluorescent protein (DsRed) [32] or yellow fluorescent protein (YFP) [33]. JC-1 staining of a P. falciparum culture containing both living and dead parasites demonstrated that living, dying and dead parasites can be easily distinguished (Figure 2), confirming that this staining method is suitable for our purpose.
Figure 1.

Living P. falciparum parasite stages stained with JC-1. Plasmodium falciparum blood stages (rings, trophozoites, schizonts and gametocytes) stained with JC-1. The first panel in each row shows the staining of the parasite’s nucleus with Hoechst dye (350 nm); the second panel shows JC-1 monomer visible at 488 nm (green channel), which colours the parasite’s cytoplasm; the third panel shows the JC-1 aggregate visible at 568 nm (red channel) staining the parasite’s active mitochondrion; while the last panel shows the corresponding bright field image.
Figure 2.

Living, dead and dying P. falciparum schizonts stained with JC-1. JC-1 staining of Plasmodium falciparum schizonts. The first row of images shows a living parasite as can be appreciated by the bright red signal at 568 nm indicating a functioning mitochondrion; the second row of images shows a dead parasite killed using anti-malarial drugs (no signal at 568 nm). In the third series only a faint red signal (sometimes difficult to detect) can be seen at 568 nm indicating that the mitochondrion membrane potential is disrupted and the parasite is dying.
Optimization of the JC-1 staining protocol
Next we evaluated different JC-1 staining conditions: wet, live versus dry, fixed staining, extensive versus limited washing before and after staining, different wash media (Ringers, RPMI, PBS) and different incubation times. The most optimal conditions found, measured at 5% haematocrit and 1% parasitaemia to be: i) wet slides of standard cultures (reduces the bleaching); ii) extensive washing with PBS to get rid of all serum residues, as serum interferes with the staining procedure; iii) incubation of the washed parasitized RBCs for 30 minutes at 37°C in the presence of JC-1 dye at a final concentration of 2 μM and iv) extensive washing with PBS after staining to remove excess JC-1 thereby reducing the background. For the final microscopical analysis, cells need to be re-suspended in PBS as Ringers interferes with the detection. The preparations need to be studied immediately after staining to prevent parasites in PBS dying over time or the staining fading with time and/or during prolonged exposure to fluorescent light sources. In general, the nuclear stain allows to quickly recognize parasitized RBCs; the presence of a diffuse green signal defines cytoplasmic staining of any parasite and the additional presence of a bright red signal identifies an intact mitochondrion and thus a viable parasite. The exclusive presence of a cytoplasmic green signal in the absence of a red signal identifies a dead parasite devoid of mitochondrial potential (Figure 2).
Initially, in parallel staining of drug-treated cultures and cultures left purposefully outside the incubator overnight with both Giemsa and JC-1 were carried out, where Giemsa was used as a Gold standard (GS). In brief, 2000 parasites per slide were counted each time and classified either as live or dead. Table 1 shows representative results of repeated, parallel slide counting. As is apparent, slightly more living parasites are estimated in Giemsa slides, than is actually the case when JC-1 is used as a read out. This is probably due to the fact that it is difficult to distinguish between live and dead in early parasite stages stained with Giemsa.
Table 1.
Giemsa versus JC-1 read-out
| Read-out | Amount of dead parasites | Amount of live parasites |
|---|---|---|
| Gold Standard = Giemsa |
947 |
1053 |
| JC-1 | 981 | 1019 |
JC-1 staining is applicable to a variety of plasmodium species
After establishing that different life cycle stages of P. falciparum could be successfully labelled as living, dying or dead using the JC-1 dye, the fluorescent JC-1 staining methodology was extended to in vitro or ex vivo cultured parasites infecting different types of RBC. Figure 3 shows the staining of in vitro cultured P. knowlesi (non-human primate and human malaria that has been adapted to in vitro growth in rhesus normocytes, and ex vivo P. cynomolgi (a P. vivax-type parasite infecting non-human primate reticulocytes obtained from rhesus macaque infections. In all cases JC-1 gave a sensitive and specific red signal that could be clearly distinguished from the nuclear staining signal. Living parasites were characterized by a defined red fluorescent structure; while a only diffused green signal was observed in dead parasites (Additional file 3). This demonstrates that the JC-1 staining procedure can be used for different Plasmodium species, infecting reticulocytes or normocytes and cultured in vitro or obtained from an in vivo infection.
Figure 3.

Living Plamodium knowlesi and P. cynomolgistages stained with JC-1. JC-1 staining of additional Plasmodium spp and stages. Top row: Plasmodium knowlesi in vitro cultured blood stage trophozoite. Middle row: Ex vivo harvested Plasmodium cynomolgi blood stage schizont. Bottom row: P. cynomolgi salivary gland sporozoite.
The method can thus be considered widely applicable to malaria parasite species and different developmental stages and is therefore a useful tool for quickly monitoring culture viability during the adaptation of new field strains to in vitro growth. In particular, we are now developing this methodology further for the assessment of parasite viability during the adaptation of patient-derived P. vivax to long-term in vitro culture. Furthermore, the ability to also visualize sporozoites (Figure 3, a freshly isolated P. cynomolgi sporozoite) opens the possibility to quickly assess viability e.g. following different cryo-preservation protocols, before testing subsequent hepatocyte invasion potential.
JC-1 staining can be used as read out in drug assays
As a second objective, the usefulness of the JC-1 staining as a read out to assess parasite viability in drug-treated cultures was evaluated using different prototype drugs i.e. choloroquine (CQ, acting on the food vacuole [34]), atovaquone (ATQ, acting on mitochondria [35]) and artemisinin (ARTE, likely acting through a free radical-based oxidant stress [36,37]). In addition we evaluated a compound series, which targets the parasite’s phospholipid synthesis pathways (G25 and albitiazolium) [38-42] and is currently being developed for clinical use. Both manual counting of non-fixed slides from drug-treated parasite cultures using a fluorescence microscope and manual counting of real-time pictures (25 fields per well) taken from plated, drug-treated parasite cultures by an automatic high-content imaging system (Operetta®) were used to monitor drug action. Parasite sensitivities to any of the above mentioned drugs either on P. falciparum or P. knowlesi was measured using JC-1 as a read-out with both counting methodologies and were found to yield the typical sigmoid curve indicating a linear relationship between drug concentration and parasite inhibition between the threshold dose level and the plateau dose level over a limited number of drug concentrations. Although the developed methodology is reproducible and the inter-experiment reproducibility is relatively high (Figure 4 Panel A), the manual counting of living non-fixed parasite slides using a fluorescent microscope had an important limitation. Due to the long-time needed to count each slide (3 h for a 24 well plate drug assessment), the non-treated control wells counted at the beginning and end of the procedure gave different results: up to 3-5% more dead parasites were found in the control slides counted at the end of the experiment versus the ones counted in the beginning. This is due to the fact that live parasites do not like to be kept in wet slide conditions for too long. The use of Operetta® pictures of the wells and manual counting of parasites in those pictures solved this problem: comparison of control wells at the beginning and end in each plate showed high (98-99%) consistency, due to the fact that the parasites’ condition was fixed by the pictures, which were taken in rapid succession across the whole plate and could thus not deteriorate over time. The imager procedure appears more sensitive than the fluorescent microscope counting at the low (IC20) drug concentrations and more accurate at high drug concentrations (IC90) when there are only very few living parasites left as many more fields are photographed in rapid succession per well and counted in comparison to manually counted slides in which a time-field counting balance needs to be found as the amount of dead parasites in control wells increases over time as explained above.
Figure 4.

IC50 determination using JC-1 and/or pLDH in parallel on drug treatedP. falciparumcultures. Culture P. falciparum-infected erythrocyte were incubated for 48 h in the presence of the indicated drug concentration at haematocrit of 5% and 1% parasitaemia. PanelA - shows that the inter-experiment variability between 2 separate JC-1 determinations is low (red and violet lines) and that the methodology is more accurate (IC50 is consistently lower) than the IC50 determination obtained with the pLDH methodology; PanelB - shows typical curves obtained by splitting parasites derived from a same mother culture into plates, in which they were cultured in parallel under the same environmental conditions in the presence of rising artemisinin concentrations and IC50 curves where either determined using the pLDH (violet line) or the JC-1 (blue line) methodology; PanelC – shows typical curves obtained by splitting parasites derived from a same mother culture into plates, in which they were cultured in parallel under the same environmental conditions in the presence of rising atovaquone concentrations and IC50 curves where either determined using the pLDH (violet line) or the JC-1 (blue line) methodology.
Parallel experiments using the LDH and JC-1 staining methodologies were carried out using CQ, ATQ (Figure 4 Panel B) and ARTE (Figure 4 Panel C) treated P. falciparum FCR3 and P. knowlesi cultures. Experiments were performed in triplicate for each drug concentration. With all three drugs, the JC-1 staining methodology appears to be more accurate as the JC-1 read out is a direct measure of parasite viability obtained by staining the whole parasite population (including rings) and not by measuring a surrogate parasite marker produced only by the trophozoite and schizont stages (pLDH or LDH enzyme activity). Repeated JC-1 staining experiments over once cycle (48 h for P. falciparum and 24 h for P. knowlesi) have on average yielded IC50s for the P. falciparum FCR3 and P. knowlesi H strains treated with the different drugs, which are lower than with the LDH methodology (Tables 2 and 3). These experiments were repeated several times and it appears that the gain in sensitivity over a longer life-cycle (P. falciparum: 48 h) is in general higher than over a shorter life-cycle (P. knowlesi: 24 h). This reflects the fact that despite the lethal effect exerted by the drugs, the production of the malaria-specific pLDH protein is not stopped rapidly and the measurement of the pLDH protein, which is still being synthesized by the parasite thus biases this type of test. However, the JC1 staining reflects the mitochondrial status of the parasite at the time when the test is applied.
Table 2.
IC50 ranges from in parallel experiments
|
Drug |
CQ in nM* |
ATQ in nM* |
ARTE in nM* |
|||
|---|---|---|---|---|---|---|
| Assay | LDH | JC-1 | LDH | JC-1 | LDH | JC-1 |
|
Pf FCR3 |
14.1-16.6 |
8.8-10.9 |
0.7-1.3 |
0.5-0.9 |
0.1-0.9 |
0.03-0.1 |
| Pk H | 9.38-10.9 | 7.8-9.7 | 2-2.6 | 2.1-2.7 | 1.1-1.6 | 0.8-1.2 |
*All IC50 ranges were determined on an average of 8–10 experiments where JC-1 and LDH experiments were carried out side by side.
Table 3.
IC50 average with standard deviation
| Drug in nM* | Assay | Pf FCR3 | Pk H |
|---|---|---|---|
| CQ |
LDH |
15.71 ± 1.23 |
10.4 ± 1.03 |
|
JC-1 |
10.33 ± 0.97 |
8.3 ± 0.92 |
|
| ATQ |
LDH |
1.16 ± 1.41 |
2.54 ± 0.86 |
|
JC-1 |
0.78 ± 0.72 |
2.27 ± 0.31 |
|
| ARTE |
LDH |
0.52 ± 0.83 |
1.53 ± 0.46 |
| JC-1 | 0.07 ± −0.59 | 0.95 ± 0.72 |
*Mean IC50s determined on an average of 8–10 experiments where JC-1 and LDH experiments were carried out side by side.
Choline analogues G25 and albitiazolium constitute a new class of anti-malarial drugs that target the malarial membrane biogenesis through the phospholipid metabolism. The clinical candidate albitiazolium (formerly named T3 and SAR97276) [42] is currently evaluated in clinical phase II trials to treat severe malaria by the parenteral route. Choline analogues give unreliable IC50 results, when assayed with the traditional LDH test [43]. It has been observed that P. falciparum left in the presence of G25 or albitiazolium [41,43,44] for a few hours incurred in parasite disappearance in the following life cycle. Compound potency is associated to their properties of accumulating in a non-reversible way within the intra-erythrocytic parasites, which irremediably impaired parasite viability while the parasite morphology is affected more belatedly. This indicates that the drug operates through a Trojan horse effect and its real pharmacological effect is dissociated from the effects on parasite morphology and metabolism [41,43]. Therefore, the methodology was further tested to establish whether it would be appropriate to assess these delayed parasite collapses, by exposing both P. falciparum FCR3 (Figure 5) and P. knowlesi H cultures to G25 and albitiazolium at their IC50 for 2 hours using CQ as a reference) [43] and followed the parasitaemia development into the next cycle using JC-1 as a read-out (Figure 3). The IC50s were first determined using radio-labelled hypoxanthine [13] after exposure to the drugs over 1 cycle, 48 h for P. falciparum and 24 h for P. knowlesi). IC50 for P. knowlesi were 0.36 mM (albitiazolium), 1.38 mM (G25) and 5.62 nM (CQ) respectively; while the IC50s for P. falciparum were 2.25 nM (albitiazolium), 0.6 nM (G25) and 7.82 nM (CQ) respectively. When parasite were incubated with the drugs at their IC50 for only 2 h followed by the washing out of the drugs, a potent delayed effect of G25 on P. falciparum FCR3 (Figure 5, red line) was found, since the full effect of the drug (100% inhibition calculated with the formula reported above for each time point) is observed in the second cycle (past the 24 h). Albitiazolium seems to have a similar effect, but with a delayed collapse even into the third cycle (Figure 5, green line), while as expected CQ (Figure 5, blue line) has no delayed-death effect and acts within the first cycle on trophozoites and schizonts. Results obtained with P. knowlesi H (Additional file 4) also indicate that after incubation of the parasite with the choline analogues at their IC50 for only 2 h, both G25 and albithiazolium exerted delayed pharmacological effects that are observed during the cycle following the contact with the drug. Altogether, results support the conclusion indicating that use of the JCI staining may be a suitable alternative method to LDH for assessing the IC50 of compounds suspected of having delayed death effects.
Figure 5.
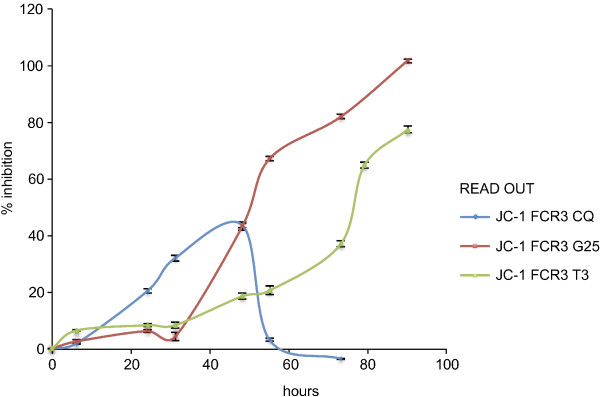
Delayed death phenotypes monitored using JC-1 on synchronized P. falciparum FCR3 cultures exposed to chloroquine and the phospholipid pathway inhibitors prototype drug. P. falciparum FCR3 culture (5% hematocrit and 0.1 starting parasitaemia) were exposed to 7.82 nM chloroquine and the phospholipid pathway inhibitors prototype drugs (2.25 nM albitiazolium and 0.6 nM G25). After two hours of contact with the drugs, cell were washed and resuspended in drug-free fresh complete medium. JC-1 staining was then used to monitor parasite viability over the course of at least 3 cycles to detect any delayed drug effects. The results are expressed as means ± SEM (n = 3). Exposure of a ring culture to the IC50 of chloroquine for 2 h has no delayed effect but acts on trophozoites and schizonts during the first cycle as expected (blue line), but exposure of the same ring culture to the IC50 of G25 and albitiazolium for only 2 hours results in a potent delayed death effect, which for G25 reaches 100% within the second parasite cycle (red line) and for albitiazolium probably within beyond the third (green line) cycle.
Conclusions
The JC-1 staining approach is a versatile methodology that can be used to assess parasite viability during the adaptation of field samples to culture and during drug treatment. On-going efforts aimed at establishing P. vivax blood stage cultures will particularly benefit from the use of the described method. In particular, this methodology appears to be more accurate and reliable than Giemsa staining of fixed parasites, which rarely gives clear quantitative evidence of whether a parasite culture is in good shape or not.
In the context of the assessment of a compound’s anti-malarial potency, this methodology offers reproducible, robust results comparable to established methods such as the LDH IC50 test, while being in general more accurate. Furthermore, a live stain such as JC-1 offers the possibility to follow compounds overtime, thus opening the way to the assessment of delayed death drug phenotype.
Abbreviations
CQ: Chloroquine; ARTE: Artemisinin; ATQ: Atovaquone; IC50: Half maximal inhibitory concentration; pLDH: Plasmodium lactate dehydrogenase.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EP and CK designed the research; DvdI and EP performed the research; HV contributed reagents; EP and CK analysed the data and wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
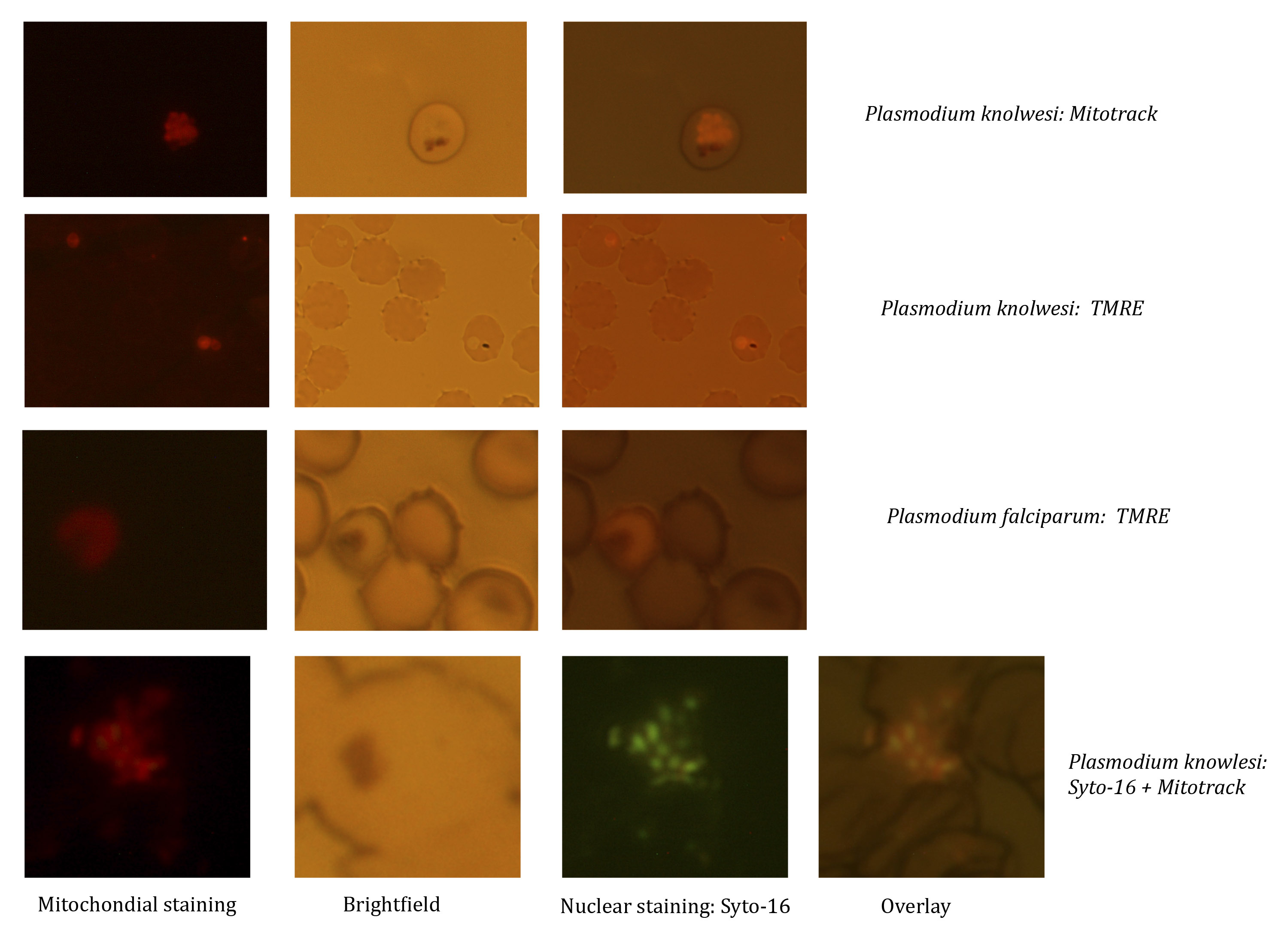
P. falciparum and P. knowlesi parasites stained with the mitochondrial dyes TMRE and Mitotrack. The mitochondrial dye TMRE and Mitortrack appear to give rise to a very aspecific, diffused signal when compared to a well defined signal of the potential-sensitive mitochondrial dye JC-1 (Figures 1, 2, 3). Both TMRE and Mitotrack appear to stain vacuoles and the cytoplasm of the parasites. This raises the question on whether they are indeed labelling specifically the parasites’ mitochondria.
{kind=link}
Living P. falciparum and P. knowlesi parasites stained with the nuclear Syto-16 dye and co-stained with the mitochondrial Mitotrack dye. In eukaryotic cells, Syto-16 is used to distinguish live (no stain) from apoptotic /necrotic cells (nuclei stained green). The dye is able to cross the plasma membrane of the cell only if it is compromised and thus enter the cell nucleus staining it green. However, as Plasmodium schizonts due to their increased membrane permeability, the Syto-16 dyes labels living schizonts as dead by staining their nuclei green.
{kind=link}
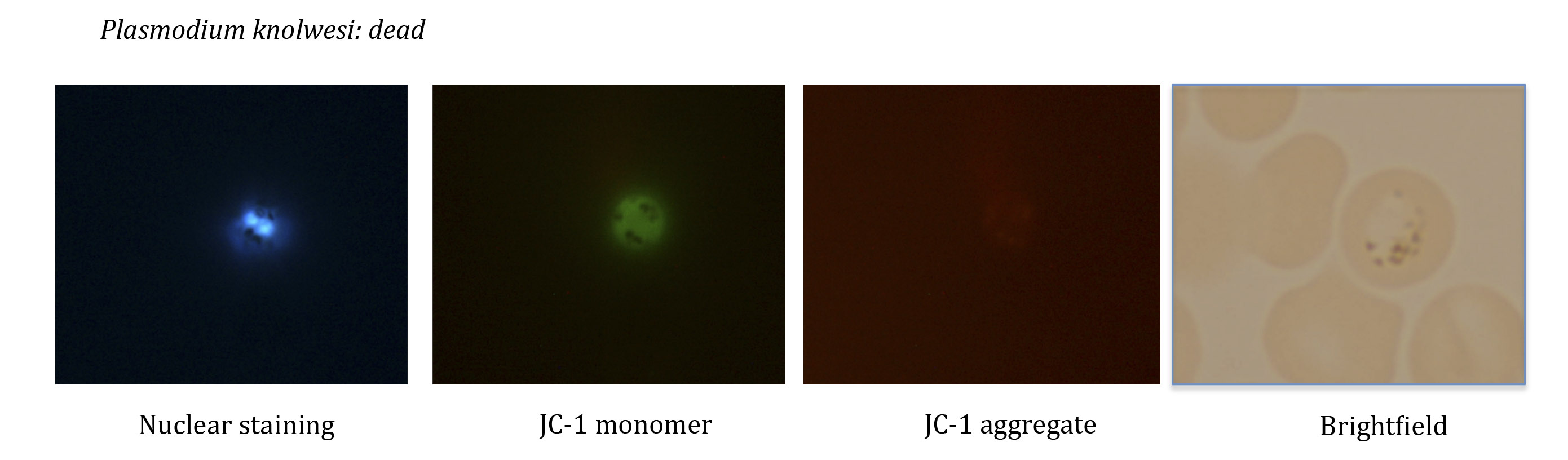
Dead P. knowlesi parasites stained with JC-1. JC-1 staining of drug-treated Plasmodium knowlesi culture showing a dead parasite as can be appreciated by the absence of signal at 568 nm.
{kind=link}
Delayed death phenotypes monitored using JC-1 on synchronized P. knowelsi H cultures exposed to chloroquine and the phospholipid pathway inhibitors prototype drug.P. knowlesi H culture (5% hematocrit and 0.1 starting parasitaemia) were exposed to 5.62 nM chloroquine and the phospholipid pathway inhibitors prototype drugs (1.38 nM albitiazolium and 0.36 nM G25). After two hours of contact with the drugs, cell were washed and resuspended in drug-free fresh complete medium. JC-1 staining was then used to monitor parasite viability over the course of at least 3 cycles to detect any delayed drug effects. The results are expressed as means ± SEM (n = 3). Exposure of a ring culture to the IC50 of chloroquine for 2 h has no delayed effect but acts on trophozoites and schizonts during the first cycle as expected (violet line), but exposure of the same ring culture to the IC50 of G25 and albitiazolium for only 2 hours results in a potent delayed death effect, which for G25 reaches 100% within the second parasite cycle (blue line) and for albitiazolium probably within beyond the third (red line) cycle.
Contributor Information
Erica M Pasini, Email: pasini@bprc.nl.
Denise van den Ierssel, Email: ierssel@bprc.nl.
Henri J Vial, Email: vial@univ-montp2.fr.
Clemens HM Kocken, Email: kocken@bprc.nl.
Acknowledgements
The authors would like to acknowledge Els Klooster and Anne-Marie Zeeman for help with the use of the Operetta. This work received funding from Antimal contract LSHP-CT-2005-018834, EVIMalaR (contract number 242095) and Gates Foundation Project OPP1023583.
References
- Murray CJ, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet. 2012;379:413–431. doi: 10.1016/S0140-6736(12)60034-8. [DOI] [PubMed] [Google Scholar]
- Hay SI, Guerra CA, Tatem AJ, Noor AM, Snow RW. The global distribution and population at risk of malaria: past, present, and future. Lancet Infect Dis. 2004;4:327–336. doi: 10.1016/S1473-3099(04)01043-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunamoorthi K. Vector control: a cornerstone in the malaria elimination campaign. Clin Microbiol Infect. 2011;17:1608–1616. doi: 10.1111/j.1469-0691.2011.03664.x. [DOI] [PubMed] [Google Scholar]
- van den Berg H, Zaim M, Yadav RS, Soares A, Ameneshewa B, Mnzava A, Hii J, Dash AP, Ejov M. Global trends in the use of insecticides for vector-borne disease control. Environ Health Perspect. 2012;120:577–582. doi: 10.1289/ehp.1104340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remarque EJ, Faber BW, Kocken CH, Thomas AW. Apical membrane antigen 1: a malaria vaccine candidate in review. Trends Parasitol. 2008;24:74–84. doi: 10.1016/j.pt.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Fernando SD, Rodrigo C, Rajapakse S. Chemoprophylaxis in malaria: drugs, evidence of efficacy and costs. Asian Pac J Trop Med. 2011;4:330–336. doi: 10.1016/S1995-7645(11)60098-9. [DOI] [PubMed] [Google Scholar]
- Gosling RD, Okell L, Mosha J, Chandramohan D. The role of antimalarial treatment in the elimination of malaria. Clin Microbiol Infect. 2011;17:1617–1623. doi: 10.1111/j.1469-0691.2011.03660.x. [DOI] [PubMed] [Google Scholar]
- Petersen I, Eastman R, Lanzer M. Drug-resistant malaria: molecular mechanisms and implications for public health. FEBS Lett. 2011;585:1551–1562. doi: 10.1016/j.febslet.2011.04.042. [DOI] [PubMed] [Google Scholar]
- Burrows JN, Chibale K, Wells TN. The state of the art in anti-malarial drug discovery and development. Curr Top Med Chem. 2011;11:1226–1254. doi: 10.2174/156802611795429194. [DOI] [PubMed] [Google Scholar]
- Kantele A, Jokiranta TS. Review of cases with the emerging fifth human malaria parasite, Plasmodium knowlesi. Clin Infect Dis. 2011;52:1356–1362. doi: 10.1093/cid/cir180. [DOI] [PubMed] [Google Scholar]
- Cox-Singh J. Zoonotic malaria: Plasmodium knowlesi, an emerging pathogen. Curr Opin Infect Dis. 2012;25:530–536. doi: 10.1097/QCO.0b013e3283558780. [DOI] [PubMed] [Google Scholar]
- Diseases WHODoCoT. In vitro micro-test (mark III) for the assessment of the response of Plasmodium falciparum to chloroquine, mefloquine, quinine, amodiaquine, sulfadoxine/pyrimethamin and artemisinin. 2001. pp. 1–21. http://www.who.int/malaria/publications/atoz/markiii.pdf.
- Desjardins RE, Canfield CJ, Haynes JD, Chulay JD. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother. 1979;16:710–718. doi: 10.1128/AAC.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JD, Dennull RA, Gerena L, Lopez-Sanchez M, Roncal NE, Waters NC. Assessment and continued validation of the malaria SYBR green I-based fluorescence assay for use in malaria drug screening. Antimicrob Agents Chemother. 2007;51:1926–1933. doi: 10.1128/AAC.01607-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noedl H, Bronnert J, Yingyuen K, Attlmayr B, Kollaritsch H, Fukuda M. Simple histidine-rich protein 2 double-site sandwich enzyme-linked immunosorbent assay for use in malaria drug sensitivity testing. Antimicrob Agents Chemother. 2005;49:3575–3577. doi: 10.1128/AAC.49.8.3575-3577.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noedl H, Krudsood S, Leowattana W, Tangpukdee N, Thanachartwet W, Looareesuwan S, Miller RS, Fukuda M, Jongsakul K, Yingyuen K, Sriwichal S, Ohrt C, Knirsch C. In vitro antimalarial activity of azithromycin, artesunate, and quinine in combination and correlation with clinical outcome. Antimicrob Agents Chemother. 2007;51:651–656. doi: 10.1128/AAC.01023-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druilhe P, Moreno A, Blanc C, Brasseur PH, Jacquier P. A colorimetric in vitro drug sensitivity assay for Plasmodium falciparum based on a highly sensitive double-site lactate dehydrogenase antigen-capture enzyme-linked immunosorbent assay. AmJTrop Med Hyg. 2001;64:233–241. doi: 10.4269/ajtmh.2001.64.233. [DOI] [PubMed] [Google Scholar]
- Makler MT, Ries JM, Williams JA, Bancroft JE, Piper RC, Gibbins BL, Hinrichs DJ. Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity. AmJTrop Med Hyg. 1993;48:739–741. doi: 10.4269/ajtmh.1993.48.739. [DOI] [PubMed] [Google Scholar]
- Noedl H, Wongsrichanalai C, Wernsdorfer WH. Malaria drug-sensitivity testing: new assays, new perspectives. Trends Parasitol. 2003;19:175–181. doi: 10.1016/S1471-4922(03)00028-X. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Tarleton RL. Transgenic parasites accelerate drug discovery. Trends Parasitol. 2012;28:90–92. doi: 10.1016/j.pt.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parapini S, Basilico N, Pasini E, Egan TJ, Olliaro P, Taramelli D, Monti D. Standardization of the physicochemical parameters to assess in vitro the beta-hematin inhibitory activity of antimalarial drugs. Exp Parasitol. 2000;96:249–256. doi: 10.1006/expr.2000.4583. [DOI] [PubMed] [Google Scholar]
- Kocken CH, Ozwara H, van der Wel A, Beetsma AL, Mwenda JM, Thomas AW. Plasmodium knowlesi provides a rapid in vitro and in vivo transfection system that enables double-crossover gene knockout studies. Infect Immun. 2002;70:655–660. doi: 10.1128/IAI.70.2.655-660.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- van der Wel AM, Tomas AM, Kocken CH, Malhotra P, Janse CJ, Waters AP, Thomas AW. Transfection of the primate malaria parasite Plasmodium knowlesi using entirely heterologous constructs. J Exp Med. 1997;185:1499–1503. doi: 10.1084/jem.185.8.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wel A, Kocken CH, Pronk TC, Franke-Fayard B, Thomas AW. New selectable markers and single crossover integration for the highly versatile Plasmodium knowlesi transfection system. Mol Biochem Parasitol. 2004;134:97–104. doi: 10.1016/j.molbiopara.2003.10.019. [DOI] [PubMed] [Google Scholar]
- Kocken CH, van der Wel A, Rosenwirth B, Thomas AW. Plasmodium vivax: in vitro antiparasitic effect of cyclosporins. Exp Parasitol. 1996;84:439–443. doi: 10.1006/expr.1996.0132. [DOI] [PubMed] [Google Scholar]
- Borel E, Mayencon M, Kaiser K, Picot S, Peyron F. Fluorogenic detection of viable Toxoplasma gondii. Parasite. 1998;5:371–373. doi: 10.1051/parasite/1998054371. [DOI] [PubMed] [Google Scholar]
- Mendiratta DK, Bhutada K, Narang R, Narang P. Evaluation of different methods for diagnosis of P. falciparum malaria. Indian J Med Microbiol. 2006;24:49–51. doi: 10.4103/0255-0857.19895. [DOI] [PubMed] [Google Scholar]
- Keiser J, Utzinger J, Premji Z, Yamagata Y, Singer BH. Acridine orange for malaria diagnosis: its diagnostic performance, its promotion and implementation in tanzania, and the implications for malaria control. Ann Trop Med Parasitol. 2002;96:643–654. doi: 10.1179/000349802125001834. [DOI] [PubMed] [Google Scholar]
- Kong HH, Chung DI. Comparison of acridine orange and Giemsa stains for malaria diagnosis. Korean J Parasitol. 1995;33:391–394. doi: 10.3347/kjp.1995.33.4.391. [DOI] [PubMed] [Google Scholar]
- Sato S, Wilson RJ. The use of DsRED in single- and dual-color fluorescence labeling of mitochondrial and plastid organelles in Plasmodium falciparum. Mol Biochem Parasitol. 2004;134:175–179. doi: 10.1016/j.molbiopara.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Tschan S, Kreidenweiss A, Stierhof YD, Sessler N, Fendel R, Mordmuller B. Mitochondrial localization of the threonine peptidase PfHslV, a ClpQ ortholog in Plasmodium falciparum. Int J Parasitol. 2010;40:1517–1523. doi: 10.1016/j.ijpara.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Zhang J, Krugliak M, Ginsburg H. The fate of ferriprotorphyrin IX in malaria infected erythrocytes in conjunction with the mode of action of antimalarial drugs. Mol Biochem Parasitol. 1999;99:129–141. doi: 10.1016/S0166-6851(99)00008-0. [DOI] [PubMed] [Google Scholar]
- Fisher N, Abd Majid R, Antoine T, Al-Helal M, Warman AJ, Johnson DJ, Lawrenson AS, Ranson H, O’Neill PM, Ward SA, Biagini GA. Cytochrome b mutation Y268S conferring atovaquone resistance phenotype in malaria parasite results in reduced parasite bc1 catalytic turnover and protein expression. J Biol Chem. 2012;287:9731–9741. doi: 10.1074/jbc.M111.324319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CM, Hoey EM, Trudgett A, Timson DJ. Artemisinins act through at least two targets in a yeast model. FEMS Yeast Res. 2011;11:233–237. doi: 10.1111/j.1567-1364.2010.00706.x. [DOI] [PubMed] [Google Scholar]
- O’Neill PM, Barton VE, Ward SA. The molecular mechanism of action of artemisinin–the debate continues. Molecules. 2010;15:1705–1721. doi: 10.3390/molecules15031705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Mamoun C, Prigge ST, Vial H. Targeting the lipid metabolic pathways for the treatment of malaria. Drug Dev Res. 2010;71:44–55. doi: 10.1002/ddr.20347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocken CH, Remarque EJ, Dubbeld MA, Wein S, van der Wel A, Verburgh RJ, Vial HJ, Thomas AW. Statistical model to evaluate in vivo activities of antimalarial drugs in a Plasmodium cynomolgi-macaque model for Plasmodium vivax malaria. Antimicrob Agents Chemother. 2009;53:421–427. doi: 10.1128/AAC.00576-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengelnik K, Vidal V, Ancelin ML, Cathiard AM, Morgat JL, Kocken CH, Calas M, Herrera S, Thomas AW, Vial HJ. A class of potent antimalarials and their specific accumulation in infected erythrocytes. Science. 2002;295:1311–1314. doi: 10.1126/science.1067236. [DOI] [PubMed] [Google Scholar]
- Vial HJ, Wein S, Farenc C, Kocken C, Nicolas O, Ancelin ML, Bressolle F, Thomas A, Calas M. Prodrugs of bisthiazolium salts are orally potent antimalarials. Proc Natl Acad Sci U S A. 2004;101:15458–15463. doi: 10.1073/pnas.0404037101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vial H, Penarete D, Wein S, Caldarelli S, Fraisse L, Peyrottes S. In: Apicomplexan Parasites: Molecular Approaches toward Targeted Drug Development. Editions B, editor. Germany: Wiley-VCH Press; 2011. Lipids as drug targets for malaria therapy; p. 23. [Google Scholar]
- Wein S, Maynadier M, Tran Van Ba C, Cerdan R, Peyrottes S, Fraisse L, Vial H. Reliability of antimalarial sensitivity tests depends on drug mechanisms of action. J Clin Microbiol. 2010;48:1651–1660. doi: 10.1128/JCM.02250-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancelin ML, Calas M, Vidal-Sailhan V, Herbute S, Ringwald P, Vial HJ. Potent inhibitors of plasmodium phospholipid metabolism with a broad spectrum of in vitro antimalarial activities. Antimicrob Agents Chemother. 2003;47:2590–2597. doi: 10.1128/AAC.47.8.2590-2597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
P. falciparum and P. knowlesi parasites stained with the mitochondrial dyes TMRE and Mitotrack. The mitochondrial dye TMRE and Mitortrack appear to give rise to a very aspecific, diffused signal when compared to a well defined signal of the potential-sensitive mitochondrial dye JC-1 (Figures 1, 2, 3). Both TMRE and Mitotrack appear to stain vacuoles and the cytoplasm of the parasites. This raises the question on whether they are indeed labelling specifically the parasites’ mitochondria.
Living P. falciparum and P. knowlesi parasites stained with the nuclear Syto-16 dye and co-stained with the mitochondrial Mitotrack dye. In eukaryotic cells, Syto-16 is used to distinguish live (no stain) from apoptotic /necrotic cells (nuclei stained green). The dye is able to cross the plasma membrane of the cell only if it is compromised and thus enter the cell nucleus staining it green. However, as Plasmodium schizonts due to their increased membrane permeability, the Syto-16 dyes labels living schizonts as dead by staining their nuclei green.
Dead P. knowlesi parasites stained with JC-1. JC-1 staining of drug-treated Plasmodium knowlesi culture showing a dead parasite as can be appreciated by the absence of signal at 568 nm.
Delayed death phenotypes monitored using JC-1 on synchronized P. knowelsi H cultures exposed to chloroquine and the phospholipid pathway inhibitors prototype drug.P. knowlesi H culture (5% hematocrit and 0.1 starting parasitaemia) were exposed to 5.62 nM chloroquine and the phospholipid pathway inhibitors prototype drugs (1.38 nM albitiazolium and 0.36 nM G25). After two hours of contact with the drugs, cell were washed and resuspended in drug-free fresh complete medium. JC-1 staining was then used to monitor parasite viability over the course of at least 3 cycles to detect any delayed drug effects. The results are expressed as means ± SEM (n = 3). Exposure of a ring culture to the IC50 of chloroquine for 2 h has no delayed effect but acts on trophozoites and schizonts during the first cycle as expected (violet line), but exposure of the same ring culture to the IC50 of G25 and albitiazolium for only 2 hours results in a potent delayed death effect, which for G25 reaches 100% within the second parasite cycle (blue line) and for albitiazolium probably within beyond the third (red line) cycle.