

Abstract
We report on a method for quantitating the distance dependence of cell-cell interactions. We employ a microchip design that permits a multiplex, quantitative protein assay from statistical numbers of cell pairs, as a function of cell separation, with a 0.15 nanoliter volume microchamber. We interrogate interactions between pairs of model brain cancer cells by assaying for 6 functional proteins associated with PI3k signaling. At short incubation times, cells do not appear to influence each other, regardless of cell separation. For 6 hour incubation times, the cells exert an inhibiting influence on each other at short separations, and a predominately activating influence at large separation. Protein-specific cell-cell interaction functions are extracted, and by assuming pairwise additivity of those interactions, the functions are shown to correctly predict the results from 3-cell experiments carried out under the identical conditions.
Cell-cell interactions contribute to processes ranging from immune system activation to the functional behaviors of healthy and diseased tissues to cellular interactions within a tumor microenvironment that can influence tumorigenesis. Such interactions are often inferred via molecular analyses at the transcriptome or proteome level of two or more co-cultured cell types, such as glioma cells and astrocytes1, 2, relative to similar analyses of pure cell cultures. In other studies, mixtures of defined cell types are utilized to seed tumors in mice, and the nature of the grown tumor is correlated back to the initial seed composition3. More quantitative studies have focused on issues such as how cell-cell contacts and soluble factor signaling influence interactions. For example, Hui and Bhatia utilized mechanically adjustable surfaces to explore the importance of contact and soluble factor signaling between colonies of epithelial and stroma cells4. Nelson and Chen utilized micropatterned surfaces to control cell contact and spreading for endothelial and smooth muscle cells, and found that cell-cell contact positively regulates proliferation5. This compares to recent optical tracking investigations of cell interactions in epithelial cell cultures, which suggest that mechanical contact and constraints in cell area inhibit cell replication6.
We take the physical approach of viewing two cells as two particles. A particle-particle interaction can be quantitated by holding two particles at a fixed separation, measuring a parameter that corresponds to the strength of the interaction, and then repeating that measurement at a different separation, etc., until the functional form is resolved7. Knowledge of such interactions, which may switch between attractive and repulsive as a function of separation, can be utilized to understand and predict the structure and other physical properties of particle assemblies8. Here we begin to extend this concept towards understanding how two cancer cells influence each other. Because these are cancer cells, we utilize quantitative measurements of the levels of functional cytoplasmic and secreted proteins associated growth factor signaling to capture how tumorigenic activity changes as the distance between two cells is varied. We then extract protein-specific cell-cell interaction functions and use them to accurately predict the protein levels as measured from similarly executed 3-cell experiments.
Single cells are finite systems. This means that a measurement of a specific property from one cell may not yield the same value when it is measured from an otherwise identical cell. Of course, the same holds for a system of two or three cells. Thus, our experiment is designed to capture and yield predictions for statistically representative data sets.
EXPERIMENTAL
The platform utilized here is conceptually similar to our previously published single cell barcode chip (SCBC)9, 10, but with major modifications. The concept is to isolate a cell within a microchamber that contains an antibody array (Figure 1). The antibody array provides the capture antibodies for a multiplex sandwich-type enzyme-linked immuno-assay of a panel of secreted, cytoplasmic, or membrane proteins (we detect all 3 types here). The concentration of a protein to be detected is determined by the copy numbers produced by the cell (or cells) within the microchamber, plus the microchamber volume.
Figure 1.

The SCBC microchip platform. (a) Photo of the microchip and a fluorescence micrograph of a 20 microchamber cellular assay unit (out of 435 total). The central channel contains cell lysate. (b) On-chip operation flow. Cells are loaded into the microchambers with the chip held in the “fully open” position. Low pressure on the microchip (Close-I state) seals most of the chamber, but leaves open a channel for lysis buffer introduction. Application of additional pressure accesses Close-II state, which completely isolates the cells within the microchambers from the adjacent microchannels. (c) The steps of the sandwich-type fluorescence immunoassay for detecting of secreted or cytoplasmic or membrane proteins. (d) Data collected from a microchamber includes numbers and positions of cells, plus the fluorescence intensity from each antibody-based protein assay. (e) Fluorescence data for secreted (IL6) and cytoplasmic (p-Erk) protein assays from an SCBC in which U87 EGFRvIIII cells were incubated on-chip for 6 hours prior to cell lysis. P values of 0.05 (*), 0.01 (**) and 0.001 (***), with 0.05 considered statistically significant. An insignificant statistical difference is denoted as NA.
The current SCBC was designed to capture how aspects of cell signaling associated with tumorigenesis are influenced by cell-cell interactions. This guided the choice of the assayed panel of proteins (discussed below), and it required many 2-cell experiments, with knowledge of cell-cell separation distance for each of those experiments. This was achieved through a new (valveless) SCBC design that contained many (8700) 0.15 nanoliter volume microchambers (Figure 1a and Figure S1, Supporting information), and by loading SCBCs with sufficient numbers of cells to ensure statistical numbers of 0, 1, 2, and 3 cell experiments. Cells are randomly loaded, and the numbers and positions of cells within a given microchamber are measured by microscopy imaging through the transparent microchip. After cell loading, the SCBC is incubated for a period of time, during which certain secreted proteins are captured by designated elements of the antibody array. The cells are then lysed (Figure 1b) and cytoplasmic or membrane proteins are similarly captured (Figure 1c). The SCBC is dissembled, and a cocktail of detection antibodies is utilized to develop the protein detection arrays. Fluorescence imaging, using an array scanner, is used to digitize the protein assay (Figure 1d). The fluorescence level of a given protein assay can be compared against a calibration curve (measured using an identical SCBC and spiked solutions of standard proteins) to convert protein levels into copy numbers. A given SCBC experiment thus provides a table of results. Each row corresponds to a microchamber address. The columns contain the numbers and locations of cells, as well as the digitized and calibrated outputs from the protein assays.
The SCBC utilized here involves two advances. First, the antibody array is patterned at significantly higher density. As with prior versions, we start with microfluidic flow-patterning to generate an n-element stripe-structured array of distinct ssDNA oligomers on polylysine coated glass. Previously, this array was converted into an n-element antibody array through DNA hybridization of appropriately labeled capture antibodies11. However, here a second DNA flow pattering step is carried out at right angles to the first to yield an n × m array, where n and m are the numbers of microchannels utilized for the two flow patterning steps (Figure S2 and Figure S3, supporting information). The flowpatterned ssDNA oligomers are designed so that each n × m array element has a unique molecular identity for localizing an appropriately labeled capture antibody (Figure S4, supporting information). See Supporting Tables S1 and S2 for the biomolecular reagents used here. This approach, which was recently reported by us for assembling single cell arrays12, allowed for the creation of a 9-element array (7 of which were used here) within each 0.15 nanoliter microchamber.
The second advance involves the molded elastomer component of the SCBC (refer to Figure 1 and Figure S1, supporting information). Previously, individual microchambers were isolated from each other, and from lysis buffer, using programmable valves. The valves take up significant chip space, and so limit the numbers of experiments per SCBC. The valve-less architecture used here permits some 30-fold more cellular assays than the previous SCBCs that required cell lysis10. When the PDMS layer is bonded to the barcode patterned slide, the primary points of contact are PDMS posts. These posts are deformable and compressible under stress. The posts support all of the microchambers in the state where the microchambers are open to the microchannel volumes. This ‘Open’ state is used for cell loading. By applying slight pressure, the PDMS posts collapse, but the rest of the PDMS structure is only slightly deformed. This Close-I state permits chemical communication between the microchannels and the microchamber, but does not allow the cells to move between those volumes. Additional pressure increases the deformation of the PDMS, completely isolating the microchambers from the microchannels. This is the Close-II state, and it is used for cell incubation. The pressure required to access these various states can be estimated by correlating an adjustment screw position with microscopic imaging. For the Open state, cells and food dye can flow between the microchambers and microchannels. For the Close-I state, only food dye can flow between these volumes.
For each microchamber, 6 array elements assayed for 6 functional proteins, with a 7th spot providing an alignment marker (Figure. 1d). The sensitivity of each protein assay is antibody limited, but typically approaches a few hundred copies per cell (see calibration and cross-reactivity data in Figure. S4, supporting information)10, which is in the range of relatively rare proteins13, 14. We previously demonstrated that the measurement error for similar single or few-cell experiments is 10% or less10, and that data sets collected across different microchips, but for otherwise identical experiments, are statistically indistinguishable9.
We assayed for 3 cytoplasmic phospho-proteins (phospho(p)-Erk, p-s6k, and p-Akt), one phosphorylated receptor tyrosine kinase (p-Epidermal Growth Factor Receptor (p-EGFR)), and two secreted proteins (interleukin(IL)-6 and Vascular Endothelial Growth Factor (VEGF)). Calibration (and cross-reactivity) curves were utilized to convert the assayed fluorescent levels for each protein (Figure S4, Figure S5, Supplementary Information) into copy numbers per cell. These proteins are associated with growth factor-induced PI3k signaling, and their levels may be interpreted as a measurement of signaling activity15, 16. We studied U87 EGFRvIII cells17, 18, which are representative of certain GBM tumors that contain the EGFR variant III oncogenic protein. EGFRvIII renders the cells constitutively active, but still responsive to growth factor stimulation10. We validated the platform using both T cells (see below) and the isogenic U87 cell line with and without Epidermal Growth Factor (EGF) stimulation. Those cells are not constitutively activated, but EGF stimulation provides a sort of surrogate for the EGFRvIII oncogene (Figure S6, S7, supporting information).
Results
A statistically significant data set of 1-, 2-, and 3-cell assays (plus 0-cell controls) can be collected in a single experiment (Figure 1e). Such data will contain around 1500, 2000, 500, and 150 0-, 1-, 2-, and 3-cell assays, respectively. The 0-cell data provides a measurement of the protein background levels. The 1-cell data yields a measurement of a single cell, unperturbed by any additional cells. For the 2-cell assays, we bin the data into 5 ranges of cell-cell separation (spanning from 0–150 μm), with ~100 experiments per range. The 3-cell data is used to test predictions from the 2-cell data.
We carried out two types of experiments. For the first, the cells are loaded onto the chip, and held just long enough (~30 min) to permit imaging to catalogue the numbers and locations of cells in each microchamber. The cells are lysed, and the proteins assayed. The second type of experiment is carried out identically, except that the cells are incubated on-chip in a CO2 incubator at 37°C for 6 hours prior to lysis. We are interrogating secreted proteins and phosphorylated kinases associated with phosphoinositide-3-kinase (PI3k) signaling. The activation (phosphorylation) of the assayed proteins, or the stimulation of protein secretion, is a relatively rapid process once the cells have been exposed to appropriate growth factors19. Based upon the cellular production of growth factors, we estimated that 6 hours was sufficient to interrogate the influences of cell-cell induced signaling. We find, for example, that VEGF and IL6 are readily detected from single cells following the 6 hour incubation, but not after a 30 minute incubation. The cells are also spread out and adherent after 6 hours, but not after 30 minutes. For proteins up to 100 kDa size (growth factors are typically < 10 kDa), diffusion times across the length of a microchamber are 150 seconds or less 20.
Molecular signatures of the distance-dependent interaction between two U87 EGFRvIII cells are presented in Figure 2a and 2b for the 6 hour and 30 minute incubation experiments. The 2-cell data is normalized by the 1-cell data. Thus, for Figure 2a and 2b the y-axis value of 1.0 corresponds to where the average amount of protein in the 2-cell experiments equals twice the 1 cell average. When average protein levels from 2 cells are below (or above) 1.0, the implication is of signaling inhibition (or activation) relative to isolated single cells. A more complete statistical representation of these data may be found in Figure S8, supporting information.
Figure 2.

Cell-separation dependent signaling activity within the PI3k signaling network. (a) Data for 2 cell, 6-hour incubation experiments. Cell distances are measured as shown in the inset. The ~500 2-cell experiments are binned according to cell separation, and the average protein level is calculated for each distance range, and then normalized to the single cell data (× 2) from the same chip. All proteins show evidence of inhibition at small separation, and 3 proteins (IL-6, p-EGFR, and p-ERK) are activated at larger separations. (b) No distance effect is observed when the cells are incubated for 30 minutes (secreted proteins are not detected at short incubation times). (c) Coordination of the PI3k signaling network for 1-cell, 2-cell (with distance dependence), and 3-cell experiments. The pie charts represent the composition of the 3 largest amplitude eigenvectors of the protein-protein-covariance matrix.
The key result of Figure 2a is that, when two U87 EGFRvIII cells are incubated for 6 hours in close proximity, the interaction is inhibitory: signaling activity is reduced by between 1.5 – 2.5 fold, depending upon the protein. At large separations (120–150 μm), the protein levels (except p-S6k) have risen to at or above those observed for two isolated cells, implying mostly activating interactions. No such distance dependence is seen for 2 cells incubated for 30 minutes (Figure. 2b). Cell contacts are not always visible from the fluorescent images (Figure 1a). However, it appears that most cells closer than 60 μm have contacts, while cells >90μm apart do not. Thus, we may be observing a form of contact inhibition. IL-6, which exhibits the largest amplitude changes, is a growth and survival factor in human GBM cells, and can promote their invasiveness21. Thus, when the cells are out of contact, IL-6 may be produced to promote cell growth. This is consistent with the strong increase in p-EGFR levels, which likely indicates increased levels of growth factor signaling (EGF secretion) at large separation distances. Secreted proteins, from non-adherent T-cells incubated for 6 hours, were similarly assayed (Figure S7, supporting information) as a control experiment. Those cells move within their respective microchambers during the course of the experiment, and so would not be expected to yield a distance-dependence in the assayed protein levels, which is what was found.
We can quantitate how protein interactions are influenced by cell separation. The assayed proteins are associated with a common signaling network, and play various roles as upstream or downstream regulators of each other. Since we are measuring the whole panel of proteins from each microchamber, we have a direct measurement of protein-protein correlations (Figure S6; Table S3, supporting information). For the 30 min incubation assay, the proteins are not highly correlated; only 1 pair has a correlation coefficient above 0.4. For the 6 hour assay almost all protein-protein interactions, regardless of cell separation, exhibit correlation coefficients above 0.6. Those correlations increase by about 10% with increasing separation, likely indicating growth factor signaling. The amplitude and composition of the eigenvectors of the protein-protein covariance matrix can be calculated to reflect the signaling network coordination. In Figure. 2c are pie-charts representing the composition of the three dominant (out of 6) eigenvectors, as a function of cell-cell separation, along with a similar analysis for the 1- and 3-cell data, all representing 6 hour incubation. The eigenvectors for the single cells are largely single-component, indicating little coordinated signaling. For the 2-cell data, the eigenvectors reflect a much higher level of coordination. While the composition of those eigenvectors is similar at the narrowest and largest separations, it differs at the intermediate 60–90 μm distance, with p-S6k playing a strong role only within this distance range.
Analysis of the data from the minimum on-chip incubation time experiment (Figure S9, supporting information) reveals that the signaling network coordination exhibits little distance dependence, and the two-cell and one-cell eigenvector compositions are similar. These data imply that these cells are more strongly influenced by their time in the cell culture dish, rather than the microchambers. The opposite is true of the data from the 6 hour incubation experiment.
The distance-dependent behaviors seen in the 2-cell, 6 hour incubation experiments do not exert an influence on the locations of the cells within the microchambers (Figure S10, supporting information). In other words, the location of the cells is statistical. At longer times, such interactions would likely influence relative cell locations. For example, most densely packed cells would likely be the least proliferative, and so cell-cell interactions might ultimately influence cell organization within a culture dish. We now explore this issue by addressing whether the pairwise cell-cell interactions are additive.
Three cell interactions
Pairwise interactions between particles are often useful for predicting certain bulk properties. For example, many properties of rare gas solids can be successfully estimated from the functional form of the atom-atom interaction potential22. Knowledge of the exact origins of the interaction is not required for making accurate predictions, just the functional form. For the GBM cells, there are distinct biomolecular processes influencing the distance dependent behaviors revealed in Figure 2a. However, we choose to ignore those mechanisms, and instead explore the predictive utility of the protein-specific distance-dependent functions. To this end, we fitted the data of Figure. 2a to second order polynomials (Figure S11, supporting information). With those functions in hand, we predicted the 3-cell microchamber results. The predictions assumed, for cells A, B, and C, that the protein levels of cell A are influenced by the product of the distance dependent interactions with B and C, B is influenced by A and C, and so on. We simulated 104 3-cell experiments, with random placement of non-overlapping cells (as supported by the results of Figure S10, supporting information). We calculated the expected levels of each protein for each simulated microchamber, and then took the average over all simulations (Figure 3). With the exception of p-S6k, the simulation correctly predicts the 3 cell experiments to within experimental noise.
Figure 3.
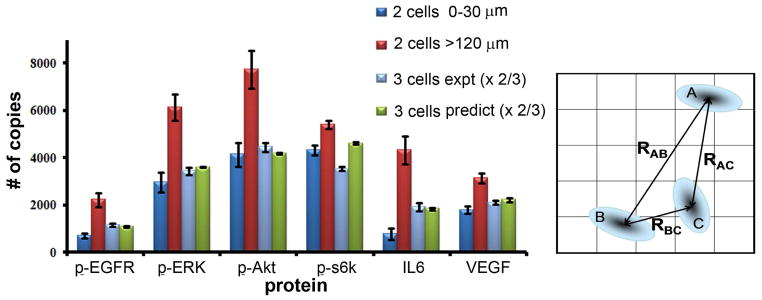
Predictions of average protein levels for 3-cell, 6 hour incubation experiments, compared with the average protein levels recorded from 155 separate 3 cell experiments. Also shown are the average values for the 2 cell experiments for both the smallest and largest of the binned distance ranges. The error bars represent standard error of mean copy number. At right is a drawing of the model used to predict the protein levels of the 3-cell experiments, from the fitted, protein-dependent interaction functions.
DISCUSSION
We interrogated the activity associated with PI3k signaling in model U87 EGFRvIII GBM cancer cells, as a function of cell-cell separation. When the cells were incubated for a short time (30 min), cell interactions were not evident. However, after incubating for 6 hours, the cells exert inhibitory influences on each other at small (< 90 μm) intercellular separations, and largely activating influences at larger separations. We fitted pairwise protein-specific cell-cell interaction functions and used them to accurately estimate the protein levels measured from statistical numbers of identically executed 3 cell experiments.
These types of measurements may provide a route towards gaining a deeper understanding of tumor architecture. At the very least, this will require measurements of a number of heterogeneous and homogeneous pairwise cellular interactions of cells culled from an actual tumor. However, it is interesting to note that the results reported here are at least consistent with what has been found for EGFRvIII+ GBM tumors. As Bonavia, et al., (2011) have written, “Paradoxically, despite EGFRvIII’s potent ability to enhance tumorigenicity (which is not shared by the wtEGFR), its expression is typically observed only in a subpopulation of cells and almost never in the entirety of the tumor.” According to the results found here, we would not expect EGFRvIII+ cells to dominate the entirety of a tumor, since, at high density, the cells would be self-inhibiting. The selection pressure appears to encourage the EGFRvIII+ cells to spread out, which is consistent with the diffuse nature of GBM tumors.
Supplementary Material
Acknowledgments
This work was funded by the National Cancer Institute (5U54 CA119347: J.R.H. PI), the Ben and Catherine Ivy Foundation, the Grand Duchy of Luxembourg, and the Jean Perkins Foundation. We acknowledge very helpful discussions from Professor Paul Mischel.
Footnotes
Detailed description of DNA array patterning, chip fabrication, cell preparation, assay quantification and statistics, simulations, figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gagliano N, Costa F, Cossetti C, Pettinari L, Bassi R, Chiriva-Internati M, Cobos E, Gioia M, Pluchino S. Oncol Rep. 2009;22(6):1349–1356. doi: 10.3892/or_00000574. [DOI] [PubMed] [Google Scholar]
- 2.Bonavia R, Inda MD, Cavenee WK, Furnari FB. Cancer Res. 2011;71(12):4055–4060. doi: 10.1158/0008-5472.CAN-11-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Nature. 2007;449(7162):557–U4. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 4.Hui EE, Bhatia SN. Proc Natl Acad Sci U S A. 2007;104(14):5722–6. doi: 10.1073/pnas.0608660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson CM, Chen CS. FEBS Lett. 2002;514(2–3):238–42. doi: 10.1016/s0014-5793(02)02370-0. [DOI] [PubMed] [Google Scholar]
- 6.Puliafito A, Hufnagel L, Neveu P, Streichan S, Sigal A, Fygenson DK, Shraiman BI. Proc Natl Acad Sci U S A. 2012;109(3):739–44. doi: 10.1073/pnas.1007809109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meiners JC, Quake SR. Phys Rev Lett. 1999;82(10):2211–2214. [Google Scholar]
- 8.Sear RP, Chung SW, Markovich G, Gelbart WM, Heath JR. Phys Rev E. 1999;59(6):R6255–R6258. doi: 10.1103/physreve.59.r6255. [DOI] [PubMed] [Google Scholar]
- 9.Shin YS, Ahmad H, Shi QH, Kim H, Pascal TA, Fan R, Goddard WA, Heath JR. Chemphyschem. 2010;11(14):3063–3069. doi: 10.1002/cphc.201000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi QH, Qin LD, Wei W, Geng F, Fan R, Shin YS, Guo DL, Hood L, Mischel PS, Heath JR. Proc Natl Acad Sci USA. 2012;109(2):419–424. doi: 10.1073/pnas.1110865109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bailey RC, Kwong GA, Radu CG, Witte ON, Heath JR. J Am Chem Soc. 2007;129(7):1959–1967. doi: 10.1021/ja065930i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vermesh U, Vermesh O, Wang J, Kwong GA, Ma C, Hwang K, Heath JR. Angew Chem Int Edit. 2011;50(32):7378–7380. doi: 10.1002/anie.201102249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang B, Wu HK, Bhaya D, Grossman A, Granier S, Kobilka BK, Zare RN. Science. 2007;315(5808):81–84. doi: 10.1126/science.1133992. [DOI] [PubMed] [Google Scholar]
- 14.Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, Aebersold R. Mol Syst Biol. 2011:7. doi: 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phung TL, Ziv K, Dabydeen D, Eyiah-Mensch G, Riveros M, Perruzzi C, Sun J, Monahan-Earley RA, Shiojima I, Nagy JA, Lin MI, Walsh K, Dvorak AM, Briscoe DM, Neeman M, Sessa WC, Dvorak HF, Benjamin LE. Cancer Cell. 2006;10(2):159–170. doi: 10.1016/j.ccr.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H, Lathia JD, Wu QL, Wang JL, Li ZZ, Heddleston JM, Eyler CE, Elderbroom J, Gallagher J, Schuschu J, MacSwords J, Cao YT, McLendon RE, Wang XF, Hjelmeland AB, Rich JN. Stem Cells. 2009;27(10):2393–2404. doi: 10.1002/stem.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang HJS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD, Huang CM, Gill GN, Wiley HS, Cavenee WK. J Biol Chem. 1997;272(5):2927–2935. doi: 10.1074/jbc.272.5.2927. [DOI] [PubMed] [Google Scholar]
- 18.Huang PH, Mukasa A, Bonavia R, Flynn RA, Brewer ZE, Cavenee WK, Furnari FB, White FM. Proc Natl Acad Sci USA. 2007;104(31):12867–12872. doi: 10.1073/pnas.0705158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barkovich KJ, Hariono S, Garske AL, Zhang J, Blair JA, Fan QW, Shokat KM, Nicolaides T, Weiss WA. Cancer Discovery. 2012;2:450–457. doi: 10.1158/2159-8290.CD-11-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu MK, Li P, Giddings JC. Protein Sci. 1993;2(9):1520–1531. doi: 10.1002/pro.5560020917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Q, Li G, Li R, Shen J, He Q, Deng L, Zhang C, Zhang J. J Neurooncol. 2010;100(2):165–76. doi: 10.1007/s11060-010-0158-0. [DOI] [PubMed] [Google Scholar]
- 22.Niebel KF, Venables JA. In: Rare Gas Solids. Klein ML, Venables JA, editors. Academic Press; New York: 1997. pp. 558–589. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.