

Abstract
We report on a novel, high-dimensional method to detect autoantibodies that are complexed with their natural autoantigens. Specifically, autoantibody-autoantigen complexes in serum or plasma are directly incubated onto a high-density antibody microarray. Detection of the bound autoantibody-antigen complex is made via fluorescently labeled anti-human immunoglobulin G or other immunoglobulin isotype secondary antibodies and quantification in a microarray scanner. Uncomplexed antibodies do not interfere with this assay. The whole process is very rapid and applicable for high-throughput screening without the need for production of proteins or immunoglobulin purification from the samples. Using these methods, we found that plasma from healthy individuals contains hundreds of autoantibodies complexed with cellular proteins. Thus, this highly sensitive, multiplex method is capable of discovering new autoantibody-antigen or circulating immune complexes many of which will likely be useful for disease detection and characterization.
Keywords: autoantigen, autoantibody, autoantigen-autoantibody complexes, circulating immune complexes, microarray, high-throughput profiling
Introduction
Autoantibodies are produced by the humoral immune response against self-molecules. By definition, autoantibodies are present in autoimmune diseases but they have also occasionally been found to be associated with other diseases including cancers.1 Autoantigens could trigger an immune response because they are over/aberrantly expressed, are mislocalized, have point mutations,2 have altered post-translational modifications (PTMs),3 are misfolded, or are truncated due to aberrant splicing or proteolytic cleavage.4 However, it is still not well known why certain proteins become autoantigens or why some individuals make autoantibodies and others do not. Furthermore, we have no idea how prevalent autoantibodies are in healthy individuals. Autoantibodies are attractive sources of potential disease biomarkers.5 Immune surveillance often occurs early during a disease process so antibodies could potentially detect a disease before overt symptoms occur particularly in chronic diseases. Antibody production can be amplified as part of an immune response, so low levels of antigen can still lead to a robust signal. Antibodies are high affinity, structurally stable reagents, reducing the need for extensive upfront purification. Thus, if immune complexes are identified that are specific to disease conditions, definitive clinical screening might be possible.
In fact, there are a several assays for antigen-antibody or circulating immune complexes (CIC) currently used in the clinic. Most of these assays have been developed for bacterial, viral or allergen antigens. In a few cases, detection of autoantigens as CIC has been proposed to enhance the sensitivity of cancer detection or prognosis monitoring utilizing prostate-specific antigen (PSA),6 squamous cell carcinoma antigen,7 carcinoembryonic antigen8 and CA-125.9 Thus, autoantigen-antibody complexes have been shown to be useful disease biomarkers, but progress in the discovery of new CICs has been extremely slow. The few current CIC assays that exist were developed based on known protein markers on a one-at-a-time basis. We believe the major impediment to more broad adoption of CIC as biomarkers is a lack of research tools to screen putative CIC in a high-throughput manner.
In contrast to CIC-related methods, there are several screening technologies for free autoantibody detection including protein or peptide arrays,10–14 reverse capture arrays,15, 16 and SEREX (serological analysis of tumor antigens by recombinant cDNA expression cloning).17–20 Protein array analysis to detect autoantigens is usually performed by printing synthetic, recombinant or natural proteins on an array surface followed by incubation with an antibody containing solution and detection of the antibody. Although high density protein arrays are commercially available now, they are costly and if bacterially produced proteins or synthetic peptides are used, it is uncertain that the printed proteins have relevant PTMs. An alternative method is nucleic acid programmable protein array (NAPPA) where an immobilized DNA template for a tagged version of the protein is translated using reticulocyte lysate and the resulting protein immobilized via an anti-tag antibody within the same spot.21 Reverse capture microarrays are another approach that usually involves fractionation of proteins from tissue lysates, cell culture, serum or plasma and spotting the fractions on a microarray. The solution containing the antibody is then incubated on the array and the antibody detected with labeled secondary anti-human antibody. The positive fractions are analyzed for the corresponding antigen. For SEREX methodology, tumor mRNA is typically converted to cDNA clones, expressed in Escherichia coli and antibodies detected via a variety of screening strategies. All of these methods require that the autoantibody antigen binding site to be available for binding, i.e., not to be complexed with their antigen.
In this paper, we propose a novel, high dimensional method to detect antibodies that are complexed with their natural autoantigens. Our protocol utilizes a high-density antibody microarray platform that we print in-house. In the examples presented here, approximately 3600 antibodies were printed in triplicate. Some of the antibodies target the same proteins at different peptide sequences. After addition of the solution containing the autoantibodies (e.g., serum, plasma or lysate), antigen-autoantibody complexes will be captured on the array as long as the epitopes for the autoantibody and the antibody on the array do not sterically hinder each other. The captured autoantibody-antigens are then detected by fluorescently labeled anti-human immunoglobulin G (IgG). Free plasma proteins not complexed to their autoanibodies will be captured on the array, but they will give no signal. Free autoantibodies not bound to antigens or complexes that do not have a corresponding antibody on the array will be washed away from the array surface. Thus, this method detects only autoantigen complexes present in the samples and represented on the antibody microarray. Clearly, larger format antibody arrays like the ones shown here with broader proteomic coverage and/or multiple antibodies to antigens of specific interest have a distinct advantage for autoantibody-antigen discovery experiments. The whole process is very rapid and can be designed for high throughput experiments. In addition to their biomarker utility, more broad characterization of the full repertoire of autoantibody-antigen immune complexes will help us study the biogenesis and characteristics of the complexes and define the nature of autoantigenic epitope.22
Experimental section
Autoantigen-Autoantibody Complex Detection
The completely de-identified plasma samples used were collected prior to colonoscopy under an Institutional Review Board approved protocol and diagnosis was confirmed by colonoscopy/pathology review. We probed these samples with our in-house printed antibody microarrays. Each array contained approximately 3600 human-protein specific antibodies in triplicate (10800 total spots) that are covalently immobilized via N-hydroxysuccinimide (NHS)-ester reactive 3-D thin film surface slides (Nexterion H slide, Schott). Printed antibodies were selected based on our differential proteomic analyses from the colon, breast, pancreas and ovarian cancer plasma proteins or general interest as signaling and cancer-related proteins. Briefly, the frozen microarray slides were equilibrated to room temperature for 30 min and hydrated in 0.5% Tween 20 in phosphate buffered saline (PBS) and then rinsed with distilled/deionized water (dd H2O). The slides were then blocked by incubation for 30 min with 0.3% (v/v) ethanolamine in 50 mM sodium borate pH 8, followed by 30 min with 1% BSA (w/v), 0.5% Tween 20 in PBS. Next, the arrays were washed with 0.5% Tween 20 in PBS, followed by dd H2O. Then, the arrays were dried by centrifugation at 500 rpm for 8 min in a swinging bucket rotor with a slide rack holder (Sorvall Legend RT). The antibody-printed area of the arrays was covered with a coverslip (mSeries Lifter Slips, 22×25×1 mm, Thermo Scientific). To detect free protein in the plasma samples, we depleted albumin and IgG and 200 μg of the remaining protein from either the case or control sample was labeled with Cy5 or from a reference sample (a pool of plasma from seven healthy individuals) was labeled with Cy3, mixed and analyzed as previously described.23, 24 To detect the presence of autoantibody-antigen complexes, 1 μl of undepleted human plasma was diluted 1:80 in 0.1% (w/v) BSA, 0.5% Tween 20 in PBS, pipetted onto the slide at the microarray/coverslip junction and incubated for 60 min at room temperature. Then, the slides were washed two times for 5 min with 0.5% Tween 20 in PBS. Human bound autoantigen-autoantibody complexes were detected after incubation with Alexa Fluor 546-goat anti-human IgG and Alexa Fluor 647-goat anti-human IgM or Alexa Flour 647-goat anti-human IgG alone (all highly cross absorbed and from Invitrogen; diluted 1:20000 in 0.1% BSA, 0.5% Tween 20 in PBS) for 60 min at room temperature. The arrays were washed two times for 5 min with 0.5% Tween 20 in PBS, followed by two times with PBS (5 min each) and once with dd H2O water followed by drying by centrifugation. As a control to determine background levels of signal, the arrays were incubated with just secondary antibody (no plasma added) and the resulting signals were used for background subtraction. Finally, the slides were scanned on a GenePix 4200A microarray scanner (Axon Instruments) to produce red (Alexa Fluor 647) and green (Alexa Fluor 546) images. Spot intensities of the scanned array images are obtained using Genepix Pro 6.0 image analysis software. For this, the raw Genepix Array List (GAL) file is aligned and resized to fit the individual spot features. The average pixel intensity within a feature is used and the median value out of triplicate is selected for the intensity calculation to reduce the effect of outliers. For determination of the number of positive autoantibody complexes, we used a threshold of 3000 arbitrary intensity units after background subtraction.
Immunoprecipation and Western blotting
Immunoprecipiation was performed as detailed in the Instructions of UltraLink Immobilized NeutrAvidin Protein (Pierce) with specific conditions as follows. Anti-LAMB1 or HIS-tag antibodies (10 μg, both Santa Cruz Biotechnology) were incubated with 10 μl of 10 mMNHS-biotin(Pierce) overnight at 4°C followed by removal of non-reacted excess biotin using centrifugal filter units (10 kD cut off, Millipore). The biotinylated LAMB1 and His-tag and unbiotinylated LAMB1 (10 μg)antibodies were each coupled to UltraLink Immobilized NeurtrAvidin Protein (Pierce) for 30 min at room temperature and the beads thoroughly washed. Plasma (20 ul) from the colon cancer patient used in Fig. 2 (20 μl diluted to 100 μl) was incubated with the 3 tubes containing NeutrAvidin beads for 30 min at room temperature. After washing, bound immune complexes were eluted with 100 mM glycine-HCl, pH 2.5 containing 0.01% n-Octyl β-D-glucopyranoside. Western blotting was performed as previously described25 with the following specific conditions. Eluted complexes were separated using non-reducing NuPAGE 4–12% Bis-Tris gel system with MOPS SDS running buffer (NOVEX).IgG detection utilized Alexa Fluor 647 anti-human IgG (H+L) (Molecular Probes) with an Odyssey imaging system (LI-COR) and associated software.
Figure 2. Protein and autoantibody-antigen complexes in control and colon cancer plasma samples.
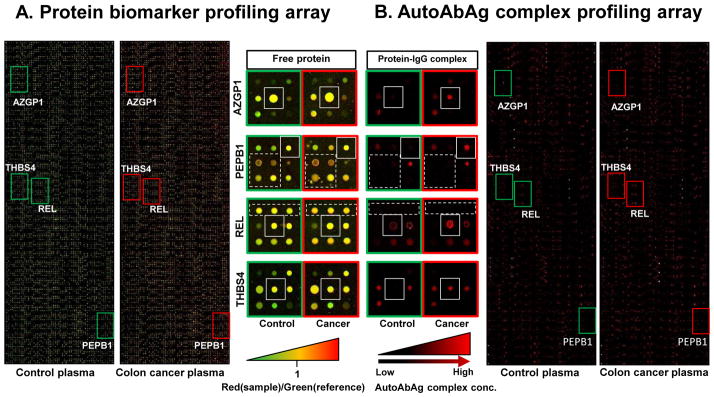
(A) Plasma protein biomarkers were profiled by our antibody microarray method. IgG and albumin depleted proteins (200 μg) from colon cancer case or control sample were labeled with Cy5 dye (red) and incubated with 200 μg of a similarly depleted pooled reference sample labeled with Cy3 dye (green). (B) Autoantibody-antigen complexes (AutoAbAg) were profiled using our novel method. Undepleted and unlabeled plasma samples from the same patients that were analyzed in A for proteomic profiling were screened for AutoAgAb complex formation as described in Fig. 1. Proteins in the solid-outlined boxed regions show no significant change in colon cancer and control plasma but their levels of immune-complex are significantly higher in cancer. Proteins within the dashed-outlined boxes are relatively abundant proteins that do not show any significant AutoAbAg complex detection.
Mass Spectrometry
Protein A precipitation of human IgG from 10 μl of a plasma sample from an individual that had colon cancer was performed using Dyna Beads Protein A(Novex, Life Technologies). Briefly, 10 μl of plasma was diluted in 200 μl PBS with 0.05 % Tween 20 and incubated with Dyna Beads for 20 min at room temperature. After several washing steps to remove unbound proteins, bound normal IgGs and auto AbAg complexes were eluted in 100 ul of SDS-PAGE sample buffer at 80°C without reductant. 20 μl of the sample was separated in 4–12 % NuPAGEBis-Trisgel using MOPS running buffer (Novex, Life Technologies). The gel was silver stained, the lane below the portion corresponding to IgG (~150 kDa) was divided into 4 pieces, and proteins were extracted and subjected to tandem mass spectrometry (MS/MS) on an Orbitrap XL (Thermo Scientific) as previously described.25
Results and Discussion
We have developed a high dimensional assay to discover complexes of antibodies and antigens. A schematic describing the assay is shown in Fig. 1. Antibodies that have been covalently linked to the array surface (Fig. 1A) are incubated with plasma that contains free plasma proteins, IgG, free autoantibodies (AutoAb), and autoantibody-antigen (Auto AbAg) complexes (Fig. 1B). After appropriate incubation (Fig. 1C, typically 1h), unbound proteins and IgG’s not bound to autoantigens present on the array are washed away leaving proteins bound to the arrayed commercial antibodies and any autoantibodies bound to these proteins (Fig. 1D). The autoantibody-autoantigen complexes (AutoAbAg) are then detected via fluorescently labeled, anti-human IgG secondary antibodies (Fig. 1E).
Figure 1. A schematic overview of the autoantibody-autoantigen complex profiling array method.

(A) Antibodies are spotted onto slides and covalently linked via N-hydroxysuccinimide (NHS)-ester reactive groups to the slide. (B) The sample containing the autoantibody complexes, free plasma proteins and IgGs is applied to the array. (C) Proteins in the serum or plasma samples are affinity captured when the appropriate commercial antibody is present on the antibody microarray surface. (D) Only plasma proteins that bind tightly to the capture antibodies on the array will remain after washing. (E) The autoantibody-antigen complex is detected by fluorescently tagged, anti-human secondary antibody.
In Fig. 2, we show images from our antibody microarray containing 3600 different antibodies to ~3000 different proteins spotted in triplicate (total features with control spots is 10800; the full list of antibodies is shown in a Supplemental Table). Plasma samples from a healthy control and from a patient who has colon cancer were analyzed both by our autoantibody-antigen profiling array method and our published antibody microarray method for proteomic profiling.23 For the latter, we labeled proteins in the colon cancer case sample or the control plasma with Cy5 and a reference sample with Cy3. Equal total protein amounts from the case or control samples are mixed with the reference and incubated on the array. In Fig. 2A, especially in the blown-up inset portions, one can see a number of labeled proteins bound to specific antibodies on the array (i.e., red or green signal is present), and a range of apparent protein concentration is evident by the different intensity of the spots. If the protein is higher in the reference sample, the spot will appear green. If the amounts are approximately equal, the spot will be yellow, and if it is higher in the control or cancer sample than the reference, it will be red. In protein biomarker profiling arrays (Fig. 2A), antibodies to AZGP1, PEBP1, REL and THBS4 (shown in insets as solid outlined boxes) had red/green (R/G) channel ratios of 1.12,0.44, 0.81 and 0.55 for the control sample and 1.59, 0.68, 1.10, 0.93 for the cancer sample, respectively, and generally appeared greenish to reddish yellow.
In Fig. 2B, we probed new arrays with diluted but unpurified plasma samples from the same subjects. These arrays show only red signal from the Alexa Fluor 647-labeled anti-human IgG secondary antibody bound to the IgG part of the antigen-autoantibody complex (see Fig. 1). Assessing the same four proteins as shown in the protein comparison, several differences in the abundance of autoantibody-antigen complexes were evident. The average arbitrary red signal intensities of the corresponding spots between the control and colon cancer case sample were increased from 938 to 5502 for AZGP1 immune-complex(5.87 fold), from 170 to 3754f or PEBP1 immune-complex(22.08 fold), from 2506 to 6067 for REL immune-complex (2.42 fold) and from 688 to 5891 for THBS4 immune-complex (8.56 fold) Many spots (e.g., those in dashed box in Fig. 2B) showed little or no signal indicating autoantibodies were not present even though the plasma concentrations of these proteins shown in dashed boxes in Fig. 2A indicated they were generally as abundant as AZGP1, PEBP1, REL and THBS4. When we examine the entire array, we conclude that there is no obvious relationship between total protein and autoantibody-antigen complex level, indicating specificity of the assay. Since most microarray scanners are capable of signal detection at multiple wavelengths, we decided to attempt to multiplex assays for both anti-human IgG and IgM in one array to determine the relative frequency of autoantigen complexes between the two isotypes and determine the specificity of the assay to distinguish the different isotypes. However, we first needed to investigate whether our assay might show spectral dye bias. Thus, a plasma sample from the cancer patient used in Fig. 2 was incubated on an array and the captured IgG immune complexes were simultaneously detected with equal amounts of goat anti-human IgG antibodies labeled with either Alexa Fluor 647 (our secondary used in most of the experiments) or Alexa Fluor 546 by the same manufacturer. After setting microarray scanning conditions to yield an ~1:1 spot intensity count ratio, the resulting overlay image of the two dyes showed that virtually all the detected spots appeared yellow. When spots greater than 3000 intensity were examined, a plot of log2 transformed 635 nm/532 nm intensity ratios for each of the spots showed a clear linear 1:1 relationship and average ratio of 1.03±0.02 (mean ± std dev)(Fig 3A). Therefore, multiplexing of different autoantibody isotypes seemed plausible.
Figure 3. Simultaneous IgG and IgM multiplex autoantibody-antigen complex array analysis.

To assess spectral bias, IgG complexes were detected simultaneously with anti-human IgG antibodies labeled with two dyes and their spot intensities were plotted on Cy5 and Cy 3 log2 coordinates (panel A). IgG complexes showing >3000 intensities are all found clustered near a slope 1 trend line. Detection of IgG- and IgM-antigen complexes using the secondary anti-human Ig reagents with no sample (panels B1–B3) or the colon cancer case sample (panels B4–B6) was performed. Panels B4 and B5 show CRB3 (spot 1: intensity 2863) and TAGLN2 (spot 2: intensity 2703) are exclusively associated with IgG autoantibodies (i.e., not visible in panel B6). Conversely, TNC (spot 3: intensity 3653) is a putative autoantigen that is complexed only with IgM autoantibodies (only visible in panels B4 and B6). Some autoantigens are associated with both IgG and IgM autoantibodies as exemplified by spot 4 (CD5L: spot intensities 7135 (IgG) and 20927 (IgM)). Panel C shows the top 50 detected putative autoantigens on a total intensity basis with the signal from the IgG- and IgM-specific secondary antibodies associated with the autoantibody-antigen specific array antibody indicated. The C2 spot intensity detected as aIgG complex is 57837. Panel D shows the presence of array identified IgG complexes in the plasma via immunoprecipitation of the discovered autoantigens followed by Western blotting detection of coimmunoprecipitated IgGs. For immunoprecipitation, the same antibody used for microarray was biotinylated and coupled to UltraLink Immobilized NeutrAvidin gel (PIERCE). LAMB1 antibody precipitated sample shows intense IgG detection at much higher levels than immunoprecipitations performed with non-botinylated LAMB1 antibody or biotinylated anti-HIS tag antibody.
Fig. 3 panels B1–B3 show the overlay and individual channels of the same portion of a control array lacking any sample but probed with anti-human IgG (green) and IgM (red) secondary antibodies. Very little signal was observed with either secondary antibody indicating their high specificity for human IgG (i.e., little cross reaction to the antibodies on the array). In panels B4–B6, the same colon cancer case sample used above was incubated, probed with the secondary antibodies and scanned at the same laser intensity settings as the no sample control. The overlay panel B4 shows some examples of green (spots 1 and 2), red (spot 3) and yellow (spot 4) signals indicating autoantigen complexes with either IgM and/or IgG. In order to see the distribution better, we graphed the signal from the average of the triplicates to the highest 50 total intensity signal for both IgG and IgM (Fig. 3C). From this histogram, we can see that few autoantigens had similar signal levels for IgG and IgM, and in about half of the 50 highest signals one channel clearly predominated (18 were complexed with IgG and 8 were with IgM). If we calculate the total intensity of immune complexes for each isotype considering all spots having greater than 3000 spot intensity, the combined total intensity of IgG complexes was 4.92 times higher than that of IgM complexes, generally consistent with the relative amounts of IgG and IgM in blood. These results indicate there is Ig-specificity in the biology of immune complex formation and that our assay can detect that specificity. It is of note that our multiplex assay provides semi-quantitative and qualitative information over immunoglobulin Ig-specificity. Following discovery of immune complexes, a modified sandwich ELISA having autoantigen or anti-human Igs as capture agents would need to be performed with human Ig standards for quantification.
Surprisingly, we found that the total number of autoantibody-antigen complexes were much higher than we expected. If we take an arbitrary value of at least 3000 spot intensity after subtraction of the negative control slide value at that spot, we have 113 putative autoantibody-antigen IgG and 84IgM complexes out of the ~3000 distinct protein-antibody spots on the colon cancer case array. A spot intensity of 3000 is 5.3x the average background value for all spots prior to background subtraction, and it is a very obvious spot (e.g. PDPB1 in Fig. 2, intensity 3754). About two-thirds of the autoantibody-antigen IgG complexes that were present were found in both the case and control samples shown while about one-third were distinct between the samples. The detection of these IgG and IgM complexes were proven to be reproducible in five independent array experiments using the same sample, and the array to array variation is presented in Table 1.
Table 1.
Reproducibility of the microarray for autoantibody-antigen assay
| a Intra-array coefficient of variation | b Inter-array coefficient of variation | |||
|---|---|---|---|---|
| IgG-complex
|
IgM-complex
|
IgG-complex
|
IgM-complex
|
|
| CV<0.1 | 86.1% | 80.0% | 59.5% | 50.3% |
| 0.1<CV<0.2 | 9.7% | 12.3% | 31.1% | 31.9% |
| 0.2<CV<0.3 | 1.3% | 4.6% | 4.5% | 8.1% |
| 0.3<CV | 2.9% | 3.2% | 5.0% | 7.3% |
Five microarrays were independently incubated with the same colon cancer plasma. Median spot intensity at each antibody position was used and only spots showing >3000 intensity were selected for CV calculation.
We next sought to see if we could detect the array-identified immune complexes with an independent method. To do this, we chose the antibody that detected LABM1 (median signal 5459) and biontinylated it (or not as a control) and an anti-HIS-tag (a second control) antibody and bound them to UltraLink Immobilized NeutrAvidin (Pierce). Plasma (20 ul) from the same cancer patient used for Fig. 3C was applied to the antibody-coupled gel and after washing, elution and Western blotting, the relative levels of human IgG were detected. As shown in Fig. 3D, human IgG detection level was high for the biotinylated LAMB1 and barely detectable in the controls.
It is possible that human antibodies in the sample or the secondary detection reagents could interact with proteins (e.g., a complement complex) captured at a spot not through its antigen binding region but rather other portions such as the Fc region. To see if this this type of interaction was prevalent, we performed array experiments in an identical manner except the captured immune complexes were probed with Cy3-labled anti-HIS tag antibodies rather than anti-human IgG. This procedure yielded only one spot (other than the anti-Cy dye control antibodies printed on the array) with greater than 3000 intensity (NTRK1-3 atintensity 12369) and four spots between 1200 and 1000 (HSPA1A at 1165, SPARC at 1149, STIP1 at 1129, GTF2A2 at 1068). We are not sure why NTRK1-3 showed a fairly high intensity (possibly a poly-histidine sequence within the antibody variable region) but it was not detected as an autoantigen-autoantibody complex in any of our assays. Among the others, STIP1 and GTF2A2 were part of the list of 113IgG-complexes with greater than 3000 intensity described above, but their intensities were far greater by detection with Alexa Fluor 546-anti human IgG (5411 versus 1129, 12369 versus 1068). This result indicates there is probably minimal interference from Ig binding at non-antigenic sites.
We next sought to see if we could detect putative autoantigen complexes in a high throughput manner via mass spectrometry. To do this, we utilized an approach similar to Ohyama et al.26 by purifying the complex from the opposite direction, i.e., precipitating the IgG with protein A and assaying for co-precipitated proteins by mass spectrometry. The protein A precipitated IgG and proteins were run on non-reduced SDS-PAGE and after silver staining, the lane area below the prominent IgG band at ~150kDa was divided into 4 pieces, destained, extracted, trypsinized and subjected to tandem mass spectrometry (MS/MS) via standard methods.25 Using relatively stringent criteria for peptide/protein identification (Peptide Prophet error rate < 0.05 and Protein Prophet Probability > 0.89), we found 34 non-IgG or keratin related proteins in the mixture (Table 2).Of the 34, 9 were present on the array (ACTA2, ACTB, APOA1, APOE, DCD, DSP, LARP1, SERPINA3, TTR) and all had signals above background. The number of identifications was similar to Ohyama et al.26 who detected 33 proteins primarily apolipoproteins (6 proteins), complement family members (11 proteins), coagulation related proteins (4 proteins) and 12 others. We both found ApoA1 and ApoE and both were also found via the array method. Aside from the low number of potential autoantigens identified, this MS/MS technique cannot readily distinguish proteins non-specifically bound to the beads without the use of extensive controls. It is also not high throughput and there is inherent variability in the protein A precipitation step, the gel extraction efficiency, and the MS/MS analysis that would affect the overall reproducibility.
Table 2.
Complete list of colon cancer plasma autoantigens identifiable by protein A affinity purification and gel-electrophoresis coupled tandem mass spectrometry.
| Gene | Protein name | IPI ID | Mass | Group | Prob | Error | Unique |
|---|---|---|---|---|---|---|---|
| ACTA1 | Actin, alpha skeletal muscle | IPI00021428 | 42051 | 27 | 0.9334 | 0.01 | 1 |
| IPI00878173 | 37824 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI00514530 | 32275 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI00414057 | 28165 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI00922693 | 38579 | 27 | 0.9334 | 0.01 | 1 | ||
|
| |||||||
| ACTA2 | Actin, aortic smooth muscle | IPI00008603 | 42009 | 27 | 0.9334 | 0.01 | 1 |
| IPI01018712 | 36807 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI00645534 | 16759 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI00921887 | 17062 | 27 | 0.9334 | 0.01 | 1 | ||
|
| |||||||
| ACTB | Actin, cytoplasmic 1 | IPI00894523 | 10979 | 27 | 0.9334 | 0.01 | 1 |
| IPI00893981 | 11394 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI00942659 | 13859 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI00893604 | 8534 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI01018925 | 37349 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI00894365 | 17975 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI01015522 | 38633 | 27 | 0.9334 | 0.01 | 1 | ||
|
| |||||||
| ACTC1 | Actin, alpha cardiac muscle 1 | IPI00930343 | 42047 | 27 | 0.9334 | 0.01 | 1 |
| IPI01011107 | 37792 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI00023006 | 42019 | 27 | 0.9334 | 0.01 | 1 | ||
|
| |||||||
| ACTG1 | Actin, cytoplasmic 2 | IPI00021440 | 41793 | 27 | 0.9334 | 0.01 | 1 |
| IPI00930226 | 39799 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI01011344 | 37407 | 27 | 0.9334 | 0.01 | 1 | ||
|
| |||||||
| ACTG2 | Actin, gamma-enteric smooth muscle | IPI00025416 | 41877 | 27 | 0.9334 | 0.01 | 1 |
| IPI00917457 | 12969 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI00917545 | 5570 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI00917820 | 16271 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI00917282 | 11403 | 27 | 0.9334 | 0.01 | 1 | ||
| IPI00916212 | 37083 | 27 | 0.9334 | 0.01 | 1 | ||
|
| |||||||
| ALB | Serum albumin | IPI00966829 | 69227 | 8-1 | 1.0000 | 0.00 | 10 |
| IPI00022434 | 71704 | 8-1 | 1.0000 | 0.00 | 10 | ||
| IPI00216773 | 45159 | 8-2 | 1.0000 | 0.00 | 6 | ||
| IPI00384697 | 47360 | 8-3 | 1.0000 | 0.00 | 7 | ||
| IPI00878517 | 56212 | 8-3 | 1.0000 | 0.00 | 7 | ||
| IPI00953582 | 47287 | 8-3 | 1.0000 | 0.00 | 7 | ||
| IPI00965074 | 52059 | 8-3 | 1.0000 | 0.00 | 7 | ||
| IPI00745872 | 69367 | 8-4 | 1.0000 | 0.00 | 11 | ||
| IPI00878282 | 22859 | 8-5 | 1.0000 | 0.00 | 3 | ||
| IPI00908876 | 59574 | 8-6 | 1.0000 | 0.00 | 9 | ||
| IPI00965913 | 51572 | 8-6 | 1.0000 | 0.00 | 9 | ||
|
| |||||||
| APOA1 | Apolipoprotein A-I | IPI00021841 | 30778 | 23 | 0.9504 | 0.01 | 1 |
| IPI00853525 | 27909 | 23 | 0.9504 | 0.01 | 1 | ||
|
| |||||||
| APOE | Apolipoprotein E | IPI00021842 | 36154 | 15 | 0.9981 | 0.00 | 3 |
| IPI00878953 | 32558 | 15 | 0.9981 | 0.00 | 3 | ||
| IPI00879456 | 24903 | 15 | 0.9981 | 0.00 | 3 | ||
| IPI00879368 | 24647 | 15 | 0.9981 | 0.00 | 3 | ||
|
| |||||||
| CASP14 | Caspase-14 | IPI00013885 | 27680 | 1 | 1.0000 | 0.00 | 2 |
|
| |||||||
| DCD | Dermcidin | IPI00027547 | 11284 | 30 | 0.8909 | 0.02 | 1 |
| IPI00847793 | 12414 | 30 | 0.8909 | 0.02 | 1 | ||
|
| |||||||
| DSP | Desmoplakin | IPI00013933 | 331774 | 6-1 | 1.0000 | 0.00 | 8 |
| IPI00217182 | 260119 | 6-1 | 1.0000 | 0.00 | 8 | ||
| IPI00746877 | 19730 | 6-2 | 1.0000 | 0.00 | 1 | ||
| IPI01009332 | 156303 | 6-3 | 1.0000 | 0.00 | 6 | ||
| IPI00969616 | 278916 | 6-1 | 1.0000 | 0.00 | 8 | ||
|
| |||||||
| ECM1 | Extracellular matrix protein 1 | IPI00003351 | 60674 | 20 | 0.9611 | 0.01 | 1 |
| IPI00006969 | 46099 | 20 | 0.9611 | 0.01 | 1 | ||
| IPI00645849 | 63563 | 20 | 0.9611 | 0.01 | 1 | ||
|
| |||||||
| FLG2 | Filaggrin-2 | IPI00397801 | 248073 | 24 | 0.9468 | 0.01 | 1 |
|
| |||||||
| HRNR | Hornerin | IPI00398625 | 282390 | 3 | 1.0000 | 0.00 | 8 |
|
| |||||||
| IDUA | Alpha-L-iduronidase | IPI00964811 | 22039 | 25 | 0.9445 | 0.01 | 1 |
|
| |||||||
| KRT77 | Keratin, type II cytoskeletal 1b | IPI01012272 | 37621 | 9-20 | 1.0000 | 0.00 | 3 |
|
| |||||||
| LARP1 | La-related protein 1 | IPI00411690 | 116464 | 31 | 0.8824 | 0.03 | 1 |
| IPI00185919 | 123510 | 31 | 0.8824 | 0.03 | 1 | ||
| IPI00975979 | 20894 | 31 | 0.8824 | 0.03 | 1 | ||
| IPI00977919 | 69903 | 31 | 0.8824 | 0.03 | 1 | ||
|
| |||||||
| PIGR | Polymeric immunoglobulin receptor | IPI00004573 | 83283 | 28 | 0.9186 | 0.02 | 1 |
|
| |||||||
| POTEE | POTE ankyrin domain family member E | IPI00479743 | 121363 | 27 | 0.9334 | 0.01 | 1 |
|
| |||||||
| POTEF | POTE ankyrin domain family member F | IPI00739539 | 121445 | 27 | 0.9334 | 0.01 | 1 |
|
| |||||||
| POTEI | POTE ankyrin domain family member I | IPI00740545 | 121282 | 27 | 0.9334 | 0.01 | 1 |
|
| |||||||
| POTEJ | POTE ankyrin domain family member J | IPI00738655 | 117390 | 27 | 0.9334 | 0.01 | 1 |
|
| |||||||
| PS1TP5BP1 | HCG15971, isoform CRA_a | IPI00021439 | 41737 | 27 | 0.9334 | 0.01 | 1 |
|
| |||||||
| SERPINA3 | Alpha-1-antichymotrypsin | IPI00847635 | 47651 | 21 | 0.9576 | 0.01 | 1 |
| IPI01025667 | 50598 | 21 | 0.9576 | 0.01 | 1 | ||
| IPI01025316 | 23406 | 21 | 0.9576 | 0.01 | 1 | ||
|
| |||||||
| SRP68 | Signal recognition particle 68 kDa protein | IPI01014848 | 41373 | 29 | 0.8984 | 0.02 | 1 |
|
| |||||||
| TTR | Transthyretin | IPI00022432 | 15887 | 22 | 0.9564 | 0.01 | 1 |
| IPI00855916 | 20199 | 22 | 0.9564 | 0.01 | 1 | ||
| IPI00940791 | 20294 | 22 | 0.9564 | 0.01 | 1 | ||
| IPI01015306 | 15120 | 22 | 0.9564 | 0.01 | 1 | ||
|
| |||||||
| TUBB | Tubulin, beta | IPI01013012 | 30693 | 5-2 | 1.0000 | 0.00 | 1 |
| IPI00908770 | 35921 | 5-2 | 1.0000 | 0.00 | 1 | ||
| IPI01015236 | 39895 | 5-2 | 1.0000 | 0.00 | 1 | ||
| IPI01019113 | 49671 | 5-1 | 1.0000 | 0.00 | 2 | ||
| IPI00645452 | 47766 | 5-1 | 1.0000 | 0.00 | 2 | ||
| IPI01025656 | 52048 | 5-1 | 1.0000 | 0.00 | 2 | ||
| IPI00953417 | 27436 | 5-1 | 1.0000 | 0.00 | 2 | ||
| IPI01020686 | 48519 | 5-1 | 1.0000 | 0.00 | 2 | ||
| IPI01022164 | 48461 | 5-1 | 1.0000 | 0.00 | 2 | ||
| IPI01022236 | 46510 | 5-1 | 1.0000 | 0.00 | 2 | ||
| IPI00647896 | 41742 | 5-2 | 1.0000 | 0.00 | 1 | ||
| IPI01018768 | 30693 | 5-2 | 1.0000 | 0.00 | 1 | ||
|
| |||||||
| TUBB2A | Tubulin beta-2A chain | IPI00013475 | 49907 | 5-1 | 1.0000 | 0.00 | 2 |
| IPI00930715 | 49897 | 5-1 | 1.0000 | 0.00 | 2 | ||
| IPI01015204 | 39993 | 5-2 | 1.0000 | 0.00 | 1 | ||
|
| |||||||
| TUBB2B | Tubulin beta-2B chain | IPI00031370 | 49953 | 5-1 | 1.0000 | 0.00 | 2 |
| IPI01014387 | 39896 | 5-2 | 1.0000 | 0.00 | 1 | ||
|
| |||||||
| TUBB2C | Tubulin, beta 2C | IPI00007752 | 49831 | 5-2 | 1.0000 | 0.00 | 1 |
|
| |||||||
| TUBB3 | Tubulin beta-3 chain | IPI00013683 | 50433 | 5-1 | 1.0000 | 0.00 | 2 |
| IPI01026194 | 88381 | 5-1 | 1.0000 | 0.00 | 2 | ||
| IPI00640115 | 42433 | 5-2 | 1.0000 | 0.00 | 1 | ||
| IPI01025600 | 16432 | 5-3 | 1.0000 | 0.00 | 1 | ||
| IPI01024745 | 18288 | 5-3 | 1.0000 | 0.00 | 1 | ||
| IPI01024873 | 13583 | 5-3 | 1.0000 | 0.00 | 1 | ||
|
| |||||||
| TUBB4 | Tubulin beta-4 chain | IPI00956241 | 26908 | 5-2 | 1.0000 | 0.00 | 1 |
| IPI00926685 | 40567 | 5-2 | 1.0000 | 0.00 | 1 | ||
| IPI00023598 | 49585 | 5-2 | 1.0000 | 0.00 | 1 | ||
| IPI01015284 | 50373 | 5-1 | 1.0000 | 0.00 | 2 | ||
| IPI01013705 | 50405 | 5-1 | 1.0000 | 0.00 | 2 | ||
|
| |||||||
| TUBB6 | cDNA FLJ11352 fis | IPI00930130 | 44602 | 5-2 | 1.0000 | 0.00 | 1 |
Concern about these issues led us to further determine the performance characteristics of our array-based assay for autoantibody-antigen complexes both within the triplicates of a single array and between five arrays incubated with the same sample. Within an array, when the 3 replicate spots were analyzed, 86.1% of the signal from the IgG-specific secondary and 80.0% of the IgM-specific with an intensity greater than 3000 had a coefficient of variation (CV) of less than 0.10 (see Table 1). Between array comparisons on the same sample yielded 59.5% and 50.3% of the IgG and IgM-specific spots with coefficients of variation less than 0.1 (Table 1). Thus, even though some of the spots showed levels of variation that would not be acceptable for a specific clinical assay, as a whole they indicate that the platform is certainly reproducible enough for discovery purposes and that the signals we are observing are consistent within and between arrays.
Biomarker studies need to be performed in more than one set of samples in order to be believable, and a single study would not be sufficient to make any claims about specific autoantibody complexes being good biomarkers. However, we decided to perform a small study with 13 villous adenomas, 10 invasive cancers and 7 controls diagnosed during surveillance colonoscopy to see if any differences were apparent. In fact, all 3 groups showed statistically different IgG complexes at the p value level as reported in Table 3. However, when the high dimensionality of the array is taken into account with multiple comparison considerations and a so-called q value it taken into account, few remain significant. This result is not particularly surprising since we are assaying for over 3000 possible autoantibody complexes with a handful of samples. Thus, this data is far too preliminary and the sample set too small to make specific claims that these complexes could be good biomarkers. However, this demonstration supports that idea that differences can be found between samples (i.e., people show differences in their autoantibody-autoantigen complexes) and the technique might ultimately yield good candidate biomarkers that can be individually tested in larger sample sets to avoid multiple comparison testing issues.
Table 3.
Log2 ratios, p and q values of autoantgen-antibody complexes showing statistical significance in plasma samples from patients with villous adenoma or invasive colon cancer
| IgG complex | Villous | Invasive | ||||
|---|---|---|---|---|---|---|
| Coef | p | q | Coef | p | q | |
|
|
|
|
||||
| ADAMTS1 3 |
1.67 | <0.01 | 0.28 | - | - | - |
| BCL3 | - | - | - | 1.79 | <0.01 | 0.04 |
| BIRC5 | 1.61 | 0.02 | 0.33 | - | - | - |
| CCSP-1 | - | - | - | 1.89 | <0.01 | 0.03 |
| CD2 | 3.72 | <0.01 | 0.22 | - | - | - |
| CD86 | 3.91 | <0.01 | 0.11 | - | - | - |
| ENO1 | - | - | - | 3.35 | <0.01 | 0.03 |
| ENO2 | 2.69 | <0.01 | 0.11 | 1.49 | 0.05 | 0.10 |
| F5 | 4.02 | <0.01 | 0.11 | - | - | - |
| HIF3A | - | - | - | 4.34 | <0.01 | 0.02 |
| KEAP1 | 3.08 | 0.02 | 0.36 | 1.79 | <0.01 | 0.05 |
| KIAA1199 | - | - | - | 0.99 | 0.08 | 0.11 |
| LMN1 | 0.94 | 0.05 | 0.49 | 1.75 | <0.01 | 0.03 |
| MYC | - | - | - | 7.61 | <0.01 | 0.02 |
| NOTCH2 | 2.26 | <0.01 | 0.11 | - | - | - |
| PLA2G5 | 2.59 | <0.01 | 0.11 | - | - | - |
| PRDX6 | 3.47 | <0.01 | 0.11 | - | - | - |
| PSMB6 | 2.34 | 0.01 | 0.29 | - | - | - |
| SERPINE1 | - | - | - | 1.17 | 0.08 | 0.11 |
| TCEB3 | - | - | - | 1.20 | 0.07 | 0.11 |
| TLN1 | 7.04 | <0.01 | 0.19 | 4.95 | 0.08 | 0.11 |
| VCP | 2.90 | <0.01 | 0.11 | - | - | - |
| VWF | 5.39 | <0.01 | 0.11 | - | - | - |
Conclusions
We present a novel method to profile autoantibody-antigen complexes present in blood or bodily fluid samples. Our platform appears to be specific for Ig molecules and has reproducibility sufficient for discovery purposes. Any potential biomarkers discovered could certainly be readily adapted to well controlled single analyte assays with standards and calibration to yield low coefficients of variation. This protocol detects endogenous autoantigens that are captured on high density antibody arrays and requires very minimal sample preparation. If deemed interesting, two or more isotypes of immunoglobulins (e.g., IgG and IgM) can be analyzed simultaneously in multiplex assay using two or more scanner wavelengths (e.g., Cy3 and Cy5). We conclude that in the colon cancer case and control samples tested, there were more autoantigen-IgG than –IgM complexes and that different autoantigens usually have strong preference for one over the other. We also found different autoantibody complexes in the samples from the colon cancer patients than in the plasma from healthy individuals. While it is interesting to speculate that the disease is the cause of this difference and that specific autoantibody complexes may be diagnostic for colon cancer, we need to perform this assay with many more samples from multiple sets of colon cancer patients before we can make any claims about specific complexes being diagnostic. Finally, we speculate that the free autoantigen concentration in combination with the amount of autoantigen-antibody complex and free autoantibody might even be more informative in terms of disease status than a single biomarker alone.
Supplementary Material
Supplemental Table. Complete list of antibodies that are present on our high-density array.
Acknowledgments
Funding Sources
This work was funded in part by grant U01 CA152746 from the National Institutes of Health.
References
- 1.Desmetz C, Mange A, Maudelonde T, Solassol J. Autoantibody signatures: progress and perspectives for early cancer detection. J Cell Mol Med. 2011;15(10):2013–24. doi: 10.1111/j.1582-4934.2011.01355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soussi T. p53 Antibodies in the sera of patients with various types of cancer: a review. Cancer Res. 2000;60(7):1777–88. [PubMed] [Google Scholar]
- 3.von Mensdorff-Pouilly S, Petrakou E, Kenemans P, van Uffelen K, Verstraeten AA, Snijdewint FG, van Kamp GJ, Schol DJ, Reis CA, Price MR, Livingston PO, Hilgers J. Reactivity of natural and induced human antibodies to MUC1 mucin with MUC1 peptides and n-acetylgalactosamine (GalNAc) peptides. Int J Cancer. 2000;86(5):702–12. doi: 10.1002/(sici)1097-0215(20000601)86:5<702::aid-ijc16>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 4.Ulanet DB, Torbenson M, Dang CV, Casciola-Rosen L, Rosen A. Unique conformation of cancer autoantigen B23 in hepatoma: a mechanism for specificity in the autoimmune response. Proc Natl Acad Sci U S A. 2003;100(21):12361–6. doi: 10.1073/pnas.2035245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson KS, LaBaer J. The sentinel within: exploiting the immune system for cancer biomarkers. J Proteome Res. 2005;4(4):1123–33. doi: 10.1021/pr0500814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beneduce L, Prayer-Galetti T, Giustinian AM, Gallotta A, Betto G, Pagano F, Fassina G. Detection of prostate-specific antigen coupled to immunoglobulin M in prostate cancer patients. Cancer Detect Prev. 2007;31(5):402–7. doi: 10.1016/j.cdp.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Makrantonakis P, Pectasides D, Aggouridakis C, Visvikis A, Daniilidis J, Fountzilas G. Squamous cell carcinoma antigen, circulating immune complexes, and immunoglobulins in monitoring squamous cell carcinoma of head and neck: a study of the hellenic co-operative oncology group (HeCOG) Am J Clin Oncol. 1999;22(6):542–9. doi: 10.1097/00000421-199912000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Fuchs C, Krapf F, Kern P, Hoferichter S, Jager W, Kalden JR. CEA-containing immune complexes in sera of patients with colorectal and breast cancer--analysis of complexed immunoglobulin classes. Cancer Immunol Immunother. 1988;26(2):180–4. doi: 10.1007/BF00205613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cramer DW, O’Rourke DJ, Vitonis AF, Matulonis UA, Dijohnson DA, Sluss PM, Crum CP, Liu BC. CA125 immune complexes in ovarian cancer patients with low CA125 concentrations. Clin Chem. 2010;56(12):1889–92. doi: 10.1373/clinchem.2010.153122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larman HB, Zhao Z, Laserson U, Li MZ, Ciccia A, Gakidis MA, Church GM, Kesari S, Leproust EM, Solimini NL, Elledge SJ. Autoantigen discovery with a synthetic human peptidome. Nat Biotechnol. 29(6):535–41. doi: 10.1038/nbt.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Babel I, Barderas R, Diaz-Uriarte R, Martinez-Torrecuadrada JL, Sanchez-Carbayo M, Casal JI. Identification of tumor-associated autoantigens for the diagnosis of colorectal cancer in serum using high density protein microarrays. Mol Cell Proteomics. 2009;8(10):2382–95. doi: 10.1074/mcp.M800596-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y, Zhou Y, Qiu S, Wang K, Liu S, Peng XX, Li J, Tan EM, Zhang JY. Autoantibodies to tumor-associated antigens combined with abnormal alpha-fetoprotein enhance immunodiagnosis of hepatocellular carcinoma. Cancer Lett. 289(1):32–9. doi: 10.1016/j.canlet.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qiu J, LaBaer J. Nucleic acid programmable protein array a just-in-time multiplexed protein expression and purification platform. Methods Enzymol. :500151–63. doi: 10.1016/B978-0-12-385118-5.00009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson KS, Sibani S, Wallstrom G, Qiu J, Mendoza EA, Raphael J, Hainsworth E, Montor WR, Wong J, Park JG, Lokko N, Logvinenko T, Ramachandran N, Godwin AK, Marks J, Engstrom P, Labaer J. Protein microarray signature of autoantibody biomarkers for the early detection of breast cancer. J Proteome Res. 10(1):85–96. doi: 10.1021/pr100686b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ehrlich JR, Qin S, Liu BC. The ‘reverse capture’ autoantibody microarray: a native antigen-based platform for autoantibody profiling. Nat Protoc. 2006;1(1):452–60. doi: 10.1038/nprot.2006.66. [DOI] [PubMed] [Google Scholar]
- 16.Tang L, Yang J, Ng SK, Rodriguez N, Choi PW, Vitonis A, Wang K, McLachlan GJ, Caiazzo RJ, Jr, Liu BC, Welch WR, Cramer DW, Berkowitz RS, Ng SW. Autoantibody profiling to identify biomarkers of key pathogenic pathways in mucinous ovarian cancer. Eur J Cancer. 46(1):170–9. doi: 10.1016/j.ejca.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tureci O, Usener D, Schneider S, Sahin U. Identification of tumor-associated autoantigens with SEREX. Methods Mol Med. 2005:109137–54. doi: 10.1385/1-59259-862-5:137. [DOI] [PubMed] [Google Scholar]
- 18.Kiyamova R, Garifulin O, Gryshkova V, Kostianets O, Shyian M, Gout I, Filonenko V. Preliminary study of thyroid and colon cancers-associated antigens and their cognate autoantibodies as potential cancer biomarkers. Biomarkers. 2012;17(4):362–71. doi: 10.3109/1354750X.2012.677476. [DOI] [PubMed] [Google Scholar]
- 19.Heller A, Zornig I, Muller T, Giorgadze K, Frei C, Giese T, Bergmann F, Schmidt J, Werner J, Buchler MW, Jaeger D, Giese NA. Immunogenicity of SEREX-identified antigens and disease outcome in pancreatic cancer. Cancer Immunol Immunother. 2010;59(9):1389–400. doi: 10.1007/s00262-010-0870-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang K, Xu X, Nie Y, Dai L, Wang P, Zhang J. Identification of tumor-associated antigens by using SEREX in hepatocellular carcinoma. Cancer Lett. 2009;281(2):144–50. doi: 10.1016/j.canlet.2009.02.037. [DOI] [PubMed] [Google Scholar]
- 21.Miersch S, LaBaer J. Nucleic Acid programmable protein arrays: versatile tools for array-based functional protein studies. Curr Protoc Protein Sci. Chapter 27(Unit 27):2. doi: 10.1002/0471140864.ps2702s64. [DOI] [PubMed] [Google Scholar]
- 22.Corper AL, Sohi MK, Bonagura VR, Steinitz M, Jefferis R, Feinstein A, Beale D, Taussig MJ, Sutton BJ. Structure of human IgM rheumatoid factor Fab bound to its autoantigen IgG Fc reveals a novel topology of antibody-antigen interaction. Nat Struct Biol. 1997;4(5):374–81. doi: 10.1038/nsb0597-374. [DOI] [PubMed] [Google Scholar]
- 23.Loch CM, Ramirez AB, Liu Y, Sather CL, Delrow JJ, Scholler N, Garvik BM, Urban ND, McIntosh MW, Lampe PD. Use of high density antibody arrays to validate and discover cancer serum biomarkers. Mol Oncol. 2007;1(3):313–20. doi: 10.1016/j.molonc.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramirez AB, Loch CM, Zhang Y, Liu Y, Wang X, Wayner EA, Sargent JE, Sibani S, Hainsworth E, Mendoza EA, Eugene R, Labaer J, Urban ND, McIntosh MW, Lampe PD. Use of a single-chain antibody library for ovarian cancer biomarker discovery. Mol Cell Proteomics. 2010;9(7):1449–60. doi: 10.1074/mcp.M900496-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solan JL, Marquez-Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J Cell Biol. 2007;179(6):1301–9. doi: 10.1083/jcb.200707060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohyama K, Ueki Y, Kawakami A, Kishikawa N, Tamai M, Osaki M, Kamihira S, Nakashima K, Kuroda N. Immune complexome analysis of serum and its application in screening for immune complex antigens in rheumatoid arthritis. Clin Chem. 57(6):905–9. doi: 10.1373/clinchem.2010.157776. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table. Complete list of antibodies that are present on our high-density array.