

Abstract
Synaptic and extrasynaptic activation of the N-methyl-D-aspartate receptor (NMDAR) has distinct consequences on cell signaling and neuronal survival. Since conantokin (con)-G antagonism is NR2B-selective, which is the key subunit involved in extrasynaptic activation of the receptor, its ability to specifically elicit distinct signaling outcomes in neurons with synaptically or extrasynaptically-activated NMDARs was evaluated. Inhibition of Ca2+ influx through extrasynaptic NMDAR ion channels was neuroprotective, as it effectively enhanced levels of activated extracellular signal-regulated kinase 1/2 (ERK1/2), activated cAMP response element binding protein (CREB), enhanced mitochondrial viability, and attenuated the actin disorganization observed by extrasynaptic activation of NMDARs. Conversely, the pro-signaling pathways stimulated by synaptically-induced Ca2+ influx were abolished by con-G. Furthermore, subunit non-selective con-T was unable to successfully redress the impairments in neurons caused by extrasynaptically-activated NMDARs, thus indicating that NR2B-specific antagonists are beneficial for neuron survival. Neurons ablated for the NR2B subunit showed weak synaptic Ca2+ influx, reduced sensitivity to MK-801 blockage, and diminished extrasynaptic current compared to WT and NR2A−/− neurons. This indicates that the NR2B subunit is an integral component of both synaptic and extrasynaptic NMDAR channels. Altogether, these data suggest that con-G specifically targets the NR2B subunit in the synaptic and extrasynaptic locations, resulting in the opposing action of con-G on differentially activated pools of NMDARs.
Keywords: NMDA receptor subunits, Conantokin inhibitors, NMDAR inhibition, Synaptic and extrasynaptic NMDA receptors, Neuron signaling, Actin organization, Knockout mice
1. Introduction
It is documented that neuronal development and survival is dependent on activation of concerted pathways elaborated by receptor signaling. Neurotransmitters, such as glutamate, physiologically stimulate signals that promote pro-survival pathways and suppress apoptosis (Verhage et al., 2000). However, under certain conditions, pro-apoptotic signaling pathways are activated, triggering neuronal death. Factors that determine the pro- or anti-survival outcomes of the signaling pathways involved include the type of receptors activated, their location, and their intensity of stimulation. The N-methyl-D-aspartate receptors (NMDARs) belong to the family of ionotropic receptors coactivated by glutamate/glycine, and participate in activity-dependent processes involving long-term potentiation (LTP) and long-term depression (LTD), thus effectuating neuronal plasticity, learning, and memory (Malenka and Bear, 2004; Rebola et al., 2010). Release of excess glutamate during a variety of neuropathologies results in over-activation of the NMDARs, followed by neuronal death (Prorok and Castellino, 2007). Therefore, intervention with NMDAR inhibitors would support neuron survival (Palmer, 2001).
The first generation of NMDAR inhibitors that were neuroprotective included high-affinity NMDAR ion channel blockers, e.g., phencyclidine and ketamine (Jentsch and Roth, 1999; Olney et al., 1999), but these drugs were unsuccessful in clinical trials due to unacceptable side-effects (Kemp and McKernan, 2002). This has been attributed to their inability to antagonize specific NMDAR subunits, thus allowing indiscriminate inhibition of total NMDAR activity. The NMDARs are also referred to as glutamate receptor NMDAR (GluN) and three subunits of NMDARs have been identified: NR1 (GluN1), composed of eight splice variants (a–h); NR2 (GluN2), encompassing four different gene products (A–D); and NR3 (GluN3) that are products of two different genes, A and B (Chatterton et al., 2002; Ciabarra et al., 1995; Hollmann and Heinemann, 1994; Moriyoshi et al., 1991). A functional NMDAR is a heterotetramer composed of two ubiquitous NR1 subunits and two NR2 subunits (Paoletti and Neyton, 2007; Ulbrich and Isacoff, 2008), and different subunit compositions result in unique pharmacological properties of the assembled receptor (Cull-Candy and Leszkiewicz, 2004). Several reports have suggested the involvement of the NR2B subunit in mediation of seizures (Moddel et al., 2005), ethanol dependence (Nagy and Laszlo, 2002; Narita et al., 2000; Pawlak et al., 2005), pain modulation (Wei et al., 2001), and Huntington’s disease (Arzberger et al., 1997). Additionally, the cellular location of the NMDAR has been reported to modulate signaling pathways. NMDARs located in the postsynaptic densities (PSD) constitute the synaptic NMDARs and their activation promotes cAMP response element binding protein (CREB)-mediated signaling, with anti-apoptotic effects. When extrasynaptic NMDARs are activated, phosphorylation of extracellular signal-regulated kinases 1/2 (ERK1/2) and CREB are dampened, and mitochondrial membrane dysfunction and neuronal apoptosis (Hardingham and Bading, 2002; Ivanov et al., 2006) are also observed. The pro-survival response associated with synaptic NMDAR activation upregulates several pro-survival proteins, e.g., BCL6 and BTG2, with down-regulation of pro-death proteins, e.g., CASP8AP2, DID01. On the other hand over-stimulation of extrasynaptic NMDARs induces up-regulation of the CLCA1 channel, and activation of p38 protein kinase, both of which contribute to neuronal death (Zhang et al., 2007). It is noted that in the mature hippocampus, NR2B receptors are predominantly present in extrasynaptic locations, while NR2A receptors preferentially occupy the synapses (Tovar and Westbrook, 1999).
Taken together, it is surmised that excitotoxicity is elicited by signaling pathways coupled to NR2B-containing NMDARs that are extrasynaptically located. Thus, NR2B-specific antagonists have been employed to antagonize the ion channel of this receptor. Limited clinical success has been achieved, due to poor bioavailability and a limited pharmacokinetic profile of existing agents (Kew and Kemp, 2005). Ifenprodil, a NR2B-specific antagonist blocked ischemia-induced excitoxicity in hippocampal neurons (Hardingham et al., 2002). Other NR2B-selective compounds, e.g., Co101244 and Ro25-6981, also combatted NMDA- or glutamate-induced apoptosis (Stanika et al., 2009). Another group of compounds, the benzamidines, have high NR2B affinity with improved efficacy and pharmacokinetic profiles in a rodent hyperalgesia model (Claiborne et al., 2003; Curtis et al., 2003).
Conantokin-G (con-G), which belongs to the conantokin family of γ-carboxyglutamic acid (Gla)-containing polypeptides, is a NR2B-selective antagonist of NMDAR activated ion channels (Donevan and McCabe, 2000). This peptide has been tested for neuroprotection in rat and mouse models of stroke (Williams et al., 2000); neuropathic pain (Malmberg et al., 2003), morphine withdrawal (Wei et al., 2006), and epilepsy (Barton et al., 2004), among others, with favorable outcomes in animal models. Nonetheless, mechanisms of con-G-mediated neuroprotection, and its differential activity on synaptically- and extrasynaptically-activated NMDARs, are largely unknown. It has been demonstrated that administration of con-G, following ischemia in rats, increased Bcl-2 levels, with a concomitant decrease in levels of c-fos, a signaling pathway that is capable of providing neuroprotection (Williams et al., 2003). More recently, we have shown that con-G affected NMDA-induced phosphorylation of CREB(P-Ser133) in a developmental manner in rat hippocampal neurons (Huang et al., 2010), also signaling a cell survival mechanism. Based on this latter work, we assessed the effects of con-G on NR2B-based survival signaling pathways and on neuron cytoskeletal organization, with emphasis on synaptic and extrasynaptic pathways. The results of these studies are reported herein.
2. Materials and methods
2.1. Reagents
Tetrodotoxin (TTX), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), dizocilpine (MK-801), glycine, bicuculline, nifedipine, strychnine, N-(2,6-dimethylphenylcar bamoylmethyl)triethylammonium bromide (QX314), and 4-aminopyridine (4-AP) were purchased from Sigma (St. Louis, MO). D-(−)-2-amino-5-phosphonopentanoic acid (D-APV) was obtained from Acros Organics (Morris Plains, NJ) and NMDA was purchased from Tocris Bioscience (Ellisville, MO). Poly-L-lysine for coating dishes was a product of Peptides International (Louisville, KY).
2.2. Conantokin synthesis
The amino acid sequences of the conantokins that were chemically synthesized for these studies are provided below. The methods used for the syntheses have been published earlier (Prorok et al., 1996).
con-G: GEγγL5QγNQγ10LIRγK15SN(NH2)
con-T: GEγγY5QKMLγ10NLRγA15EVKKN20A(NH2)
Solid phase peptide synthesis was employed using an Applied Biosystems (Foster City, CA) Model 433A peptide synthesizer.
2.3. Mice
WT C57Bl/6 mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and were used as controls. The NR2A−/− mice have been previously described (Sakimura et al., 1995) and were obtained from Dr. Gary Westbrook, Oregon Health and Science University. The NR2B+/− mice (Kutsuwada et al., 1996) were obtained from National Institutes of Health/National Institute of Alcohol Abuse and Alcoholism. Both the NR2A−/− and NR2B+/− mice were fully backcrossed in the C57Bl/6 background. Animal handling and experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Notre Dame (Protocol numbers 11-096, approved on 11/24/08, and 14-086, approved on 11/2/11).
2.4. Cell culture
Primary rat hippocampal neuron cultures were prepared from embryonic day (ED)-18 Sprague-Dawley rat embryos. The hippocampal neurons were dissociated and maintained as described previously (Huang et al., 2010). Neurons were dissociated using 2 mg papain and plated on 14 mm glass-bottomed microwell dishes (Mat Tek, Ashland, MA) or 35 mm tissue culture-treated dishes (Corning Life Sciences, Lowell, MA) coated with poly-L-lysine in Neurobasal medium (Invitrogen, Carlsbad, CA), supplemented with 2% B27 (Invitrogen)/1% L-glutamine. Cell cultures were maintained at 37 °C in a humidified atmosphere with 5% CO2.
Cortical neurons were dissociated with 1 mg papain from embryos of ED-18 WT, NR2A−/−, and NR2B+/− mice. To obtain NR2B−/− embryos, mice heterozygous for the NR2B allele were bred and embryos at ED-18 were harvested and genotyped for the double NR2B−/− alleles. NR2B−/− embryos were used for neuron culture.
2.5. Activation of synaptic, extrasynaptic, and total NMDARs
Mature neurons at DIV 13–15 were utilized for differential activation of the NMDARs, which was performed with slight modifications of a published procedure (Ivanov et al., 2006). Neurons plated at a density of 2.5 × 105 cells/ml, on poly-L-lysine-coated 14 mm glass-bottom microwell dishes, were utilized for this purpose. At a time of 24 h prior to the experiment, one-half of the culture medium was changed with Neurobasal medium containing 2% B27. Three hours before stimulation, the following antagonists were added to the neurons: 1 μM TTX/40 μM CNQX/100 μM D-AP5/5 μM nifedipine. The synaptic pool of NMDARs were activated by briefly rinsing the neurons in culture medium containing 10 μM bicuculline/10 μM glycine/5 μM nifedipine and then incubating the cells for 5 min at 37 °C in the same solution. The extrasynaptic pool of NMDARs were activated by rinsing the neurons in medium containing 50 μM MK-801/5 μM nifedipine, followed by incubation for 2 min in medium containing 50 μM MK-801/10 μM bicuculline/10 μM glycine/5 μM nifedipine. The NMDAR open channel blocker, MK-801, was added to irreversibly block synaptic NMDAR ion channels. The neurons were then briefly rinsed in medium containing 1 μM TTX/40 μM CNQX/5 μM nifedipine and transferred to bath medium containing 50 μM NMDA/10 μM glycine/40 μM CNQX/1 μM TTX/5 μM nifedipine for 10 min. To activate the total NMDAR population, the neurons were rinsed in culture medium containing 1 μM TTX/40 μM CNQX/5 μM nifedipine, followed by incubation with medium containing 50 μM NMDA/10 μM glycine/40 μM CNQX/1 μM TTX/5 μM nifedipine for 10 min. To assay the antagonists, after the last step of activation, the neurons were incubated with medium containing either 40 μM con-G or 40 μM con-T for 10 min. After the various treatments, the neurons were washed 3× in PBS, fixed in 4% formaldehyde/4% sucrose/PBS for 30 min at 4 °C, and washed with PBS.
A summary of these steps as used to pharmacologically isolate different receptor populations is provided in the following scheme:
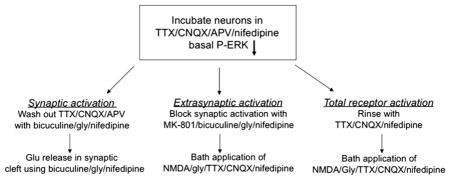
2.6. Immunostaining of neurons
Primary antibodies employed for immunoflourescent staining were: rabbit anti-P-ERK1/2(P-Thr202/Tyr204) (Cell Signaling Technology, Danvers, MA), mouse monoclonal MAP2 (Sigma), and rabbit anti-P-CREB(P-Ser133) (Cell Signaling Technology), and Phalloidin-conjugated Alexa Fluor 647 for actin stains (Invitrogen, Carlsbad, CA). The fixed neurons were stained according to manufacturer’s protocols and dilutions. Where required, goat-anti-rabbit IgG or goat-anti-mouse IgG conjugated to Alexa Flour 488 or Alexa Fluor 647 were used as secondary antibodies (Invitrogen). After labeling, the slides were rinsed and coverslipped. Coverslips were mounted with ProLong Gold antifade reagent containing DAPI (Invitrogen). Images were captured using a 40× oil objective on a Nikon Eclipse TE 2000-S microscope and acquired using the NIS-Elements AR 3-2 software (Nikon Instruments, Melville, NY). For intensity measurements of ERK(P-Thr202/Tyr204) and CREB(P-Ser133), the neurons were co-immunostained with MAP2 and visualized using Alexa Flour 647. Neuron images that were positive for MAP2 were analyzed for P-ERK(P-Thr202/Tyr204) and P-CREB(P-Ser133) intensity. Images of 3–5 random fields were acquired and fluorescent intensities of the whole image were recorded. Fluorescence intensities were calculated as percent of synaptic activation of controls.
2.7. Mitochondrial viability
After stimulation and treatment with con-G or con-T, the neurons were stained live with 100 nM MitoTracker Red CMXRos (Invitrogen), which was diluted in pre-warmed culture medium for 30 min at 37 °C. The cells were washed once with medium and then fixed at 37 °C for 15 min in culture medium containing 3.7% formaldehyde. Following fixation, the cells were rinsed in PBS and images acquired using a 40× oil objective.
2.8. Real-time calcium imaging
Measurements of Ca2+ influx through synaptic or extrasynaptic NMDARs were performed as described earlier (Hardingham et al., 2002). Neurons at DIV 13–15 were incubated with 1 μM fura-2-acetoxymethyl ester (Fura-2/AM, Invitrogen) in ACSF (artificial cerebrospinal fluid: 140 mM NaCl/5 mM KCl/2 mM CaCl2/10 mM Hepes/24 mM glucose, pH 7.2) as described (Huang et al., 2010). Synaptic activation was facilitated by application of 50 μM bicuculline/2.5 mM 4-AP/5 μM nifedipine, which was blocked either by 40 μM con-G or con-T, to evaluate the inhibitory effect of these antagonists on synaptic Ca2+ influx. For extrasynaptic activation of NMDARs, neurons were sequentially treated with 50 μM bicuculline/2.5 mM 4-AP/5 μM nifedipine, after which synaptically-activated NMDARs were irreversibly blocked with 10 μM MK-801. This was followed by application of 50 μM NMDA/10 μM glycine to stimulate extrasynaptic NMDARs. For blockage of extrasynaptic Ca2+ influx, 50 μM NMDA/10 μM glycine, containing either 40 μM con-G or con-T was added to the neurons. Imaging was performed using a Nikon Eclipse TE 2000-S microscope (Nikon) and ratiometric (340/380 nm) traces were generated using the NIS-Elements program AR 3-2 (Nikon).
2.9. Electrophysiology
Electrophysiological recordings were taken with WT, NR2A−/−, and NR2B−/− mouse cortical neurons (DIV 16–19). For isolation of extrasynaptic NMDA receptor currents, the NMDA currents were recorded after the neurons were rinsed with the extracellular bath solution (without TTX) containing 50 μM MK-801, 10 μM bicuculline, and 10 μM glycine for 1 min in order to block synaptic NMDA receptors (Ivanov et al., 2006). NMDA-related sEPSCs were recorded by bath application of 20 μM bicuculline/10 μM glycine/1 μM strychnine/20 μM CNQX. TTX was omitted from the extracellular bath solution. Membrane impermeable N-(2,6-dimethylphenylcarbamoylmethyl) triethylammonium bromide (QX314; 2 mM) was added to the intracellular solution to block the voltage-gated Na+ currents. Currents were recorded using an Axopatch 200B amplifier (Axon Instruments, Union City, CA), filtered at 2 kHz, and digitized at 5 kHz. The data were analyzed using Clampfit software (Axon Instruments, Union City, CA). Recordings of 6–10 neurons were averaged. Mg2+ block experiments were performed by applying a voltage ramp (−100 to +30 mV, within 2 s) to obtain current–voltage (I–V) curves. First, the leakage I–V curves were obtained by application of 10 μM glycine. Then, the same solution containing 100 μM NMDA was applied, and a test I–V curve was recorded. Leakage I–V curves were subtracted from test I–V curves. Normalized I–V curves were calculated relative to the value at −100 mV of the Mg2+ free solution.
2.10. Data analysis
Data are presented as mean ± S.E.M of ≥3 independent experiments. The Student’s t-test was used to examine the statistical significance of the differences between data sets. Significance was assigned at p < 0.05.
3. Results
3.1. Con-G restores survival signaling via extrasynaptic NMDARs
It is believed that the activation of the synaptic pool of NMDARs result in phosphorylation of ERK1/2 and CREB, thus contributing to neuron survival mechanisms. Conversely, stimulation of the extrasynaptic population of NMDARs prevents activation of ERK1/2 and CREB, and thereby induces neuronal death (Hardingham et al., 2002; Krapivinsky et al., 2003). Since, in mature rat hippocampal neurons, the NR2B subunits primarily occupy extrasynaptic sites, con-G, an NR2B-selective antagonist, should preferentially modulate signaling molecules via receptors containing NR2B. To examine this hypothesis, synaptic, extrasynaptic, or total NMDARs of DIV 13–15 hippocampal neurons were stimulated, and were then treated with 40 μM con-G or 40 μM con-T. This strategy allowed a comparison of the efficacy of NR2B-selective con-G and the non-subunit selective con-T on activation of ERK1/2 and CREB, as well as on mitochondrial viability. Initial data showed that basal P-ERK levels after preincubation with 1 μM TTX/40 μM CNQX/100 μM D-AP5/5 μM nifedipine were undetectable, and since our main focus was to observe the effect of con-G/T on extrasynaptic activation in comparison to synaptic activation, P-ERK levels are represented as percent of synaptic activation (taken as 100%). The data of Fig. 1A show that ERK1/2 activation primarily occurred via synaptic NMDARs (control). Addition of con-G (+G) effectively attenuated extrasynaptic-induced dephosphorylation of ERK1/2, resulting in robust P-ERK1/2 levels. On the other hand, addition of con-T (+T) only weakly maintained levels of phosphorylated ERK1/2 (Fig. 1A,B). Both antagonists effectively diminished levels of phospho (P)-ERK1/2 mediated by activation of the synaptic pool of NMDARs. Activation of both synaptic and extrasynaptic NMDARs (total) led to attenuated levels of P-ERK1/2, indicating that the effects of extrasynaptic activation on P-ERK were predominant under these conditions. Treatment of the neurons with con-G or con-T increased the P-ERK1/2 levels in the total NMDAR population, but not to the same extent as was observed when con-G was added to extrasynaptically-activated NMDARs. Qualitatively similar results were obtained with the downstream product of ERK1/2 activation, CREB (not shown), consistent with previous data (Hardingham et al., 2002).
Fig. 1.

Con-G specifically increases levels of P-ERK in neurons with extrasynaptically-activated NMDARs. A. P-ERK immunostaining of cultured rat hippocampal neurons at DIV 13–15 that were treated to selectively activate synaptic, extrasynaptic, or total NMDARs. Neurons with differentially activated subpopulations of the NMDARs were untreated (C) or treated with 40 μM con-G (+G) or 40 μM con-T (+T). B. The histogram, displaying mean ± SEM of P-ERK intensity from three independent experiments. P-ERK levels are relative to synaptic activation, taken as 100%. Synaptic (white bars); extrasynaptic, (gray bars); total (black bars). *p < 0.05, compared with the control (C) within each group. ^p < 0.05, compared with the synaptic control (C).
It had previously been shown that exposure to glutamate mediated mitochondrial membrane depolarization in cultured forebrain neurons (White and Reynolds, 1996), and this glutamate toxicity was blocked by addition of mitochondrial inhibitors and uncouplers (Stout et al., 1998). In addition, it was reported that exposure to glutamate disrupted mitochondrial membrane potential via activation of extrasynaptic NMDARs, but not activation of synaptic NMDARs (Hardingham et al., 2002). Thus, we investigated whether differential activation of the NMDAR would affect mitochondrial viability, and whether conantokin-derived NMDAR antagonists effectively counteracted this function. As anticipated, application of bicuculline-induced near-continuous bursting, did not affect mitochondrial viability (synaptic activation), but bath application of 50 μM NMDA (extrasynaptic activation) significantly decreased mitochondrial viability (Fig. 2). Activation of total NMDARs also decreased mitochondrial viability. The presence of antagonists to synaptically-activated neurons slightly diminished mitochondrial viability, but not to such an extent as was observed for ERK1/2 activation. Con-G was more effective than con-T in restoring mitochondrial viability in extrasynaptically-activated neurons (Fig. 2). Activation of total NMDARs decreased mitochondrial viability, but this loss was reestablished most successfully by the NR2B-selective con-G, than by the non-subunit selective con-T. This implies that loss of mitochondrial function is coupled to pathological activation of the NR2B subunit.
Fig. 2.

Con-G restores mitochondrial function in neurons with extrasynaptically-activated NMDARs. DIV 13–15 rat hippocampal neurons after activation of synaptic, extrasynaptic, or total NMDARs were untreated (C), or treated with 40 μM con-G (+G) or 40 μM con-T (+T), and analyzed for mitochondrial viability by staining live with Mitotracker Red. The neurons were fixed and then imaged. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2. The NR2B subunit is required for extrasynaptic activation: role in signaling and cytoskeleton organization
It has been suggested that NR2A-containing NMDARs, irrespective of their neuronal locations, enhanced the LTP and neuronal plasticity, thus promoting neuronal survival (Liu et al., 2007). However, it was recently reported that NR2B is also involved in neuronal survival and synaptic potentiation mechanisms (Martel et al., 2009). To better pinpoint the involvement of either NR2A or NR2B in con-G-mediated neuroprotection, DIV 13–15 cortical neurons from embryos of wild-type (WT), NR2A−/−, and NR2B−/− mice were employed (all fluorescence is normalized to synaptic activation). The trends of P-ERK1/2 levels of WT mouse cortical neurons, subsequent to synaptic or extrasynaptic activation of NMDARs, with or without treatment with 40 μM con-G or con-T (Fig. 3A), were very similar to those observed with rat hippocampal neurons (Fig. 1B). Surprisingly, the absence of the NR2A subunit did not prevent bicuculline-mediated synaptic ERK1/2 phosphorylation, which was robustly inhibited by con-G (Fig. 3B). Con-G treatment restored P-ERK1/2 levels in extrasynaptically-activated, as well as in total activated, NMDARs (Fig. 3B). In this regard, NR2A−/− neurons functioned similarly to WT mouse cortical and rat hippocampal neurons. However, when NR2B−/− neurons were used (Fig. 3C), P-ERK1/2 levels, via synaptic NMDARs, were not effectively inhibited by con-G. This indicated that the NR2B subunit was also present in NMDARs in synapses, through which con-G limited P-ERK1/2 formation. No significant dampening of P-ERK1/2 levels was observed via activation of extrasynaptic NMDARs. Consequently, addition of con-G had no effect on this result. Similarly, activation of total NMDARs had no demonstrable effect on P-ERK1/2 levels in the presence or absence of con-G in these cells (Fig. 3C).
Fig. 3.
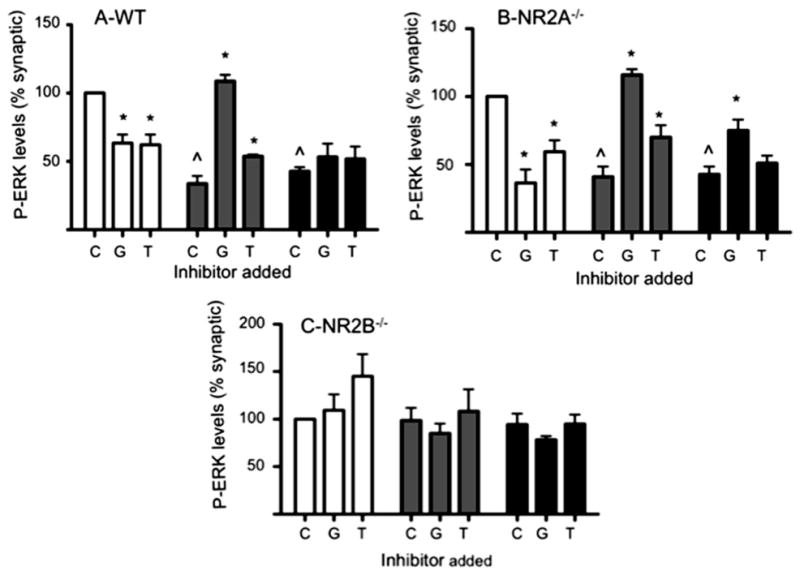
NR2B is required for extrasynaptic activation. Quantification of the effect of 40 μM con-G (G) and 40 μM con-T (T) on P-ERK1/2 levels with DIV 13–15 mouse cortical neurons, along with an untreated control (C). A. WT mouse cortical neurons. B. Mouse cortical NR2A−/− neurons. C. Mouse cortical NR2B−/− neurons. The neurons were treated to activate synaptic (white bars), extrasynaptic (gray bars), or total (black bars) NMDARs. P-ERK levels are relative to synaptic activation, taken as 100%. Quantitation of P-ERK staining shown as mean ± SEM from three independent experiments. *p < 0.05, compared with a control (C) within each group. ^p < 0.05, compared with the synaptic control (C).
The NR2A and NR2B subunits, via their C-terminal regions, directly interact with the PDZ domain of PSD-95 (Kornau et al., 1995), which, in turn, interacts with F-actin (Akashi et al., 2009; Wyszynski et al., 1997). Thus, the effects of activation of extra-synaptic NMDARs on actin organization were examined in WT, NR2A−/−, and NR2B−/− murine cortical neurons. WT cortical neurons with synaptically-activated NMDARs displayed normal actin organization, whereas extrasynaptically-activated NMDARs showed perturbations in actin organization (Fig. 4A,B). After addition of con-G to activated synaptic NMDARs (Fig. 4C), cytoskeletal actin became disorganized, suggesting that inhibition of synaptic NR2B-containing NMDARs is not beneficial to the cell, but inhibition of extrasynaptic NR2B by con-G restored cell cytoskeletal integrity (Fig. 4D), thus elaborating its protective effect. To further support this conclusion, activation of synaptic NR2B in NR2A-inactivated NMDARs (Fig. 4E), showed well-organized neuron cytoskeletal actin, which was disrupted in similar extrasynaptic NR2B-containing activated NMDARs (Fig. 4F). Con-G administration was not favorable to NR2B-containing NMDARs during synaptic activation (Fig. 4G), but restored organization to the cytoskeleton in extrasynaptically-activated NMDARs containing the NR2B subunit (Fig. 4H). In NR2B−/−-inactivated cortical neurons (Fig. 4I,J), extrasynaptic activation of the NMDARs did not lead to cytoskeleton disruption, in agreement with the con-G data for WT and NR2B−/− neurons. These data demonstrated that NR2B is the principal subunit influencing neuronal integrity when extrasynaptic NMDARs are activated.
Fig. 4.
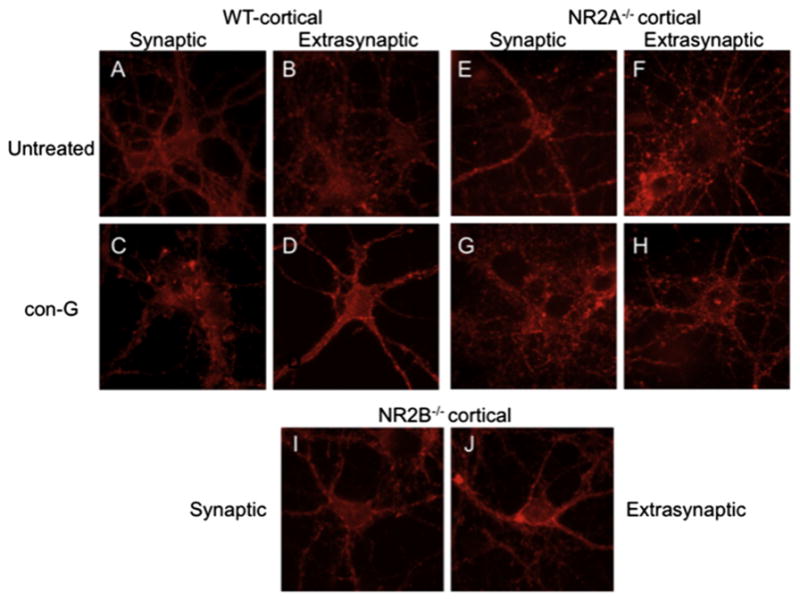
Cytoskeletal actin organization depends on NR2B. A, B. Changes in cytoskeletal actin organization using confocal fluorescence images of DIV 13–15 WT mouse cortical neurons treated to activate synaptic (A) or extrasynaptic (B) NMDARs in the absence of con-G, or in the presence (C, D) of 40 μM con-G. E–H. Actin organization of DIV 13–15 NR2A−/−-derived mouse cortical neurons with synaptic (E) and extrasynaptic (F) activated NMDARs in the absence of con-G, and in the presence (G, H) of 40 μM con-G. I, J. Actin organization of DIV 13–15 NR2B−/−-derived mouse cortical neurons with synaptic (I) and extrasynaptic (J) activated NMDARs in the absence of con-G.
3.3. Conantokin-mediated inhibition of intracellular (i) Ca2+ influx
We previously demonstrated that con-G, con-T, and another conantokin, con-R[1–17], attenuated influx of whole cell iCa2+ transients, an effect that was dependent on the maturity of the neuron (Huang et al., 2010). We further evaluated the impact of con-G and con-T on iCa2+ influx via synaptic or extrasynaptic NMDARs. With DIV 13–15 rat hippocampal neurons it was observed that con-G and con-T effectively inhibited bicuculline-induced activation of synaptic NMDARs (Fig. 5A). Very similar results were obtained with WT mouse cortical neurons (data not shown). To determine the inhibitory effect of con-G and con-T on Ca2+ influx via extrasynaptic NMDARs, bicuculline-induced synaptic Ca2+ influx was first inhibited with the open channel blocker, MK-801. This was followed by sequential bath application of NMDA/glycine to elicit Ca2+ entry via the extrasynaptic NMDARs, following which con-G or con-T was added. It was observed that both con-G and con-T inhibited the spike resulting from Ca2+ influx through extrasynaptic NMDAR channels in DIV 13–15 rat hippocampal neurons (Fig. 5B).
Fig. 5.
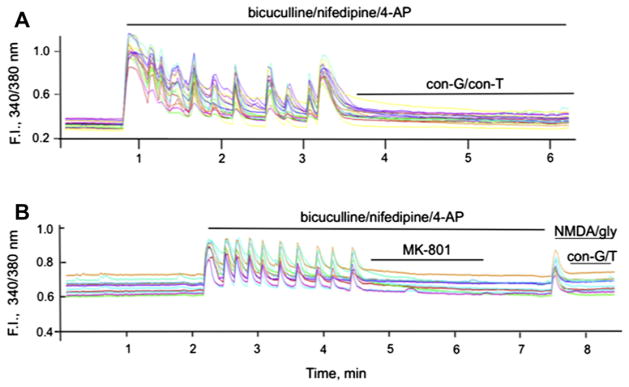
Conantokins inhibit iCa2+ uptake via synaptic or extrasynaptic NMDARs in rat hippocampal neurons. A. Single cell Ca2+ transients measured as 340/380 fluorescence intensity (F.I.) ratio in DIV 13–15 rat hippocampal neurons triggered by activation (50 μM bicuculline/5 μM nifepidine/2.5 mM 4-AP) of the synaptic NMDARs, followed by application of 40 μM con-G or 40 μM con-T. The pulses of synaptic activity were blocked by both conantokins. B. Measure of iCa2+ influx via extrasynaptic NMDARs. Synaptic NMDARs stimulated by 50 μM bicuculline/5 μM nifepidine/2.5 mM 4-AP were first irreversibly blocked by 10 μM MK-801. After a wash period, extrasynaptic NMDARs were activated by bath application of 50 μM NMDA/10 μM glycine, leading to a spike in iCa2+ influx from extrasynaptic NMDARs that was effectively inhibited by the presence of 40 μM con-G or 40 μM con-T. In each case, individual data are shown for 9–10 separate neurons at DIV 13–15.
Since con-G and con-T blocked synaptic and extrasynaptic influx of Ca2+ in these neurons, we next sought to determine whether loss of the NR2A and NR2B subunits would affect synaptic or extrasynaptic Ca2+ entry. Towards this end, cultured cortical neurons from NR2A−/− mice embryos were first utilized. Inhibition of synaptic Ca2+ influx by con-G, in DIV 13–15 NR2A−/− rat hippocampal neurons, was similar to that observed in WT mouse cortical neurons (Fig. 6A). The NR2A−/− neurons showed similar behavior as the rat hippocampal neurons and WT mouse cortical neurons when synaptic Ca2+ influx was inhibited by MK-801, except that the oscillations were not inhibited immediately upon addition of MK-801 (Fig. 6B). Subsequent bath application of NMDA/glycine-induced extrasynaptic Ca2+ influx, which was inhibited by con-G or con-T. However, when synaptic activation was stimulated in DIV 13–15 NR2B−/− neurons, only weak oscillatory pulses of synaptic Ca2+ influx were observed. Addition of 10 μM MK-801 did not noticeably diminish the amplitudes, however the pulse frequency of Ca2+ influx was slightly delayed (Fig. 6C). This implied that NR2B−/− neurons were insensitive to MK-801 blockage. These weak oscillatory pulses continued after bath application of NMDA/glycine, indicating that extrasynaptic activation was eliminated in the absence of the NR2B subunit and suggested that atypical ion channels, that are incapable of robust synaptic activation, are formed in the absence of the NR2B subunit. Overall, the data demonstrated that NR2B is required for strong synaptic activation, inhibition of synaptic Ca2+ influx, as well as for robust extrasynaptic NMDAR activation.
Fig. 6.

Effect of the NMDAR subunit composition on inhibition of synaptic and extrasynaptic iCa2+ uptake in mouse cortical neurons. A. Uptake of Ca2+ by synaptic NMDARs is inhibited by 40 μM con-G in NMDARs of NR2A−/− neurons. B. After blocking synaptic NMDARs by MK-801, extrasynaptic NMDARs were activated by 50 μM NMDA/10 μM glycine, resulting in a spike of iCa2+ resulting from extrasynaptic transport in NR2A−/− neurons. Both 40 μM con-G and 40 μM con-T inhibited this process. C. In a similar experiment with NR2B−/− neurons, 10 μM MK-801 was a poor inhibitor of synaptic currents and NMDA/gly did not activate extrasynaptic NMDAR ion channels. In each case, individual data are shown for 9–10 separate neurons at DIV 13–15.
3.4. Electrophysiology
We have previously demonstrated that inhibition of NMDA-evoked whole cell currents by con-G and ifenprodil decreased as the neurons matured from DIV 7 to DIV 19 (Huang et al., 2010). This observation was attributed to the relative decrease in expression of NR2B subunits, and increase in NR2A subunits, as the neurons aged. On the other hand, inhibition of whole cell currents by non-subunit selective conantokins, viz., con-T and con-R[1–17], did not vary with the in vitro age of the neurons. Similarly, inhibition of spontaneous excitatory postsynaptic currents (sEPSCs) in DIV 14–19 neurons, by con-G, was attenuated compared to con-T or con-R, suggesting that expression of NR2A at the synapses increased as a function of neuronal development (Huang et al., 2010). In the present study, we compared the sensitivity of sEPSCs and extrasynaptic currents to con-G and con-T in WT, NR2A−/−, and NR2B−/− cortical neurons (Fig. 7A–I). The absence of either the NR2A or the NR2B subunit did not affect amplitude or frequency of sEPSCs, as compared to WT neurons. However, the NR2A−/− neurons were more sensitive to con-G inhibition compared to WT or NR2B−/− neurons, as revealed by the decreased amplitude and frequency. This could be indicative of higher expression of the NR2B subunit in the absence of NR2A. Inhibition of sEPSCs by con-T was similar between WT, NR2A−/−, and NR2B−/− neurons, which is commensurate with its non-selective subunit characteristics. Extrasynaptic currents in NR2B−/− neurons were relatively decreased, compared to WT and NR2A−/− neurons, indicating a reduced sensitivity to MK-801 blockage and that NR2B is a key component of extrasynaptic NMDARs.
Fig. 7.

Inhibition of synaptic and extrasynaptic currents by con-G and con-T. Comparison of con-G and con-T on sEPSCs (A) and extrasynaptic currents (B) in DIV 13–15 WT mouse cortical neurons. For sEPSCs, bicuculline and CNQX were added to eliminate the fast (early) sEPSCs from AMPA/kainate channels, thus leading to the slow sEPSCs that originate from NMDARs. Inhibition of sEPSCs was achieved by 3 μM con-G or 3 μM con-T. For recording extrasynaptic currents, synaptic NMDARs were blocked with an extracellular bath solution containing bicuculline/gly/MK-801 for 1 min. The current was inhibited with 40 μM con-G or 40 μM con-T. C, D. Comparison of inhibition of sEPSCs (C) and extrasynaptic currents (D) by 3 μM con-G or 3 mM con-T in DIV 13–15 mouse NR2A−/− cortical neurons. E, F. Electrophysiological traces of inhibition by 40 μM con-G or 40 μM con-T of sEPSCs (E) and extrasynaptic currents (F) in DIV 13–15 mouse NR2B−/− cortical neurons. G. Inhibition of peak amplitudes of sEPSC in the absence (control-white bars) or presence of con-G (black bars) or con-T (horizontal striped bars) in WT, NR2A−/−, and NR2B−/− neurons. Data are plotted as the amplitudes normalized to controls (set at 1.0) for each group of neurons, using 6–8 neurons from 3 to 5 independent experiments. H. Inhibition of frequency of sEPSC in the absence (control-white bars) or presence of con-G (black bars) or con-T (horizontal striped bars) in WT, NR2A−/−, and NR2B−/− neurons. Data are plotted as the amplitudes normalized to controls (set at 1.0) for each group of neurons, using 6–8 neurons from 3 to 5 independent experiments. I. Inhibition of extrasynaptic currents in the absence (control-white bars) or presence of con-G (black bars) or con-T (horizontal striped bars) in WT, NR2A−/−, and NR2B−/− neurons. Data are plotted as the currents normalized to controls (set at 1.0) for each group of neurons, using 6–8 neurons from 3 to 5 independent experiments. *p < 0.05, compared with the control (C) within each group.
Comparing inhibition of sEPSC and extrasynaptic currents by con-G and con-T showed that inhibition by con-G of extrasynaptic currents (71.5 ± 6.9%) was higher compared to inhibition of synaptic currents (45.2 ± 6.5%) in DIV 13–15 WT mouse cortical neurons. Con-T inhibited both extrasynaptic (85.0 ± 5.0%) and synaptic (83.3 ± 5.9%) currents to a similar extent. The lack of NR2B subunits severely attenuated the inhibition by con-G of both extrasynaptic (<1%) and synaptic (3.9 ± 3.9%) currents in DIV 16 NR2B−/− mouse cortical neurons. On the other hand, con-T effectively blocked both extrasynaptic (79.3 ± 6.9%) and synaptic (65.6 ± 8.1%) currents in NR2B−/− cortical neurons, in agreement with its function as a non-selective subunit antagonist.
We further assessed the integrity of channels formed in the NR2B−/− neurons by determining the efficacy of channel blockage by Mg2+. NMDA/glycine-induced current flow was measured in the presence or absence of 100 μM Mg2+ at various holding potentials of the cell. It was observed that in WT, NR2A−/−, and NR2B−/− cortical neurons (Fig. 8) current flow varied linearly with changes in the holding potential of the cells in the absence of Mg2+. However, when Mg2+ was in present the extracellular solution, the NMDAR channels were blocked at high negative potentials (ca., −20 mV) by the Mg2+. At positive potentials, of up to 40 mV, the currents recorded in the presence or absence of Mg2+ were identical in neurons of all three genotypes. These results demonstrated that a total deficiency of either NR2A or NR2B does not affect, at least with regard to the binding of Mg2+, the channels that are formed in the NR2A−/− or NR2B−/− neurons.
Fig. 8.
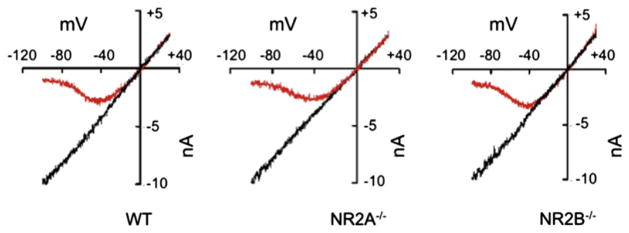
Voltage-dependent whole cell NMDA-induced current block by Mg2+. Representative current–voltage (I–V) curves recorded in the absence of Mg2+ (black lines) and in the presence of 100 μM Mg2+ (red lines) in WT, NR2A−/−, and NR2B−/− mouse cortical neurons. The curves were normalized to the current at −100 mV in Mg2+-free solution. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
The purpose of this study was to exploit the NR2B selectivity of con-G and discern its antagonist function on NMDARs that were pharmacologically activated in their synaptic or extrasynaptic locations in neurons. The effects of con-G on survival pathways and cytoskeleton architecture have provided insights regarding these mechanisms of antagonism, thus providing a basis for drugs designed to minimize indiscriminate NMDAR subunit inhibition.
4.1. Location is important for NMDAR-mediated survival signaling
There is compelling evidence from this study, and the work of others, that synaptic or extrasynaptic cellular location of the NMDARs play a vital role in maintaining neuronal homeostasis (Hardingham et al., 2002; Hetman and Kharebava, 2006). Ca2+ influx via the synaptic receptors that are rich in NR1/NR2A channels (Stocca and Vicini, 1998) is known to trigger ERK1/2 (Ivanov et al., 2006) and CREB (Hardingham and Bading, 2002) activation, thus mediating LTP and activity-dependent neuroprotection. In the current study, the studies with synaptic and extrasynaptic NMDARs that were pharmacologically isolated led to the conclusion that activation of extrasynaptic NMDARs resulted in dampened ERK1/2 and CREB activations. Mitochondrial viability was also diminished in neurons with extrasynaptically-activated NMDARs. Activation of both synaptic and extrasynaptic NMDARs (total) resulted in diminished signals for neuronal survival, demonstrating that extrasynaptic activation dominates synaptic activation. The presence of con-G systematically increased levels of P-ERK1/2, P-CREB, and the mitochondrial viability of neurons in which extrasynaptic NMDARs were selectively activated, indicating that therapeutically targeting the NR2B pool of receptors was a key feature that promoted neuronal survival. Although stimulus intensity affects NMDAR-elicited responses, activation of neuronal signaling pathways through influx of Ca2+ depends on the route of entry and is independent on the Ca2+ load. This conclusion has been experimentally justified, as we have observed that inhibition of extrasynaptic Ca2+ influx by con-G was sufficient and necessary to activate ERK1/2 and CREB. Inhibition of extrasynaptic Ca2+ influx by non-subunit selective con-T did not increase activation of ERK1/2 or CREB or mitochondrial viability. This suggested that subunit selective antagonists are more effective than pan-NMDAR antagonists. However, the NR2B-specific pharmacological inhibitor, ifenprodil, at levels that inhibit extrasynaptic Ca2+ influx to the same extent as con-G, was unable to increase ERK1/2 activation or mitochondrial viability to the same extent as for con-G (data not shown). This indicated that interaction of the receptor with con-G or ifenprodil elicited different downstream signaling events. When con-G or con-T inhibited synaptically-induced Ca2+ bursts, ERK1/2 and CREB activations were inhibited, and decreased synaptic-mediated mitochondrial viability was observed. It was also observed that the beneficial effects of con-G were more specific for neurons with extrasynaptically-activated NMDARs than for neurons in which both synaptic and extrasynaptic (total) NMDARs were activated. These results implied that while the harmful effects of extrasynaptic activation override synaptic activation, a requirement for con-G as an effective drug depends on precise targeting of the extrasynaptic receptors.
While location of the various NR subunits is important, subunit composition in extrasynaptic and synaptic NMDARs also plays an important role. Although, the NR2B subunit is mainly considered to be present at extrasynaptic sites as they develop early in neurons, there is evidence to show that NR2B also occupies synaptic sites (Thomas et al., 2006). We have observed that NR2A−/− neurons were able to elicit robust synaptic activity and were very similar to WT neurons in eliciting extrasynaptic activity. In fact, activation of ERK1/2, and the effect of con-G/T on NR2A−/− neurons that had synaptic, extrasynaptic, or total NMDARs activated, was not different from WT neurons. This indicates that functional channels in the synaptic and extrasynaptic spaces were formed in the absence of the NR2A subunit. Electrophysiology measurements also revealed that sEPSC and extrasynaptic current and inhibition by con-G/T in NR2A−/− neurons were similar to WT cells.
The bicuculline-induced synaptic Ca2+ oscillations observed in NR2B−/− neurons were not as robust as observed in WT or NR2A−/− neurons, and this activity was not blocked by MK-801 or con-G/con-T. This weak synaptic Ca2+ influx was not inhibited by the NR2A-specific inhibitor, NVP-AAM077, even at concentrations of 400 nM in the NR2B−/− neurons (data not shown). Interestingly, NVP-AAM077, at concentrations of 30–60 nM, wherein it is specific for inhibition of NR2A activity, did not inhibit synaptic Ca2+ influx in DIV 12–14 rat hippocampal neurons, although at a non-subunit discriminating concentration of 400 nM (Auberson et al., 2002), this agent inhibited synaptic Ca2+ influx (data not shown). This indicates that NR2B is an integral part of the synapses, and that in its absence weak synaptic activity is retained, but antagonist-specific targeting does not occur. A rather weak extrasynaptic activity by both real-time Ca2+ imaging and electrophysiology with NR2B−/− neurons was observed, indicating that the presence of NR2B is critical for extrasynaptic activation. However, ablation of the NR2B subunit did not affect the voltage dependency of the Mg2+ block, indicating that NMDAR channels formed in NR2B−/− neurons were ion permeable. This discrepancy, that neither non-subunit specific con-T or MK-801 was unable to block the weak synaptic Ca2+ influx, but at the same time the channels formed in NR2B−/− neurons were Mg2+ permeable, cannot be fully explained with the present data. It has been reported that irrespective of their locations, NR2B-containing NMDARs caused excitotoxicity. On the other hand, activation of either NR2A-containing synaptic or extrasynaptic NMDARs resulted in neuroprotection (Liu et al., 2007). Another study has reported that NR2B-containing NMDARs mediated neuronal survival and synaptic potentiation, as well as cell death, in dissociated hippocampal neurons (Martel et al., 2009). One possible explanation for this dichotomous signaling is that the NR2A and NR2B subunits interact with different agents that promote their synaptic insertion (Prybylowski et al., 2005; Sans et al., 2000).
In a mouse model, wherein NR2B was ablated from the hippocampal CA3 pyramidal cells, loss of NMDAR-mediated synaptic current and LTP, with concomitant decrease in F-actin, was observed (Akashi et al., 2009). Furthermore, a NMDAR-mediated LTP also caused increased actin concentration in the activated spines (Fukazawa et al., 2003). Conversely, increased F-actin stability enhanced NMDAR-evoked currents (Offenhauser et al., 2006). The mechanism as to how NMDAR activation regulates actin organization, and vice versa, is unclear. Nonetheless, this study is the first to demonstrate that extrasynaptic, but not synaptic activity, disrupts neuronal actin organization that is dependent upon activation of NMDARs containing the NR2B subunit. Activation of both synaptic and extrasynaptic NMDARs (total) also perturbs actin organization indicating that extrasynaptic activation predominates over synaptic activation. Perturbations in actin organization were observed in both WT and NR2A−/− neurons upon activation of extrasynaptic NMDARs, showing that NR2A-directed downstream signaling does not significantly contribute to neuron survival. In fact, when synaptic activity was blocked by con-G in WT and NR2A−/− neurons, actin disorganization was observed. This could mean that the heteromeric NMDAR channels containing NR1 and other NR2 subunits are able to function in their synaptic and extrasynaptic capacities in NR2A−/− neurons. The fact that lack of the NR2B subunit eliminated the extrasynaptic-induced actin disorganization signifies its functional importance in activity-dependent usage but also for maintaining neuronal architecture.
4.2. NR2B-directed therapeutic agents
The main reason why several NMDAR-directed drugs failed at the clinical trials was due to their inability to differentiate between the synaptic and extrasynaptic NMDARs. Drugs that are able to block the pro-death extrasynaptic activation, but allow the beneficial neuroprotective synaptic activation to continue, might be more successful. This leads investigators to concentrate on subunit selective NMDAR antagonists. Several NR2B-specific drugs, including ifenprodil, eliprodil, and traxoprodil, have been tested in animal ischemic models and in Parkinson’s Disease, but, because of their antagonism towards the α1-adrenergic and serotonin receptors, administration of these compounds led to cardiovascular complications (Gogas, 2006; Nash et al., 1999). Ifenprodil and its derivatives have also been tested in treating Parkinson’s disease in different animal models (Nash et al., 1999). The NR2B subunit has been found to be involved in Parkinson’s disease as well as in early onset of neurodegeneration in Huntington’s disease (Kuppenbender et al., 2000), and might greatly benefit from NR2B-directed antagonists. The presence of P-NR2B plays an important role in inflammatory and neuropathic pain, and siRNA-induced ablation of NR2B abolished formalin-induced pain in rats (Tan et al., 2005). Overexpression of forebrain NR2B receptors caused enhanced pain in mice (Wei et al., 2001), and ifenprodil, traxoprodil, and Ro25-6981 were effective in treating inflammatory and neuropathic pain (Chizh and Headley, 2005). It should be pointed out that although ifenprodil and con-G are NR2B-selective, their binding properties to NMDARs are different from each other, and thus functional responses to these agents may not be the same with NR2B-containing receptors. Ifenprodil acts as a non-competitive, voltage-independent antagonist that binds to the polyamine site on the receptor and displays use-dependence (Carter et al., 1990). This agent also increases the sensitivity of the receptor to proton blockage (Legendre and Westbrook, 1991). On the other hand, inhibition by con-G appears to be competitive or non-competitive at the glutamate binding site of NR2B (Haack et al., 1993; Hammerland et al., 1992; Skolnick et al., 1992; Wittekindt et al., 2001). In murine hippocampal neurons con-G inhibited NMDA-elicited responses in a non-competitive and voltage-independent manner (Klein et al., 1999).
Taken together, these observations suggest that NR2B-specific class of antagonists are better tolerated than pan-NMDAR blockers and that further research into understanding signaling and molecular mechanisms of NMDAR-related functions would enable design of better drugs.
Acknowledgments
We thank the technical staff of the Freimann Life Sciences Center for husbandry of the mouse lines and Juan Fu for genotyping. We also acknowledge Ms. Allison Ditmars for neuron culturing, Mayra Sandoval-Cooper for technical assistance with immunostaining and image acquisition, and Deborah Donahue for assistance in animal surgery.
Footnotes
Support: This work was supported by grant HL019982 from the NIH (to FJC). The supporting agency played no part in the design of the study or decision to publish.
Author contributions
RB, NL, DW-A, and LH performed the experimental portions of this work and developed methodology for this study. FJC designed and supervised the project. All authors edited the final draft.
References
- Akashi K, Kakizaki T, Kamiya H, Fukaya M, Yamasaki M, Abe M, Natsume R, Watanabe M, Sakimura K. NMDA receptor GluN2B (GluR epsilon 2/NR2B) subunit is crucial for channel function, postsynaptic macromolecular organization, and actin cytoskeleton at hippocampal CA3 synapses. J Neurosci. 2009;29:10869–10882. doi: 10.1523/JNEUROSCI.5531-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arzberger T, Krampff K, Leimgruber S, Weindl A. Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington’s disease–an in situ hybridization study. J Neuropathol Exp Neurol. 1997;56:440–454. doi: 10.1097/00005072-199704000-00013. [DOI] [PubMed] [Google Scholar]
- Auberson YP, Allgeier H, Bischoff S, Lingenhoehl K, Moretti R, Schmutz M. 5-phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg Med Chem Lett. 2002;12:1099–1102. doi: 10.1016/s0960-894x(02)00074-4. [DOI] [PubMed] [Google Scholar]
- Barton ME, White HS, Wilcox KS. The effect of CGX-1007 and CI-1041, novel NMDA receptor antagonists, on NMDA receptor-mediated EPSCs. Epilepsy Res. 2004;59:13–24. doi: 10.1016/j.eplepsyres.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Carter CJ, Lloyd KG, Zivkovic B, Scatton B. Ifenprodil and SL 82.0715 as cerebral antiischemic agents. III Evidence for antagonistic effects at the polyamine modulatory site within the N-methyl-D-aspartate receptor complex. J Pharmacol Exp Ther. 1990;253:475–482. [PubMed] [Google Scholar]
- Chatterton JE, Awobuluyi M, Premkumar LS, Takahashi H, Talantova M, Shin Y, Cui JK, Tu SC, Kevin ASK, Nakanishi N, Tong G, Lipton SA, Zhang DX. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature. 2002;415:793–798. doi: 10.1038/nature715. [DOI] [PubMed] [Google Scholar]
- Chizh BA, Headley PM. NMDA antagonists and neuropathic pain–multiple drug targets and multiple uses. Curr Pharm Des. 2005;11:2977–2994. doi: 10.2174/1381612054865082. [DOI] [PubMed] [Google Scholar]
- Ciabarra AM, Sullivan JM, Gahn LG, Pecht G, Heinemann S, Sevarino KA. Cloning and characterization of chi-1: a developmentally regulated member of a novel class of the ionotropic glutamate receptor family. J Neurosci. 1995;15:6498–6508. doi: 10.1523/JNEUROSCI.15-10-06498.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claiborne CF, McCauley JA, Libby BE, Curtis NR, Diggle HJ, Kulagowski JJ, Michelson SR, Anderson KD, Claremon DA, Freidinger RM, Bednar RA, Mosser SD, Gaul SL, Connolly TM, Condra CL, Bednar B, Stump GL, Lynch JJ, Macaulay A, Wafford KA, Koblan KS, Liverton NJ. Orally efficacious NR2B-selective NMDA receptor antagonists. Bioorg Med Chem Lett. 2003;13:697–700. doi: 10.1016/s0960-894x(02)01061-2. [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- Curtis NR, Diggle HJ, Kulagowski JJ, London C, Grimwood S, Hutson PH, Murray F, Richards P, Macaulay A, Wafford KA. Novel N1-(benzyl) cinnamamidine derived NR2B subtype-selective NMDA receptor antagonists. Bioorg Med Chem Lett. 2003;13:693–696. doi: 10.1016/s0960-894x(02)01060-0. [DOI] [PubMed] [Google Scholar]
- Donevan SD, McCabe RT. Conantokin G is an NR2B-selective competitive antagonist of N-methyl-D-aspartate receptors. Mol Pharmacol. 2000;58:614–623. doi: 10.1124/mol.58.3.614. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- Gogas KR. Glutamate-based therapeutic approaches: NR2B receptor antagonists. Curr Opin Pharmacol. 2006;6:68–74. doi: 10.1016/j.coph.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Haack JA, Parks TN, Olivera BM. Conantokin-G antagonism of the NMDA receptor subtype expressed in cultured cerebellar granule cells. Neurosci Lett. 1993;163:63–66. doi: 10.1016/0304-3940(93)90229-e. [DOI] [PubMed] [Google Scholar]
- Hammerland LG, Olivera BM, Yoshikami D. Conantokin-G selectively inhibits N-methyl-D-aspartate-induced currents in Xenopus oocytes injected with mouse brain mRNA. Eur J Pharmacol. 1992;226:239–244. doi: 10.1016/0922-4106(92)90067-6. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Coupling of extrasynaptic NMDA receptors to a CREB shut-off pathway is developmentally regulated. Biochim Biophys Acta. 2002;1600:148–153. doi: 10.1016/s1570-9639(02)00455-7. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hetman M, Kharebava G. Survival signaling pathways activated by NMDA receptors. Curr Top Med Chem. 2006;6:787–799. doi: 10.2174/156802606777057553. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Huang L, Balsara RD, Sheng Z, Castellino FJ. Conantokins inhibit NMDAR-dependent calcium influx in developing rat hippocampal neurons in primary culture with resulting effects on CREB phosphorylation. Mol Cell Neurosci. 2010;45:163–172. doi: 10.1016/j.mcn.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Physiol. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20:201–225. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Kemp JA, McKernan RM. NMDA receptor pathways as drug targets. Nat Neurosci. 2002;5:1039–1042. doi: 10.1038/nn936. [DOI] [PubMed] [Google Scholar]
- Kew JN, Kemp JA. Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology. 2005;179:4–29. doi: 10.1007/s00213-005-2200-z. [DOI] [PubMed] [Google Scholar]
- Klein RC, Galdzicki Z, Castellino FJ. Inhibition of NMDA-induced currents by conantokin-G and conantokin-T in cultured embryonic murine hippocampal neurons. Neuropharmacology. 1999;38:1819–1829. doi: 10.1016/s0028-3908(99)00065-9. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy M, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, Ben-Ari Y, Clapham DE, Medina I. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron. 2003;40:775–784. doi: 10.1016/s0896-6273(03)00645-7. [DOI] [PubMed] [Google Scholar]
- Kuppenbender KD, Standaert DG, Feuerstein T, Penney JB, Young AB, Landwehrmeyer GB. Expression of NMDA receptor subunit mRNAs in neurochemically identified projection and interneurons in the human striatum. J Comp Neurol. 2000;419:407–421. doi: 10.1002/(sici)1096-9861(20000417)419:4<407::aid-cne1>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E, Natsume R, Watanabe M, Inoue Y, Yagi T, Aizawa S, Arakawa M, Takahashi T, Nakamura Y, Mori H, Mishina M. Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor epsilon 2 subunit mutant mice. Neuron. 1996;16:333–344. doi: 10.1016/s0896-6273(00)80051-3. [DOI] [PubMed] [Google Scholar]
- Legendre P, Westbrook GL. Ifenprodil blocks N-methyl-D-aspartate receptors by a two-component mechanism. Mol Pharmacol. 1991;40:289–298. [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Gilbert H, McCabe RT, Basbaum AI. Powerful anti-nociceptive effects of the cone snail venom-derived subtype-selective NMDA receptor antagonists conantokins G and T. Pain. 2003;101:109–116. doi: 10.1016/s0304-3959(02)00303-2. [DOI] [PubMed] [Google Scholar]
- Martel MA, Wyllie DJ, Hardingham GE. In developing hippocampal neurons, NR2B-containing N-methyl-D-aspartate receptors (NMDARs) can mediate signaling to neuronal survival and synaptic potentiation, as well as neuronal death. Neuroscience. 2009;158:334–343. doi: 10.1016/j.neuroscience.2008.01.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moddel G, Jacobson B, Ying Z, Janigro D, Bingaman W, Gonzalez-Martinez J, Kellinghaus C, Prayson RA, Najm IM. The NMDA receptor NR2B subunit contributes to epileptogenesis in human cortical dysplasia. Brain Res. 2005;1046:10–23. doi: 10.1016/j.brainres.2005.03.042. [DOI] [PubMed] [Google Scholar]
- Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N, Nakanishi S. Molecular cloning and characterization of the rat NMDA receptor. Nature. 1991;354:31–37. doi: 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- Nagy J, Laszlo L. Increased sensitivity to NMDA is involved in alcohol-withdrawal induced cytotoxicity observed in primary cultures of cortical neurones chronically pre-treated with ethanol. Neurochem Int. 2002;40:585–591. doi: 10.1016/s0197-0186(01)00131-0. [DOI] [PubMed] [Google Scholar]
- Narita M, Soma M, Mizoguchi H, Tseng LF, Suzuki T. Implications of the NR2B subunit-containing NMDA receptor localized in mouse limbic forebrain in ethanol dependence. Eur J Pharmacol. 2000;401:191–195. doi: 10.1016/s0014-2999(00)00428-3. [DOI] [PubMed] [Google Scholar]
- Nash JE, Hill MP, Brotchie JM. Anti-Parkinsonian actions of blockade of NR2B-containing NMDA receptors in the reserpine-treated rat. Exp Neurol. 1999;155:42–48. doi: 10.1006/exnr.1998.6963. [DOI] [PubMed] [Google Scholar]
- Offenhauser N, Castelletti D, Mapelli L, Soppo BE, Regondi MC, Rossi P, D’Angelo E, Frassoni C, Amadeo A, Tocchetti A, Pozzi B, Disanza A, Guarnieri D, Betsholtz C, Scita G, Heberlein U, Di Fiore PP. Increased ethanol resistance and consumption in Eps8 knockout mice correlates with altered actin dynamics. Cell. 2006;127:213–226. doi: 10.1016/j.cell.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33:523–533. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- Palmer GC. Neuroprotection by NMDA receptor antagonists in a variety of neuropathologies. Curr Drug Targets. 2001;2:241–271. doi: 10.2174/1389450013348335. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Pawlak R, Melchor JP, Matys T, Skrzypiec AE, Strickland S. Ethanol-withdrawal seizures are controlled by tissue plasminogen activator via modulation of NR2B-containing NMDA receptors. Proc Natl Acad Sci U S A. 2005;102:443–448. doi: 10.1073/pnas.0406454102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prorok M, Castellino FJ. The molecular basis of conantokin antagonism of NMDA receptor function. Curr Drug Targets. 2007;8:633–642. doi: 10.2174/138945007780618481. [DOI] [PubMed] [Google Scholar]
- Prorok M, Warder SE, Blandl T, Castellino FJ. Calcium binding properties of synthetic γ-carboxyglutamic acid containing marine cone snail “sleeper” peptides, conantokin-G and conantokin-T. Biochemistry. 1996;35:16528–16534. doi: 10.1021/bi9621122. [DOI] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebola N, Srikumar BN, Mulle C. Activity-dependent synaptic plasticity of NMDA receptors. J Physiol. 2010;588:93–99. doi: 10.1113/jphysiol.2009.179382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S, Inoue Y, Sugiyama H, Mishina M. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature. 1995;373:151–155. doi: 10.1038/373151a0. [DOI] [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos J, Hell JW, Wenthold RJ. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20:1260–1271. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skolnick P, Boje K, Miller R, Pennington M, Maccecchini ML. Noncompetitive inhibition of N-methyl-D-aspartate by conantokin-G: evidence for an allosteric interaction at polyamine sites. J Neurochem. 1992;59:1516–1521. doi: 10.1111/j.1471-4159.1992.tb08468.x. [DOI] [PubMed] [Google Scholar]
- Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters C, Andrews SB. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc Natl Acad Sci U S A. 2009;106:9854–9859. doi: 10.1073/pnas.0903546106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol. 1998;507:13–24. doi: 10.1111/j.1469-7793.1998.013bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat vNeurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- Tan PH, Yang LC, Shih HC, Lan KC, Cheng JT. Gene knockdown with intrathecal siRNA of NMDA receptor NR2B subunit reduces formalin-induced nociception in the rat. Gene Ther. 2005;12:59–66. doi: 10.1038/sj.gt.3302376. [DOI] [PubMed] [Google Scholar]
- Thomas CG, Miller AJ, Westbrook GL. Synaptic and extrasynaptic NMDA receptor NR2 subunits in cultured hippocampal neurons. J Neurophysiol. 2006;95:1727–1734. doi: 10.1152/jn.00771.2005. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrich MH, Isacoff EY. Rules of engagement for NMDA receptor subunits. Proc Natl Acad Sci U S A. 2008;105:14163–14168. doi: 10.1073/pnas.0802075105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC. Synaptic assembly of the brain in the absence of neuro-transmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Wei F, Wang GD, Kerchner GA, Kim SJ, Xu HM, Chen ZF, Zhuo M. Genetic enhancement of inflammatory pain by forebrain NR2B overexpression. Nat Neurosci. 2001;4:164–169. doi: 10.1038/83993. [DOI] [PubMed] [Google Scholar]
- Wei J, Dong M, Xiao C, Jiang F, Castellino FJ, Prorok M, Dai Q. Conantokins and variants derived from cone snail venom inhibit naloxone-induced withdrawal jumping in morphine-dependent mice. Neurosci Lett. 2006;405:137–141. doi: 10.1016/j.neulet.2006.06.040. [DOI] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AJ, Dave JR, Phillips JB, Lin Y, McCabe RT, Tortella FC. Neuroprotective efficacy and therapeutic window of the high-affinity N-methyl-D-aspartate antagonist conantokin-G: in vitro (primary cerebellar neurons) and in vivo (rat model of transient focal brain ischemia) studies. J Pharmacol Exp Ther. 2000;294:378–386. [PubMed] [Google Scholar]
- Williams AJ, Ling G, Berti R, Moffett JR, Yao C, Lu XM, Dave JR, Tortella FC. Treatment with the snail peptide CGX-1007 reduces DNA damage and alters gene expression of c-fos and bcl-2 following focal ischemic brain injury in rats. Exp Brain Res. 2003;153:16–26. doi: 10.1007/s00221-003-1566-6. [DOI] [PubMed] [Google Scholar]
- Wittekindt B, Malany S, Schemm R, Otvos L, Maccecchini ML, Laube B, Betz H. Point mutations identify the glutamate binding pocket of the N-methyl-D-aspartate receptor as major site of Conantokin-G inhibition. Neuropharmacology. 2001;41:753–761. doi: 10.1016/s0028-3908(01)00112-5. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M. Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Steijaert MN, Lau D, Schutz G, Delucinge-Vivier C, Descombes P, Bading H. Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death. Neuron. 2007;53:549–562. doi: 10.1016/j.neuron.2007.01.025. [DOI] [PubMed] [Google Scholar]