

Abstract
Peptide amphiphiles (PAs) provide a versatile platform for the design of complex and functional material constructs for biomedical applications. The hierarchical self-assembly of PAs with biopolymers is used to create robust hybrid membranes with molecular order on the micron scale. Fabrication of membranes by assembling hyaluronic acid with positively charged PA nanostructures containing anti-cancer PAs bearing a (KLAKLAK)2 peptide sequence is reported here. Changes in nanoscale membrane morphology as the positively charged PA nanostructures vary from cylindrical nanofibers to spherical aggregates are characterized. Results indicate that formation of highly aligned fibrous membranes requires a threshold concentration of nanofibers in solution. Additionally, variation of PA nanostructure morphology from spherical aggregates to cylindrical nanofibers allows membranes to act either as reservoirs for sustained release of cytotoxicity upon enzymatic degradation or as membranes with surface-bound cytotoxicity, respectively. Thus, the self-assembly processes of these PA-biopolymer membranes can be potentially used to design delivery platforms for anti-cancer therapeutics.
Keywords: breast cancer, drug delivery, membranes, peptide amphiphiles, self-assembly
1. Introduction
Efficacious treatment of cancer without debilitating side effects to the body as a whole continues to be a significant challenge in the development of therapeutic delivery systems. The highly toxic side effects associated with systemic drug administration have been mitigated by incorporating conventional therapies within wafers or films for sustained localized delivery.[1-6] Such materials have shown promise in preventing local tumor recurrence,[7] and carmustine-releasing polyanhydride wafers (GLIADEL®) have been FDA-approved as an implantable adjunct to surgery and radiation for malignant glioma.[8,9] However, drug-eluting materials have thus far employed biologically inert components with simple erosion or elution kinetics, and research investigating anti-cancer materials with more complex and environmentally responsive behaviors is limited.[10,11]
Our laboratory has demonstrated the rational design of functional biomaterials based on self-assembling peptide amphiphiles (PAs).[12-14] These PAs typically consist of a bioactive oligopeptide sequence linked to a β-sheet forming sequence and an alkyl tail. Hydrophobic collapse of PAs in aqueous solution results in molecular aggregation, and β-sheet formation leads to one-dimensional, high-aspect-ratio supramolecular nanostructures shaped as cylindrical fibers,[15] giant nanobelts,[16] ribbons,[17] or twisted helices.[18] PAs have shown to form nanofiber gel scaffolds upon exposure to physiologically relevant conditions,[19,20] and PAs can also form monodomain aligned gels after specific thermal treatment.[21] Furthermore, bioactive hybrid PA-polymer materials have been formed by coating poly(glycolic acid) scaffolds with branched PA.[22] These supramolecular biomaterials have shown efficacy in applications such as angiogenesis,[23,24] islet transplantation,[25] neural regeneration,[26,27] hard tissue regeneration,[28,29] cartilage regeneration,[30] and therapeutic protein delivery.[31]
The complex self-assembly of PAs with biopolymers into hierarchically structured membranes was previously demonstrated by our laboratory.[32-35] Capito et al. reported on the self-assembly of robust and highly ordered membranes by a non-bioactive, positively charged PA with high molecular weight hyaluronic acid (HA).[32] Formation of an ordered fibrous membrane that grows in time results from an initial event of electrostatic complexation between PA nanostructures and HA macromolecules, which creates a diffusion barrier in the plane of liquid-liquid contact. Subsequent reptation and outward diffusion of HA chains results in the growth of nanofiber bundles aligned perpendicular to the interface. This self-assembly phenomenon is a dynamic process that can be used to form planar sheet membranes or membrane-enclosed sacs. Membrane properties such as permeability and mechanical strength can be tuned by varying HA solution concentration or incubation time in PA.[33] Bioactivity can also be incorporated, as recently demonstrated by pro-angiogenic membranes incorporating a heparin-binding PA.[34]
We explore here the self-assembly of hierarchically structured HA-PA membranes that include a PA with the amphipathic α-helical sequence KLAKLAKKLAKLAK (KLAK PA). KLAK PA has been previously shown to induce cell death by membrane lysis at low concentrations and to exhibit some specificity towards breast cancer cells over non-cancerous breast epithelial cells,[36] while HA has been shown to bind cancer cells and experience accelerated degradation in tumor environments.[37,38] These membranes with potential therapeutic function toward cancer could ultimately biodegrade into biological building blocks, and the enhanced degradation of HA by cancer cells could furthermore generate a therapy that is responsive to a cancerous environment. We investigate here the structure of membranes using electron microscopy, small angle x-ray scattering, and confocal microscopy techniques. We also assay the in vitro behavior of MDA-MB-231 and SKBR-3 breast cancer cells upon contact with the membrane surface or when incubated with membrane degradation products.
2. Results and Discussion
2.1. Characterization of PA Nanostructure
2.1.1. Aqueous Morphology of KLAK PA and K3PA
As previously discussed, the PAs chosen for this study were a non-bioactive positively charged K3 PA, which has been shown to assemble with anionic polyelectrolytes into robust and ordered membranes, [32-35] and KLAK PA, which has been shown to be cytotoxic against breast cancer cells at very low concentrations.[36] As expected, TEM revealed the self-assembly of pure K3 PA into high-aspect-ratio nanofibers whereas pure KLAK PA appeared to self-assemble into small spherical aggregates (Figure 1). Guinier region analysis of the K3 PA scattering profile revealed a −1 slope, indicating cylindrical nanofiber morphology (Figure 2). Previous work indicated that such cylindrical nanofibers consist of a hydrophobic core with PA molecules arranged in twisted β-sheets running down the length of the fiber.[39-41] Thus, a cylindrical core-shell model was used to fit the scattering profile, and the nanofiber diameter was determined to be 10 nm and seemed to be invariant with concentration. In contrast, the scattering profile of KLAK PA indicated spherical morphology but exhibited a structure factor in the low q range, suggesting aggregation of the nanostructures in solution. Fitting the data to a core-shell sphere model revealed diameters that varied with concentration, ranging from 6 nm to 9 nm (Figure S1). This finding indicates that KLAK PA forms non-ideal spherical aggregates in water. Since we expect denser packing of molecules in cylindrical nanofibers to spherical aggregates, the inability of KLAK PA to form nanofibers in water is likely due to the high charge and steric bulkiness of the (KLAKLAK)2 sequence.
Figure 1.

Transmission electron micrographs of PA nanostructures at various co-assembly compositions. High aspect-ratio nanofibers are predominant in aqueous solutions of (a) 100% K3 PA and (b) 2% KLAK PA. Solutions of (c) 20% KLAK PA display a significant number of small spherical aggregates, while solutions of (d) 50% KLAK PA and (e) 100% KLAK PA contain predominantly spherical aggregates with few short nanofibers. (f) Heating a 50% KLAK PA solution at 80 °C for 8 h leads to nanofiber morphology. Total PA concentration is 5 mm for all samples. Labels indicate the percentage of KLAK PA, with K3 PA comprising the remaining balance.
Figure 2.

Small angle x-ray scattering profiles of various co-assembly compositions. (a) The slope of the Guinier region at low q tends towards 0 as the percentage of KLAK PA is increased, indicating shift towards spherical aggregate nanomorphology. (b-c) Comparisons of co-assembled mixtures with the sum of their individual components suggest mixing on a molecular level and also highlight the bias towards spherical morphology when KLAK PA is present.
2.1.2. Effect of PA Co-Assembly
Previous work demonstrated the ability to alter the properties of bioactive PAs via co-assembly with non-active PAs.[42] K3 PA and KLAK PA differ in charge and molecular dimensions, and mixture of the two PAs in various ratios may therefore impact both membrane microstructure and bulk bioactivity. Thus, K3 PA and KLAK PA were co-assembled at various ratios by dissolving each PA individually at 5 mm in water and mixing solutions such that total PA concentration remained constant at 5 mm. TEM imaging suggested that mixing small amounts of KLAK PA with K3 PA does not noticeably affect PA morphology (Figure 1B). However, a noticeable shift away from nanofiber morphology towards spherical morphology was observed when the percentage of KLAK PA in the mixture was increased. When the KLAK PA contribution was increased to 20%, TEM showed disproportionately more spherical aggregates than would be expected given the low KLAK/K3 PA ratio (Figure 1C). At 50% KLAK PA, TEM micrographs demonstrate that PAs exist predominantly as spherical aggregates with few short nanofibers (Figure 1D). SAXS confirmed the observation that co-assembling KLAK PA with K3 PA disrupts cylindrical nanofiber formation, as the slope of the low q Guinier region approaches 0 with increasing KLAK PA content (Figure 2A).
Scattering profiles of PA mixtures exhibited much shallower Guinier region slopes as compared to the sum of the scattering of individual mixture components, indicating that the nanostructure population is disproportionately skewed towards spherical aggregate morphology even with small amounts of KLAK PA (Figure 2B-C). Additionally, these discrepancies between the actual and theoretical scattering of mixtures further suggest molecular level mixing of KLAK PA and K3 PA within a nanostructure. This molecular mixing is likely driven by energy minimization through the dilution of bulky, highly charged (KLAKLAK)2 headgroups by K3 PA. As the amount of KLAK PA is gradually increased, the surface density of (KLAKLAK)2 groups within a given nanostructure no longer favors cylindrical nanofiber morphology and PAs preferentially assemble into spherical aggregates. Both SAXS and TEM reveal that PA mixtures contain populations of nanostructures that are inhomogeneous in terms of size and shape (Figure 1 and Figure 2). Thus, in solutions exhibiting mixed nanostructure populations, it would be expected that spherical aggregates are relatively rich in KLAK PA compared to nanofibers. This hypothesis was tested by repeatedly filtering a 20% KLAK PA solution through 0.2 μm syringe filters to create an aqueous solution enriched with spherical aggregates. LCMS showed that this enriched solution indeed has a higher KLAK/K3 PA ratio than the original unfiltered solution (Figure S2). Furthermore, incubating spherical aggregates in an 80 °C water bath for 6 h appeared to drive nanostructures back towards nanofiber morphology (Figure 1F), possibly by melting nanostructures and facilitating molecular separation of KLAK PA and K3 PA upon cooling.
2.2. Hierarchical Self-Assembly of Membranes
2.2.1. Membrane Microstructure
As mentioned previously, membranes are formed by self-assembly of K3 PA with oppositely charged biopolymers such as HA through a mechanism that involves charge complexation of molecules to create a diffusion barrier between the polymer and PA solutions and subsequent outward reptation of HA templates the growth of PA nanofiber bundles perpendicular to the interface.[32] Studies have indicated that charge interaction and osmotic pressure are key components of this self-assembly.[33] Other previous studies have also hinted at the importance of PA nanostructure in dictating the mechanism of self-assembly.[34]
In order to probe the effect of PA nanostructure on membrane morphology, SEM was used to image the structure of membranes assembled using pure and co-assembled PA solutions (Figure 3). Membranes formed with pure K3 PA were mechanically robust and exhibited the characteristic multi-region structure comprising of a dense contact layer and an aligned perpendicular fiber layer (Figure 3A). Interestingly, membranes formed with pure KLAK PA were significantly weaker and showed an atypical structure with no obvious contact layer or perpendicular fiber growth (Figure 3E). As KLAK PA is more charged than K3 PA, the lack of an initial contact layer is not due to inadequate charge interaction between PA and HA but is likely due to the difference in PA nanostructure morphology instead. Previous studies have suggested that the presence of high aspect ratio PA nanofibers in solution is required for rapid diffusion barrier formation and perpendicular fiber growth.[34] Without a diffusion barrier, membrane growth should be based on PA spherical micelle diffusion into the HA solution resulting in a drastically different structure. We found that solutions with as much as 20% KLAK PA form the characteristic membrane structure (Figure 3C), while solutions with 50% KLAK PA are mechanically weak and exhibit a non-aligned, featureless structure (Figure 3D). Thus, a threshold amount of supramolecular nanofibers is likely required for the formation of a diffusion barrier and the consequent diffusion of stretched chains of HA resulting in the aligned fibrous microstructure. Heating 50% KLAK PA to 80 °C for 6 h and slow cooling the solution restored nanofiber morphology (Figure 1F), and membranes formed with the heat treated solution accordingly exhibited the characteristic diffusion barrier and perpendicular fiber membrane morphology (Figure 3F).
Figure 3.

Scanning electron micrographs of membranes assembled from hyaluronic acid and various PA co-assemblies. Evidence of diffusion barrier formation and perpendicular fiber growth is seen in the case of (a) 100% K3 PA, (b) 2% KLAK PA, (c) 20% KLAK PA, and (f) heat treated 50% KLAK PA. Non-fibrous, featureless membrane microstructure is seen for (d) 50% KLAK PA and (e) 100% KLAK PA.
2.2.2. PA Distribution Within Membranes
Because solutions of co-assembled PA exhibit a mixed population of nanostructures varying in size, shape, and composition, homogenous incorporation of KLAK PA and K3 PA within the membrane is not likely. Confocal microscopy was used to probe the distribution of PAs in the membrane by labeling K3 PA and KLAK PA with FITC and RBITC, respectively (Figure 4). Because fluorophores may affect the self-assembly of PA molecules, small quantities of fluorescent PA were co-assembled with unlabeled PA in order to maintain the self-assembly properties as much as possible. By taking orthogonal views of the confocal z-stack, the distribution of KLAK PA and K3 PA across the membrane cross-section could also be reconstructed. For membranes made with 20% KLAK PA, the KLAK PA and K3 PA distribution appeared to be homogeneous in the planar direction (Figure 4A). However, images of the cross-section showed a KLAK PA-rich layer at the membrane base (HA side) and K3 PA-rich layer above it. This PA segregation may result from more rapid diffusion of KLAK-rich spherical aggregates towards the HA compartment upon interfacial self-assembly. Surprisingly, membranes assembled with 50% KLAK PA exhibited a network of KLAK-rich “veins” that run planar to the membrane and span the membrane cross-section (Figure 4B). These large discrepancies in PA distribution between membranes formed by various co-assembly ratios correlate with the different microstructure regimes seen in SEM, suggesting a complex relationship between PA self-assembly on the nanoscale and hierarchical assembly on the microscale. Furthermore, we expected that such structural differences would inherently affect the properties and bioactivity of membranes as a macro-scale therapeutic object.
Figure 4.
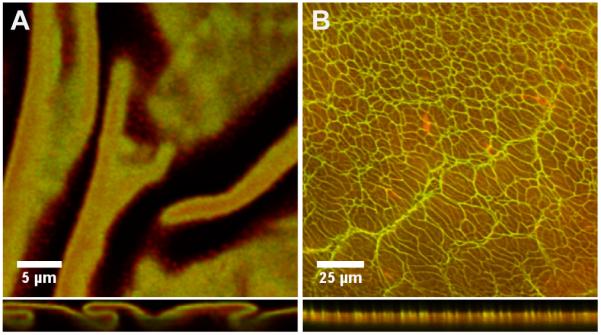
Confocal micrographs showing the distribution of RBITC-labeled K3 PA and FITC-labeled KLAK PA within the (a) 20% KLAK PA membrane and (b) 50% KLAK PA membrane. Membrane cross-sections are shown in the bottom panels, with the top of the panel corresponding to the side of the membrane that faced biopolymer compartment.
2.3. Anti-cancer Activity of Membranes
2.3.1. Degradation-Dependent Cytotoxicity
HA is naturally biocompatible and has been shown to experience accelerated degradation by cancer cells in vivo by hyaluronidase.[37,38] Thus, we investigated the ability of membranes incorporating KLAK PA to serve as reservoirs for sustained and localized release of cytotoxic PA by enzymatic degradation. Crosslinking HA after membrane formation helps to stabilize assembled structures against osmotic pressure and ionic changes upon introduction of saline or cell culture media. Thus, membranes were formed using a tyramine-functionalized HA that undergoes oxidative crosslinking in the presence of horseradish peroxidase and H2O2 to form a hydrogel. These mild crosslinking conditions have been shown to be biocompatible with in vitro and in vivo cell studies.[43] Furthermore, the level of tyramine substitution is low in order to maintain the native properties of HA. Hyaluronidase was used to enzymatically degrade membranes made with various co-assembled PA solutions, and degradation products were introduced to two breast cancer cell lines as well as one non-cancerous breast epithelial cell line. MTS assays were used to assess cytotoxicity and viability results were normalized to the degradation products of HA-Tyr gels. We observed that membranes assembled using 100% K3 PA, 10% KLAK PA, and 20% KLAK PA did not produce cytotoxic degradation products even after 4 days of enzymatic degradation, while membranes assembled with 50% KLAK PA produced potent cytotoxic products after 2 days of enzymatic degradation (Figure 5). Furthermore, membranes synthesized with 50% KLAK PA did not produce cytotoxic products in fetal bovine serum (FBS), demonstrating that cytotoxic release is enzymatically triggered. In agreement with previous studies showing selectivity of KLAK PA for cancer cells, non-cancerous MCF10a breast epithelial cells were found to be less sensitive to cytotoxic degradation products.
Figure 5.

Viability of (a) MDA-MB-231 breast adenocarcinoma cells, (b) SKBR-3 breast adenocarcinoma cells, and (c) MCF10a breast epithelial cells 24 h after addition of membrane degradation products. Membranes assembled with 50% KLAK PA solution do not produce cytotoxic products in FBS but do produce significantly cytotoxic products after two days of enzymatic degradation by hyaluronidase. Non-cancerous MCF10a breast epithelial cells are less sensitive to the cytotoxic effects of degradation products. No other membrane exhibits toxic degradation products even after 4 days of enzymatic degradation. Viability is normalized to degradation products of HA-Tyr gels. Data points represent mean and standard error for n=3 (***P<0.001).
Because viability studies indicate that co-assembling KLAK PA with K3 PA does not decrease its cytotoxic potency in solution (Figure S3), membranes that were assembled with 10% or 20% KLAK PA but did not produce cytotoxic degradation products likely exhibited much slower PA release than membranes assembled from 50% KLAK PA. In order to separate the effect of varying KLAK PA concentration in the assembly solution from varying membrane microstructure, membranes fabricated with heated and unheated 50% KLAK PA were compared. We observed that membranes containing heat-treated 50% KLAK PA solution not only exhibited the same type of microstructure as membranes formed with lower KLAK concentrations (Figure 3), but similarly did not produce cytotoxic degradation products. We therefore hypothesize that the difference in degradation and cytotoxic release is due to the structural variation between membranes assembled with PA solutions containing cylindrical nanofibers vs. spherical aggregates. Membranes formed with spherical aggregates may release more KLAK PA upon degradation due to the accessibility of KLAK PA to enzymes along the KLAK-rich “veins” that span the cross-section (Figure 4B). Also, membranes made with spherical aggregates may more readily release cytotoxic products upon degradation due to ease of releasing spherical shapes when HA chains are cleaved as compared to releasing cylindrical shapes.
2.3.2. Surface-Localized Cytotoxicity
In addition to using membranes incorporating KLAK PA as reservoirs for localized cytotoxic release, such membranes can also act as a unique construct for displaying surface-localized cytotoxicity over a large surface area. This cytotoxic membrane could potentially be utilized in breast cancer treatment as a highly localized adjuvant therapy implanted post lumpectomy to eliminate residual tumor cells around the surgical margin. In order to characterize surface-bound cytotoxicity, MB-MDA-231 breast cancer cells stably transfected to produce fluorescent mcherry protein were seeded onto membranes and imaged in vitro (Figure 6). Cell activity over time was visualized and live cells were manually counted in order to quantify death rate (Figure 6C). Cells were observed to adhere to membranes but cell spreading was not evident. Significant cell death was observed for both 20% KLAK PA membranes and 50% KLAK PA membranes over the course of 2 h. Because no hyaluronidase was added and because degradation products of 20% KLAK PA membranes were not cytotoxic in previous experiments, the observed cytotoxicity is likely due to extremely localized cellular interaction with KLAK PA on the membrane surface. This conclusion is further supported by the observation that media from dishes containing toxic membranes is non-toxic on its own. Furthermore, membranes made with heat-treated 50% KLAK PA were less cytotoxic than untreated counterparts, suggesting that structure still plays a role on membrane-cell interactions.
Figure 6.
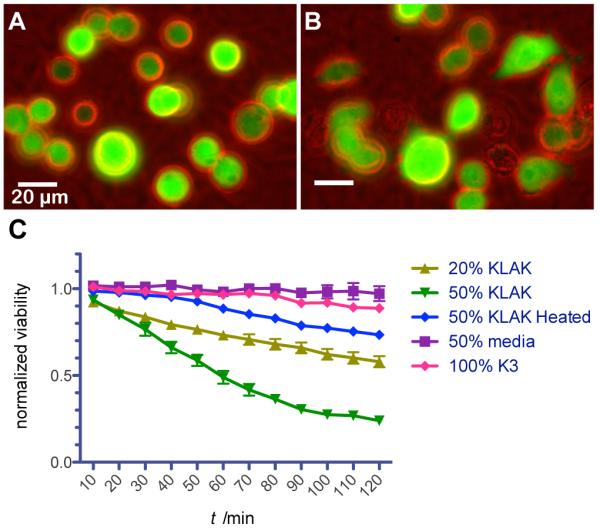
Behavior and viability of fluorescent MB-MDA-231 breast cancer cells seeded onto membranes. Cells on membranes assembled from 20% KLAK PA solution attach to the surface and are rounded (a) immediately after seeding but (b) migrate and eventually die after at later time points. (c) Membranes assembled from 20% KLAK PA and 50% KLAK PA solutions are significantly cytotoxic after only 2 h, while the associated membrane-conditioned media is not cytotoxic.
3. Conclusions
We have demonstrated that nanoscale supramolecular morphology of PAs in solution can dictate the resulting microstructure and properties of hierarchically self-assembled membranes. We have further demonstrated that this hierarchical self-assembly process can be used to incorporate anti-cancer bioactivity through specific PAs into membranes. The membranes prepared can act either as reservoirs for sustained release of cytotoxicity upon enzymatic degradation or as a material with highly localized surface cytotoxicity. Thus, the systems investigated are a versatile and potentially environmentally responsive platform for breast cancer therapy.
4. Experimental Section
PA Synthesis and Co-Assembly: Positively charged, non-bioactive K3 PA with the sequence C16-VVVAAAKKK-NH2 and KLAK PA with the sequence C16-VVVAAAGGKLAKLAKKLAKLAK-NH2 were synthesized using standard fluoren-9-ylmethoxycarbonyl (Fmoc) solid-phase peptide synthesis and purified using reversed-phase high-performance liquid chromatography in an acidic acetonitrile/water gradient containing trifluoroacetic acid (0.1%). After lyophilization of purified PAs, residual trifluoroacetic acid counterions were exchanged by sublimation from HCl (10 mm) in order to improve biocompatibility. PAs were then resolubilized in deionized water, lyophilized, and stored at − 20 °C until used. The peptide content of lyophilized PAs was analyzed (AIBioTech) in order to establish a conversion between mass concentration and molarity. Fluorescently labeled K3 PA (G-K3) and KLAK PA (R-KLAK) were synthesized by incorporating a C-terminus lysine with N-methyltrityl (MTT) side group protection, orthogonally deprotecting the side group, and reacting with fluorescein isothiocyanate (FITC) or rhodamine-b isothiocyanate (RBITC) on resin.
KLAK PA (5 mm) and K3 PA (5 mm) were co-assembled by dissolving PAs individually in deionized water and then mixing the freshly dissolved PA solutions at various ratios such that the total PA concentration always remained at 5 mm. Solutions were then sonicated for 1 h and left at room temperature overnight prior to use.
Transmission Electron Microscopy of PA Solutions
Samples for transmission electron microscopy (TEM) were prepared from pure and co-assembled solutions of K3 PA and KLAK PA (preparation described above) by placing each solution (10 μL) onto a 300 mesh copper grid with an amorphous carbon support film (Electron Microscopy Sciences). The solution was wicked after 2 min and allowed to dry for 10 min before staining with uranyl acetate (2%). Imaging was performed on a JEOL 1230 TEM with a Hamamatsu ORCA camera at an accelerating voltage of 100 kV.
Small Angle X-ray Scattering of PA Solutions
Small angle x-ray scattering (SAXS) was used to characterize the nanostructure morphology of pure and co-assembled PAs in solution (preparation described above). SAXS measurements were performed using beam line 5ID-D in the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) Synchrotron Research Center at the Advanced Photon Source at Argonne National Laboratory. Using a double-crystal monochromator to select an energy of 15 keV (corresponding to a wavelength of 0.83 A-1), scattering data was collected using a CCD detector (MAR) positioned 245 cm behind the sample. Scattering intensity was recorded in the interval 0.008< q < 0.25 A-1 and the wave vector was defined as q = (4 π / λ) sin(θ / 2), where θ is the scattering angle.
Solution samples were loaded in quartz capillaries (1.5 mm diameter). The x-ray exposure times were 2 - 8 s depending on the sample. The 2-D SAXS images collected were azimuthally averaged to produce 1-D profiles of intensity using the 2-D data reduction program Fit2D. Background scattering of the capillary with only deionized water was also collected and was subtracted from sample data.
Scanning Electron Microscopy of Membrane Cross-Section
Scanning electron microscopy (SEM) was used to image the cross-section of planar membranes formed by self- assembly of pure and co-assembled PAs with HA. In order to stabilize membranes against changes in solution ionic strength and osmotic pressure upon introduction to in vitro or in vivo environments, crosslinkable tyramine-substituted HA (1.5% substitution, MW 910 kDa, Lifecore Biomedical) was used for all membrane studies. Tyramine-substituted HA (HA-Tyr, 10 mg/mL) was dissolved in deionized water containing horseradish peroxidase type II (HRP, 10 U/mL). Membranes were fabricated by layering PA solution (50 μL) on top of HA-Tyr solution (50 μL) in the cap of a microcentrifuge tube (0.5 mL volume) and incubating at room temperature for 3 h before washing with deionized water. Samples were then fixed in glutaraldehyde (2%) with sucrose (3%) overnight at room temperature prior to dehydration using a stepwise gradient of ethanol in water (20% to 100%). In order to best preserve the hydrated structure of samples for electron microscopy, critical point drying was performed with a Tousimis SAMDRI-795 critical point dryer. Dried samples were manually torn to expose the cross-section and were mounted to SEM stubs using carbon tape. Finally, samples were coated with 8 nm of osmium using an osmium plasma coater (Structure Probe, Inc.) and imaged by a LEO 1525 field emission SEM using an accelerating voltage of 3 kV.
Confocal Microscopy of Membrane
Confocal microscopy was used to probe the distribution of KLAK PA and K3 PA within the membrane. G-K3 and R-KLAK were incorporated into co-assembled solutions by first individually dissolving unlabeled K3 PA (5 mm) and KLAK PA (5 mm) in aqueous stock solutions of the corresponding fluorescent PA (1 mg/mL). Solutions were then mixed at 1:5 or 1:2 KLAK PA to K3 PA, sonicated for 1 h, and left in darkness at room temperature overnight prior to use. Membranes were formed inside circular polyurethane washers (0.2 in inner diameter, McMaster-Carr) by filling washers with HA solution (50 μL) and then adding PA solution (50μL) on top. Membranes were incubated at room temperature for 3 h in the dark to prevent photobleaching. After washing with water, membranes were sandwiched between glass coverslips, which were sealed to prevent dehydration during imaging.
Imaging was performed using a Zeiss LSM 510 META laser scanning confocal microscope (LSCM) with the appropriate configurations for FITC (488 nm ex. laser/505-530 nm band pass emission filter) and RBITC (540 nm ex. laser/560-615 nm band pass emission filter). Multitrack mode sequential imaging was used for red and green channels in order to prevent fluorophore bleed-through. Optical slices were captured at regular intervals to produce reconstructed z-stacks and cross-section images were compiled from orthogonal views of z-stacks using NIH ImageJ software.
Viability of Cells Upon Enzymatic Degradation
In order to examine the potential of membranes to release anti-cancer agents upon enzymatic degradation, membranes were fabricated and degraded by hyaluronidase in non-tissue culture treated U-bottom 96 well plates. Using pure and co-assembled PA solutions (preparation previously described), membranes were formed by dropping HA-Tyr solution (10 μL, preparation previously described) into each well and adding PA solution (40 μL) on top. Membranes were incubated in PA solution at room temperature for 3 h prior to washing off excess PA with deionized water. A solution of H2O2 (0.1%) was then added to each membrane for 15 min in order to crosslink HA-Tyr by HRP-catalyzed oxidation of tyramine moieties, resulting in a membrane- enclosed hemisphere of HA-Tyr gel. Samples were then washed thoroughly with deionized water and were incubated in Hank’s balanced salt solution (HBSS) overnight to remove any residual H2O2.
Bovine testicular hyaluronidase (Sigma) was dissolved in HBSS (50 U/mL) and was added to each well. Degradation products were collected at 24, 48, 72, and 96 h by carefully aspirating the solution above membranes without further agitating or disturbing the membrane. Degradation products of HA-Tyr gels alone were also collected and were used as controls.
SKBR-3 breast cancer cells, MDA-MB-231 breast cancer cells, and MCF-10a breast epithelial cells were plated in flat bottom tissue-culture treated 96 well plates (Corning) at 10,000, 5000, and 10,000 cells per well respectively. Cells were incubated overnight at 37 °C before HBSS containing membrane degradation products (50 uL) was added to each well already containing cell culture media (50 uL). Cell viability was assessed after 24 h by adding CellTiter 96 AQueous One Solution MTS Proliferation Assay (Promega, 20 μL) to each well and incubating for 1 h at 37 °C before reading the absorbance at 490 nm using a SpectraMax M5 plate reader (Molecular Devices).
Live Cell Imaging Cell-Membrane Interaction
Membranes were fabricated in glass- bottom Petri dishes (MatTek, 7 mm) by layering PA solution (50 μL) onto HA-Tyr solution (50 μL) for 3 h. Excess PA was washed off with deionized water and excess HA was removed from underneath membranes using a pipette. Membranes were then crosslinked for 15 min using H2O2 (0.1%) and were subsequently washed thoroughly with deionized water. Membranes were additionally washed in cell culture media and left to dry overnight. Using cell culture media, membranes were then reconstituted and washed prior to seeding mcherry- expressing MDA-MB-231 breast cancer cells onto the membranes. Cells were imaged in vitro at 37 °C using a Biostation IM live cell recorder (Nikon). Quantification of cell viability on membranes was performed by counting intact cells every 5 mins from 15 fields per sample. Additionally, media from membranes assembled using 50% KLAK PA was aspirated from the dish after imaging and was added to a fresh dish containing cells seeded directly onto glass in order to characterize the bulk toxicity of the resultant media.
Statistics
Error bars for degradation cytotoxicity and live cell imaging indicate the standard error of the mean. Differences between groups were determined using a two-way analysis of variance (ANOVA) with a Bonferroni multiple comparisons post-hoc test using GraphPad Prism 5.0 software.
Supplementary Material
Acknowledgements
This work was supported by a grant from the NIH National Cancer Institute Center for Cancer Nanotechnology Excellence (No. 5U54CA151880-02). Characterization of peptide amphiphile and membrane structure was also supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division, of the U.S. Department of Energy (No. DE-FG02-00ER45810). R.H.Z. was supported by an NSF Graduate Research Fellowship. Experiments made use of the following facilities at Northwestern University: EPIC Facilities of the NUANCE Center, the Biological Imaging Facility, the Cell Imaging Facility, and the Institute for BioNanotechnology in Medicine. The NUANCE Center is supported by the NSF-NSEC, NSF-MRSEC, Keck Foundation, the State of Illinois, and Northwestern University. X-ray scattering studies were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by E.I. DuPont de Nemours & Co., The Dow Chemical Company, and Northwestern University. Use of the APS, an Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. The authors thank Dr. Ronit Bitton for advice in x-ray scattering data analysis and Dr. Daniel Toft for providing all cell lines used.
Footnotes
Supporting Information
Supporting Information is available online from the Wiley Online Library or from the author.
Contributor Information
R. Helen Zha, Department of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, Institute for BioNanotechnology in Medicine, Northwestern University, Chicago, IL 60611, hellz@northwestern.edu.
Shantanu Sur, Institute for BioNanotechnology in Medicine, Northwestern University, Chicago, IL 60611, s-sur@northwestern.edu.
Prof. Samuel I. Stupp, Department of Materials Science and Engineering, Department of Chemistry, Northwestern University, Evanston, IL 60208, Department of Medicine, Institute for BioNanotechnology in Medicine, Northwestern University, Chicago, IL 60611.
References
- [1].Tamada J, Langer R. J. Biomater. Sci., Polym. Ed. 1992;3:315. doi: 10.1163/156856292x00402. [DOI] [PubMed] [Google Scholar]
- [2].Domb A, Israel Z, Elmalak O, Teomim D, Bentolila A. Pharm. Res. 1999;16:762. doi: 10.1023/a:1011995728760. [DOI] [PubMed] [Google Scholar]
- [3].Brem H, Gabikian P. J. Controlled Release. 2001;74:63. doi: 10.1016/s0168-3659(01)00311-x. [DOI] [PubMed] [Google Scholar]
- [4].Chen M, Huang H, Pierstorff E, Shin E, Robinson E, Ho D. Ann. Biomed. Eng. 2009;37:2003. doi: 10.1007/s10439-009-9662-9. [DOI] [PubMed] [Google Scholar]
- [5].Lam R, Chen M, Pierstorff E, Huang H, Osawa E, Ho D. ACS Nano. 2008;2:2095. doi: 10.1021/nn800465x. [DOI] [PubMed] [Google Scholar]
- [6].Ma G, Liu Y, Peng C, Fang D, He B, Nie J. Carbohydr. Polym. 2011;86:505. [Google Scholar]
- [7].Liu R, Wolinsky JB, Walpole J, Southard E, Chirieac LR, Grinstaff MW, Colson YL. Ann. Surg. Oncol. 2010;17:1203. doi: 10.1245/s10434-009-0856-z. [DOI] [PubMed] [Google Scholar]
- [8].Dang W, Daviau T, Ying P, Zhao Y, Nowotnik D, Clow C, Tyler B, Brem H. J. Controlled Release. 1996;42:83. [Google Scholar]
- [9].Perry J, Chambers A, Spithoff K, Laperriere N. Current Oncology. 2007;14:189. doi: 10.3747/co.2007.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Serpe MJ, Yarmey KA, Nolan CM, Lyon LA. Biomacromolecules. 2005;6:408. doi: 10.1021/bm049455x. [DOI] [PubMed] [Google Scholar]
- [11].Kim JK, Anderson J, Jun HW, Repka MA, Jo S. Mol. Pharmaceutics. 2009;6:978. doi: 10.1021/mp900009n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cui H, Webber MJ, Stupp SI. Biopolymers. 2010;94:1. doi: 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Webber MJ, Kessler JA, Stupp SI. J. Intern. Med. 2010;267:71. doi: 10.1111/j.1365-2796.2009.02184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Matson JB, Zha RH, Stupp SI. Curr. Opin. Solid State. Mater. Sci. 2011;15:225. doi: 10.1016/j.cossms.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hartgerink JD, Beniash E, Stupp SI. Proc. Natl. Acad. Sci. USA. 2002;99:5133. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cui H, Muraoka T, Cheetham AG, Stupp SI. Nano Lett. 2009;9:945. doi: 10.1021/nl802813f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Goldberger JE, Berns EJ, Bitton R, Newcomb CJ, Stupp SI. Angew. Chem. Int. Ed. 2011;50:6292. doi: 10.1002/anie.201100202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Muraoka T, Cui H, Stupp SI. J. Am. Chem. Soc. 2008;130:2946. doi: 10.1021/ja711213s. [DOI] [PubMed] [Google Scholar]
- [19].Stendahl JC, Rao MS, Guler MO, Stupp SI. Adv. Funct. Mater. 2006;16:499. [Google Scholar]
- [20].Beniash E, Hartgerink JD, Storrie H, Stendahl JC, Stupp SI. Acta Biomater. 2005;1:387. doi: 10.1016/j.actbio.2005.04.002. [DOI] [PubMed] [Google Scholar]
- [21].Zhang S, Greenfield MA, Mata A, Palmer LC, Bitton R, Mantei JR, Aparicio C, Olvera de La Cruz M, Stupp SI. Nat. Mater. 2010;9:594. doi: 10.1038/nmat2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Harrington DA, Cheng EY, Guler MO, Lee LK, Donovan JL, Claussen RC, Stupp SI. J. Biomed. Mater. Res. 2006;78A:157. doi: 10.1002/jbm.a.30718. [DOI] [PubMed] [Google Scholar]
- [23].Rajangam K, Behanna H, Hui MJ, Han X, Hulvat JF, Lomasney JW, Stupp SI. Nano Lett. 2006;6:2086. doi: 10.1021/nl0613555. [DOI] [PubMed] [Google Scholar]
- [24].Rajangam K, Arnold MS, Rocco MA, Stupp SI. Biomaterials. 2008;29:3298. doi: 10.1016/j.biomaterials.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chow LC, Wang LJ, Kaufman DB, Stupp SI. Biomaterials. 2010;31:6154. doi: 10.1016/j.biomaterials.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Silva G, Czeisler C, Niece K, Beniash E, Harrington DA, Kessler JA, Stupp SI. Science. 2004;303:1352. doi: 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]
- [27].Angeloni NL, Bond CW, Tang Y, Harrington DA, Zhang S, Stupp SI, McKenna KE, Podlasek CA. Biomaterials. 2011;32:1091. doi: 10.1016/j.biomaterials.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- [29].Palmer LC, Newcomb CJ, Kaltz SR, Spoerke ED, Stupp SI. Chem. Rev. 2008;108:4754. doi: 10.1021/cr8004422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shah RN, Shah NA, Del Rosario Lim MM, Hsieh C, Nuber G, Stupp SI. Proc. Natl. Acad. Sci. USA. 2010;107:3293. doi: 10.1073/pnas.0906501107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bond CW, Angeloni NL, Harrington DA, McKenna KE, Podlasek CA. J. Sex. Med. 2011;8:78. doi: 10.1111/j.1743-6109.2010.02001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Capito RM, Azevedo HS, Velichko YS, Mata A, Stupp SI. Science. 2008;319:1812. doi: 10.1126/science.1154586. [DOI] [PubMed] [Google Scholar]
- [33].Carvajal D, Bitton R, Mantei JR, Velichko YS, Stupp SI, Shull KR. Soft Matter. 2010;6:1816. [Google Scholar]
- [34].Chow LW, Bitton R, Webber MJ, Carvajal D, Shull KR, Sharma AK, Stupp SI. Biomaterials. 2011;32:1574. doi: 10.1016/j.biomaterials.2010.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Velichko YS, Mantei JR, Bitton R, Carvajal D, Shull KR, Stupp SI. Adv. Funct. Mater. 2012;22:369. doi: 10.1002/adfm.201101538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Standley SM, Toft DJ, Cheng H, Soukasene S, Chen J, Raja SM, Band V, Band H, Cryns VL, Stupp SI. Cancer Res. 2010;70:3020. doi: 10.1158/0008-5472.CAN-09-3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Madan AK, Yu K, Dhurandhar N, Cullinane C, Pang Y, Beech DJ. Oncol. Rep. 1999;6:607. doi: 10.3892/or.6.3.607. [DOI] [PubMed] [Google Scholar]
- [38].Stern R. Pathol. Biol. 2005;53:372. doi: 10.1016/j.patbio.2004.12.021. [DOI] [PubMed] [Google Scholar]
- [39].Velichko YS, Stupp SI, Olvera de la Cruz M. J. Phys. Chem. B. 2008;112:2326. doi: 10.1021/jp074420n. [DOI] [PubMed] [Google Scholar]
- [40].Pashuck ET, Cui H, Stupp SI. J. Am. Chem. Soc. 2010;132:6041. doi: 10.1021/ja908560n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jiang H, Guler MO, Stupp SI. Soft Matter. 2007;3:454. doi: 10.1039/b614426h. [DOI] [PubMed] [Google Scholar]
- [42].Storrie H, Guler MO, Abu-Amara SN, Volberg T, Rao M, Geiger B, Stupp SI. Biomaterials. 2007;28:4608. doi: 10.1016/j.biomaterials.2007.06.026. [DOI] [PubMed] [Google Scholar]
- [43].Darr A, Calabro A. J. Mater. Sci.: Mater. Med. 2009;20:33. doi: 10.1007/s10856-008-3540-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.