

Abstract
Acetaminophen (APAP), a widely-used analgesic agent, can cause liver injury through increased nitrative stress, leading to protein nitration. However, the identities of nitrated proteins and their roles in hepatotoxicity are poorly understood. Thus, we aimed at studying the mechanism of APAP-induced hepatotoxicity by systematic identification and characterization of nitrated proteins in the absence or presence of an anti-oxidant N-acetylcysteine (NAC). The levels of nitrated proteins markedly increased at 2 h in mice exposed to a single APAP dose (350 mg/kg ip), which caused severe liver necrosis at 24 h. Protein nitration and liver necrosis were minimal in mice exposed to nontoxic 3-hydroxyacetanilide or animals co-treated with APAP and NAC. Mass-spectral analysis of the affinity-purified nitrated proteins identified numerous mitochondrial and cytosolic proteins including mitochondrial aldehyde dehydrogenase, Mn-superoxide dismutase, glutathione peroxidase, ATP synthase, and 3-ketoacyl-CoA thiolase involved in anti-oxidant defense, energy supply, and fatty acid metabolism, respectively. Immunoprecipitation followed by immunoblot with anti-3-NT antibody confirmed that the aforementioned proteins were nitrated in APAP-exposed mice but not in NAC-co-treated mice. Consistently, NAC co-treatment significantly restored the suppressed activities of these enzymes. Thus, we demonstrate a new mechanism by which many nitrated proteins with concomitantly suppressed activities promotes APAP-induced mitochondrial dysfunction and hepatotoxicity.
Keywords: Acute liver injury, acetaminophen, CYP2E1, 3-nitrotyrosine, protein nitration, N-acetylcysteine, mitochondrial dysfunction
Many clinically used drugs, abused substances, and toxic chemicals can cause acute liver injury in humans as well as experimental models. For instance, acetaminophen (APAP, 4-hydroxyacetanilide, 4-acetamidophenol)1, a widely-used analgesic and antipyretic agent, and troglitazone, a peroxisomal proliferator activating receptor-gamma agonist used for treating diabetic patients, are known to cause acute liver injury and often liver failure and death [1-4]. Abused substances such as alcohol (ethanol) [5], cocaine and amphetamine derivative such as 3, 4-methylenedioxymethamphetamine (MDMA, ecstasy) can cause liver injury as well as other organs such as brain, heart, and kidney [6, 7]. Some toxic chemicals such as carbon tetrachloride (CCl4), D-galatosamine/lipopolysaccharide (LPS) and cadmium cause acute liver injury in experimental models [8-11]. Despite the well-established acute hepatotoxicity, the underlying mechanisms by which these toxic agents cause liver injury are still elusive. In general, reduction of a cellular antioxidant glutathione (GSH), increased oxidative/nitrative stress, production of reactive free radical metabolites and their protein adducts formation, change in calcium homeostasis, mitochondrial dysfunction, activation of the c-Jun N-terminal kinase (JNK)-associated cell death signaling pathway and innate immune responses, activation of hypoxia-inducible factor, etc play important roles in acute tissue injury. It is generally accepted that each of these contributing factors works in concert to exert acute hepatotoxicity. Therefore, administration of antioxidants such as reduced GSH and N-acetylcysteine (NAC) can protect the various organs from acute injury.
APAP is a widely-used analgesic/antipyretic drug but it can cause acute hepatotoxicity with centrilobular necrosis and liver failure when it is consumed in large doses. The hepatotoxicity can be markedly increased with normal doses of APAP in certain individuals who consume alcohol through synergistic interaction between APAP and alcohol [12-15]. This is most likely through elevation of ethanol-inducible cytochrome P450 2E1 (CYP2E1), which is a major enzyme responsible for APAP metabolism [16]. Since CYP2E1 is expressed in liver and many other tissues including kidney, APAP can also cause acute kidney damage in humans [17] and experimental models [18]. Under normal conditions, APAP toxicity can be prevented by increased cellular GSH by exogenous administration of GSH-ethyl ester [19, 20] or NAC [20, 21]. When cellular GSH is depleted, APAP and its reactive electrophilic metabolite N-acetyl-p-benzoquinoneimine (NAPQI) can interact with various proteins to produce NAPQI-protein adducts. Earlier reports showed remarkable correlation between NAPQI-protein adducts and the severity of liver injury has been reported [22, 23]. However, other reports suggest that NAPQI-protein adducts may not be important in directly causing acute liver injury since non-toxic analogs of APAP such as 3-hydroxyacetanilide (AMAP) can produce similar protein adducts and that NAPQI-protein adducts were still functionally active [24-27]. On the other hand, many investigators including Jaeschke and Hinson groups independently showed the critical roles of nitrated proteins and mitochondrial dysfunction in APAP-mediated hepatotoxicity, based on strong immunohistochemical staining of the nitrated proteins [3-nitrotyrosine (3-NT)-immunoreactive proteins] in the centrilobular necrotic areas in APAP-exposed animals [25, 28]. In agreement with these reports, we also demonstrated that APAP is likely to promote liver injury through increased protein nitration, which is promoted by increased peroxynitrite levels [1]. In this report [1], we showed the role of CYP2E1 in promoting protein nitration because of no or little detection of inducible nitric oxide synthase (iNOS), which is usually induced upon exposure to cell death stimulants including ethanol (alcohol) [29-31] or in many other pathological conditions such as ischemia reperfusion injury [32]. Despite extensive studies on the roles of nitrated proteins in acute hepatotoxicity by APAP [10, 25, 28, 33-37], LPS [38], and MDMA [39], it is poorly understood which cellular (including mitochondrial) proteins are nitrated and how their functions are altered to contribute to APAP-mediated mitochondrial dysfunction and hepatotoxicity. For instance, only a few proteins such as cytosolic superoxide dismutase (Cu-Zn-SOD, SOD1) [1], catalase [37], and mitochondrial SOD2 (Mn-SOD, SOD2) [34] have been shown to be nitrated in APAP-exposed animals. However, based on numerous spots of nitrated proteins displayed on 2-D gel [1], we hypothesized that many more proteins could be nitrated and that nitrated (mitochondrial) proteins can contribute to APAP-mediated liver injury. Therefore, we aimed to systematically identify nitrated proteins from mice exposed to APAP for 2 h where liver injury and ALT levels were very low and investigate the causal relationship between protein nitration and APAP-induced mitochondrial dysfunction and liver injury with or without using an antioxidant NAC.
Materials and methods
Chemicals and other materials
APAP, a non-toxic analog AMAP, 3-[(3-cholamidopropyl)-1-dimethylammonio]-propanesulfonic acid (CHAPS), NAC, primary antibody for β-actin, and all other chemicals used in this study were obtained from Sigma Chemical (St. Louis, MO), unless indicated otherwise. Protease inhibitor and phosphatase inhibitor cocktails were obtained from Calbiochem (San Diego, CA). Agarose-coupled with anti-3-NT monoclonal antibody was purchased from Cayman Inc. (Ann Arbor, MI). Protein A/G-agarose beads were from Invitrogen Life Technologies (Grand Island, NY). Horseradish peroxidase-conjugated goat anti-rabbit and goat anti-mouse antibodies were purchased from Bio-Rad (Hercules, CA). Specific antibodies to 3-NT were from Abcam (Cambridge, MA) and mitochondrial SOD2 were from Cell Signaling Technology, Inc. (Boston, MA). Mitochondrial aldehyde dehydrogenase (ALDH2), glutathione peroxidase (Gpx), ATP synthase (complex V), and horseradish peroxidase-conjugated Protein A/G agarose beads were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Enhanced chemiluminescence reagents were obtained from Thermo Scientific (Rockford, IL).
Animal Treatment and Histological Analysis
All animal experiments were conducted in accordance with the guidelines for small animals at National Institutes of Health. Age and gender-matched (2~3 months old) young mice (on a 129/Svj background) [1] were used in this study. All mice were housed in a temperature-controlled room (23-25 °C) on a 12 h light/12 h dark cycle at the National Institute on Alcohol Abuse and Alcoholism and were fed standard rodent chow ad libitum. Handling and treatment procedures including APAP injection without or with NAC co-treatment were approved by the Institute Animal Care and Use Committee. Age-matched female mice were injected with APAP or AMAP (a single ip injection of 350 mg/kg dissolved in warm isotonic saline solution) for 2 or 24 h, as reported [1]. Control mice were treated with sterile saline solution while NAC (a single ip injection of 1200 mg/kg dissolved in warm saline) was co-treated with APAP injection [21]. The trunk blood and liver from each mouse were collected immediately after mice were euthanized by decapitation at the indicated time points. A small liver section from the largest lobe of each mouse was fixed in 10% formalin in PBS and subjected to hematoxylin and eosin (H&E) staining by American HistoLabs, Inc. (Gaithersburg, MD).
Sample preparation and Immunoblot (IB) analysis
Mouse livers within the same group from different treatments were pooled and homogenized with a plastic/glass homogenizer in cold buffer containing 150 mM sucrose, 50 mM Tris-Cl, pH 7.4 and 1 mM EDTA. Mitochondria and cytosolic fractions from pooled whole homogenates of different treatment groups (n=4-6/group) were prepared by differential centrifugation method, as described [40-42]. Mitochondria fractions were washed three times with the homogenation buffer to remove any contaminating cytosolic fractions. The buffer used in this study was freshly pre-equilibrated with nitrogen gas for 1 h to remove dissolved oxygen [41, 42].
Immuno-affinity purification of nitrated proteins
Nitrated proteins were purified by immune-affinity chromatography by following method of Song et al. [43]. To study the causal relationship between protein nitration and hepatotoxicity, we tried to identify the nitrated proteins and characterize their properties in the absence or presence of NAC co-treatment. For this purpose, we intentionally chose to purify the nitrated proteins from mice exposed to APAP for 2 h where nitrated proteins were markedly increased but no or minimal tissue damage was observed [1]. Briefly, 10 mg of solubilized cytosolic or mitochondrial proteins from APAP-exposed mice were loaded onto a small antibody-agarose column and incubated for 30 min before proteins were eluted with 15 volumes of buffer (pH 5.0). The antibody-bound nitrated proteins were eluted with a small volume of 0.1 M glycine buffer (pH 3.0) and the eluted proteins immediately neutralized to pH 7.0. These steps were repeated more than 12 times to collect sufficient amounts of nitrated proteins for mass-spectral analysis for protein sequencing. The affinity-purified proteins were then concentrated using mini-spin-column concentrators (Millipore). Aliquots of the purified nitrated proteins were separated in 1-D gels and stained with silver.
Mass-spectral analysis of affinity-purified nitrated proteins
Affinity-purified proteins were sent for mass-spectral analysis performed by Dr. Drake Zhang at ProtTech (Germantown, PA). The protein identification was carried out by using the NanoLC-ESI-MS/MS peptide sequencing technology. In brief, proteins were denatured by adding 8 M urea. The Cys residues in the solution samples were reduced with 20 mM dithiothreitol followed by alkylation with 20 mM iodoacetate. After diluting the samples to 2 M urea with 100 mM ammonium bicarbonate (pH 8.5), proteins in the solution were digested by adding sequencing grade modified trypsin (Promega, Madison, WI). The resulted peptide mixtures from digestion were desalted with a PepClean spin column (Pierce, Rockford, IL).
The dissolved peptide samples were then injected into an HPLC system (Beckman) with a 75 micrometer inner diameter reverse phase C18 column. HPLC solvent A was 97.5% water, 2% acetonitrile, 0.5% formic acid. HPLC solvent B was 9.5% water, 90% acetonitrile, 0.5% formic acid. The gradation time was 60 minutes from 2% Solvent B to 90% solvent B, plus 20 minutes for sample loading and 20 minutes for column washing. The column flow rate is around 800 nanoliter per minute after splitting. The HPLC system was on-line coupled with an ion trap mass spectrometer (LCQ DECA XP PLUS, Thermo Scientific) where a sample eluted from the HPLC column was directly ionized by an electrospray ionization (ESI) process and allowed to enter into the mass spectrometer. The mass spectrometer was set at the data-dependent mode to acquire MS/MS data via a low energy collision induced dissociation (CID) process. As default, the mass-spectrometric data acquired were used to search against the most recent non-redundant protein database (NR database, NCBI) with ProtTech’s ProtQuest software suite. The output from the database search was manually validated by a senior scientist for additional confirmation. More experimental details can be found at www.ProtTech.com.
Immunoprecipitation and immunoblot analyses
The immunoprecipitation of a few selected proteins was performed as described [1, 41]. Briefly, mitochondrial proteins from at least 4 to 6 mouse livers/group (1 mg protein) was incubated with 3 μg anti-3-NT or 3 μg of the specific antibody to each target protein overnight at 4 °C with constant head-to-tail rotation followed by addition of protein A/G-agarose (Invitrogen) for another 1 h. Proteins bound to the protein A/G-agarose were washed 4 times with PBS containing 1% CHAPS to remove nonspecifically bound proteins. After centrifugation, agarose-bound proteins were dissolved in Laemmli buffer and separated on 15% SDS-PAGE gels for immunoblot analysis using the specific antibody against 3-NT or each indicated target protein for the immunoprecipitated ALDH2, ATP synthase, 3-ketoacyl-CoA thiolase (thiolase), SOD2, and Gpx.
Measurements of mitochondrial enzyme activities
ALDH2 activity was measured by the method previously described [30, 32]. ATP synthase activity and levels (0.5 mg mitochondrial proteins) were determined by using the commercially available kit from Roche Applied Science (Indianapolis, IN) by following the manufacturer’s instruction. Thiolase activity was determined as described [30, 32]. The SOD2 or Gpx enzyme activity (0.5 mg of liver mitochondrial proteins) was measured using the commercial kit from Calbiochem (San Diego, CA) by following the manufacturer’s protocol.
Activity measurement and identification of nitrated Tyr residues of recombinant mouse SOD2
Please refer to Supplemental materials.
Data processing and statistical analysis
All data were obtained with at least three different measurements, unless otherwise stated. Data are presented as mean ± S.E.M. and statistical analyses were performed using One-Way ANOVA followed by Tukey test (Prism5 software) and p<0.05 was considered statistically significant. Other materials and methods not described here were the same as before [1, 40-43].
Results
Effects of APAP on acute liver necrosis
Consistent with our previous results [1], histopathology staining (Figs. 1A and B) showed that APAP administration (a single ip injection with 350 mg/kg) slightly increased minor necrotic foci at 2 h with a significantly elevated level of serum alanine aminotransferase (ALT) (Fig. 1D), despite being approximately 3% of that observed at 24 h (Fig. 4F). In contrast, treatment with saline (sham control) or a non-toxic analog of APAP, 3-hydroxyacetanilide (3-AMAP) (a single ip injection with 350 mg/kg) neither induced acute liver damage (Fig. 1C) nor elevated ALT level (Fig. 1D).
Fig. 1.
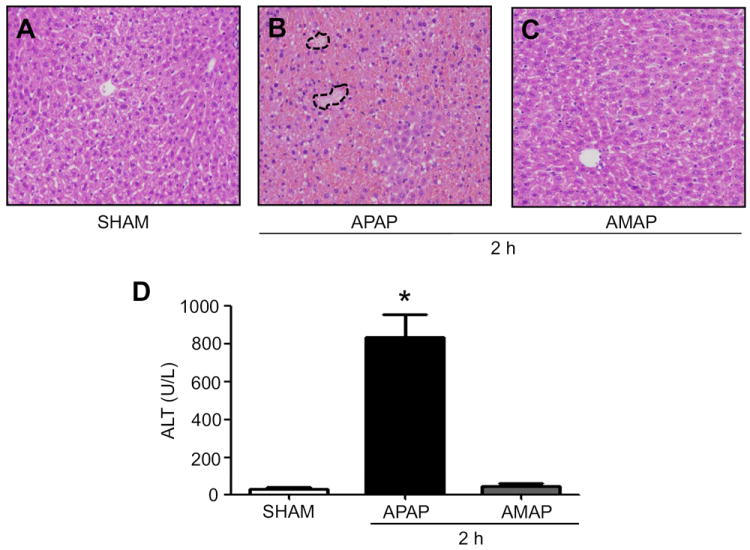
Differential effects of APAP or AMAP on liver histology and ALT levels. Hematoxylin and eosin (H&E) stained liver slides for mice treated for 2 h are presented for (A) vehicle control (SHAM), (B) APAP, and (C) AMAP (magnification 100 ×). (D) Serum ALT levels for different groups are presented. *, significantly different (*p<0.05) from the control. A few necrotic regions in APAP-exposed mice are marked with broken lines (B) (n=4-6/group).
Fig. 4.
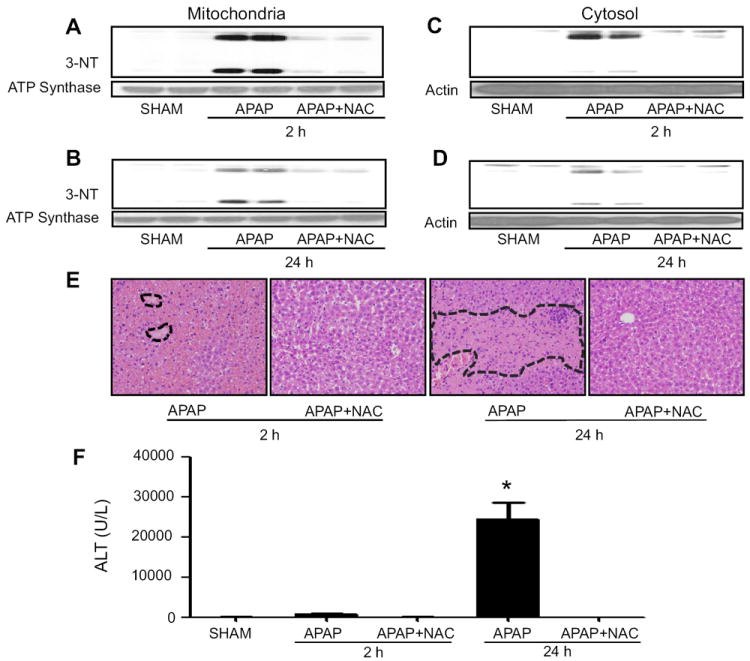
Protective effects of NAC on APAP-mediated protein nitration and liver injury. The levels of nitrated proteins in mitochondria (A and B) and cytosol (C and D) from mice exposed to APAP ± NAC for 2 or 24 h, as indicated, were evaluated with the specific anti-3-NT (upper panels). Anti-ATP synthase and actin were used for equal loading for mitochondria and cytosolic fractions, respectively (lower panels). (E) Typical H&E-stained liver slides are presented for APAP-exposed mice with or without NAC co-treatment, as indicated (magnification 100 ×). (F) Serum ALT levels for the indicated groups are presented. *, significantly different (*p<0.05) from other groups (n=4-6/group). Severe necrotic regions of APAP-exposed samples are marked with broken lines (E).
Increased levels of nitrated proteins and their purifications from APAP-exposed mice
The levels of nitrated proteins in saline-treated control group were almost negligible but their levels in both mitochondria (Fig. 2A) and cytosol (Fig. 2B) markedly elevated at 2 h in the APAP-treated mice. However, 3-AMAP did not increase the levels of nitrated proteins in mitochondrial but slightly increased the levels of nitrated proteins in cytosolic fractions (Figs. 2A and B, respectively), consistent with little hepatotoxicity (Fig. 1C).
Fig. 2.
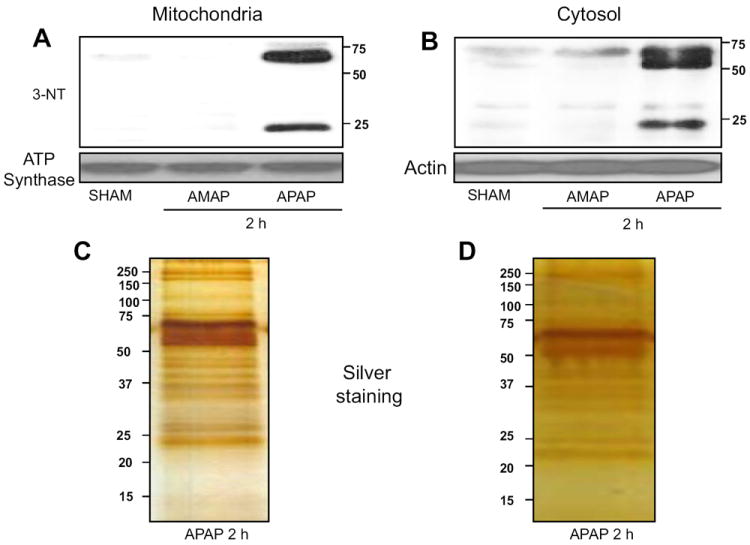
Differential effects of APAP or AMAP on protein nitration. Hepatic mitochondrial (A) and cytosolic (B) proteins from mice exposed to the indicated agents for 2 h were subjected to immunoblot analysis with the specific anti-3-NT (upper panels), while specific anti-ATP synthase and actin were used to detect equal protein loading respectively (lower panels). Silver stained images of the affinity-purified nitrated proteins from mitochondria (C) and cytosol (D) are shown (n=4-6/group). Similar results to those shown in the Figures (A-D) were observed by at least two separate experiments.
Based on these results (Fig. 2) and multiple spots of nitrated proteins on 2-D gels [1], we purified nitrated proteins in mitochondrial and cytosolic fractions from mice exposed to APAP for 2 h by specific antibody-affinity chromatography. Consistent with our previous results [1], numerous nitrated proteins stained with silver were affinity-purified from mitochondria (Fig. 2C) and cytosol (Fig. 2D), respectively. It is noteworthy to mention that many more proteins were detected with the immunopurification method (Figs. 2C and D) than the immunoblot analysis (Figs. 2A and B). The differences could possibly result from: (1) the different sensitivities between the immunopurification technique and the immunoblot method, where only a few bands can be detected especially after a short exposure time; (2) the greater amounts of protein used for immunopurification, and (3) the usage of silver staining which can detect many proteins in the nanogram ranges.
Mass-spectral analysis revealed that many mitochondrial (Table 1) and cytosolic proteins (Supplemental Table 1), respectively, were nitrated. Many nitrated proteins (summarized in Fig. 3) are involved in cellular chaperone, anti-oxidant defense, electron transport, energy production, amino acid metabolism, fat oxidation, urea detoxification, etc.
Table 1.
Summary of mass-spectral analysis of affinity-purified nitrated mitochondrial proteins in mouse liver exposed to APAP for 2 h.
| Accession number | Protein Identified | No. of peptide identified |
|---|---|---|
| 124248512 | Carbamoyl-phosphate synthase | 144 |
| 115704 | Catalase | 47 |
| 6680027 | Glutamate dehydrogenase 1 | 44 |
| 31980648 | ATP synthase subunit beta | 30 |
| 6753036 | Aldehyde dehydrogenase 2 (ALDH2) | 29 |
| 21311901 | Dimethylglycine dehydrogenase | 28 |
| 6679184 | Ornithine transcarbamylase | 22 |
| 2598562 | Grp78 | 22 |
| 29126205 | 3-Ketoacyl-CoA thiolase | 20 |
| 162461907 | Stress-70 protein | 19 |
| 6678509 | Uricase | 18 |
| 31560689 | Hydroxymethylglutaryl-CoA synthase | 17 |
| 31982186 | Malate dehydrogenase | 16 |
| 31982393 | Epoxide hydrolase 2 | 16 |
| 31560355 | 2-Hydroxyphytanoyl-CoA lyase | 15 |
| 21450129 | Acetoacetyl-CoA thiolase | 14 |
| 183396771 | 60 kDa heat shock protein | 12 |
| 22122797 | 3-Ketoacyl-CoA thiolase B | 12 |
| 464506 | Pyruvate carboxylase | 12 |
| 6680748 | ATP synthase subunit alpha | 11 |
| 19527258 | Methylmalonate-semialdehyde dehydrogenase | 11 |
| 13385310 | Propionyl-CoA carboxylase beta chain | 10 |
| 31982273 | 17-Beta-hydroxysteroid dehydrogenase | 10 |
| 31982520 | Long-chain specific acyl-CoA dehydrogenase | 9 |
| 257743052 | Esterase-31 | 9 |
| 14331135 | Carboxylesterase MH1 | 9 |
| 6755863 | Glucose regulated protein 94 | 8 |
| 20149748 | Sarcosine dehydrogenase | 8 |
| 8393866 | Ornithine aminotransferase | 7 |
| 16905127 | Acyl-coenzyme A synthetase | 7 |
| 1706111 | Carnitine-O-palmitoyltransferase II | 7 |
| 1125026 | 3-Hydroxyacyl CoA dehydrogenase | 7 |
| 18079339 | Aconitate hydratase | 7 |
| 225543103 | Aldehyde dehydrogenase 4 family (ALDH4) | 7 |
| 31981810 | 3,2-trans-enoyl-CoA isomerase | 7 |
| 111038130 | Serine-pyruvate aminotransferase | 6 |
| 6680618 | Medium-chain specific acyl-CoA dehydrogenase | 6 |
| 110225339 | Acetyl-CoA acetyltransferase | 6 |
| 188035915 | Alpha-aminoadipic semialdehyde dehydrogenase isoform b | 6 |
| 2492632 | Glutaryl-CoA dehydrogenase | 6 |
| 254540162 | Propionyl-CoA carboxylase alpha chain | 6 |
| 37202121 | 4-Aminobutyrate aminotransferase | 6 |
| 31982856 | Dihydrolipoyl dehydrogenase | 6 |
| 33859811 | Trifunctional enzyme subunit alpha | 6 |
| 20330802 | Beta-1 metal-binding globulin | 6 |
| 21312260 | Aldehyde dehydrogenase 5 | 6 |
| 27532959 | 10-Formyltetrahydrofolate dehydrogenase (FTHFDH) | 6 |
| 13097375 | Electron transferring flavoprotein alpha polypeptide | 5 |
| 21312298 | Serine hydroxymethyltransferase | 5 |
| 729032 | Adenylyl cyclase-associated protein 1 | 5 |
| 45476581 | Propanoyl-CoA C-acyltransferase | 5 |
| 54607098 | Succinate dehydrogenase [ubiquinone] flavoprotein subunit (II) | 5 |
| 84871986 | Glutathione peroxidase | 5 |
| 13385942 | Citrate synthase | 5 |
| 8393343 | Fatty acid-binding protein | 5 |
| 6755114 | Peroxiredoxin-5 | 4 |
| 6678449 | Thiosulfate sulfurtransferase | 4 |
| 29789289 | Enoyl-CoA hydratase | 4 |
| 7949037 | Delta(3,5)-Delta(2,4)-dienoyl-CoA isomerase | 4 |
| 9506589 | Fructose-1,6-bisphosphatase 1 | 4 |
| 21450291 | Fructose-bisphosphate aldolase B | 4 |
| 584714 | Butyryl-CoA dehydrogenase | 4 |
| 7709990 | Betaine-homocysteine S-methyltransferase | 4 |
| 6680309 | Heat shock protein 10-kDa | 4 |
| 53450 | Superoxide dismutase 2 | 2 |
| 123787831 | (R)-3-amino-2-methylpropionate-pyruvate transaminase | 1 |
| 6754156 | Glycolate oxidase | 1 |
Fig. 3.
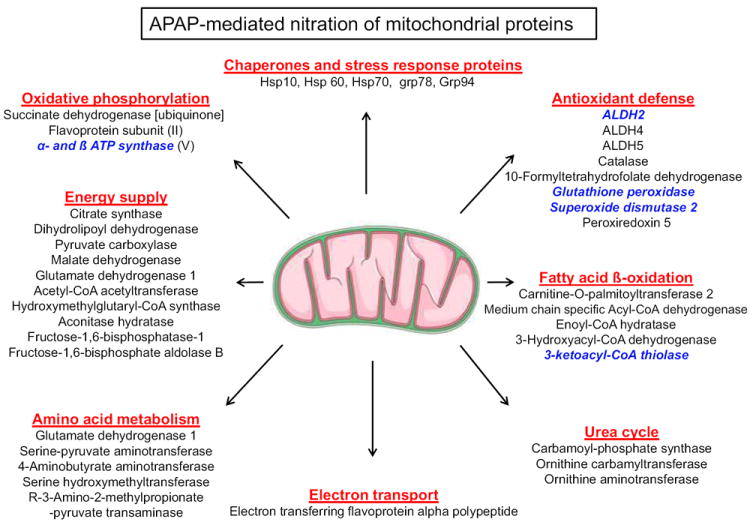
Summary of nitrated mitochondrial proteins in APAP-exposed mice. Some newly-identified nitrated mitochondrial proteins in APAP-exposed mouse liver are summarized with respect to the function of each protein identified by mass spectrometry. The five proteins written in bold and italic characters were further characterized for reversible nitration and enzyme activity changes following APAP exposure in the absence or presence of NAC co-treatment.
We then systematically evaluated whether the elevated levels of nitrated proteins and acute liver toxicity could be reversibly modulated by the co-treatment with an anti-oxidant NAC. Our results showed that the levels of protein nitration in both mitochondria (Figs. 4A and B) and cytosol (Figs. 4C and D) remained elevated for up to 24 h, although the levels of protein nitration at 24 h were markedly lower than those at 2 h post-APAP treatment, possibly due to degradation of nitrated proteins [1, 44, 45, and references within]. Increased levels of mitochondrial protein nitration in both groups returned to basal levels by NAC co-treatment. In addition, NAC co-treatment improved liver histology (Fig. 4E) and completely blocked the elevated ALT levels at 2 and 24 h post-APAP injection (Fig. 4F).
Confirmation of nitration and inactivation of nitrated proteins
Ruepp et al. [46] reported that the levels of some mitochondrial proteins such as ALDH2, ATP synthase and thiolase are decreased by APAP treatment. To test whether we have similar conditions as those described [46], we determined the amounts of five randomly-selected mitochondrial proteins out of many mitochondrial proteins (listed in Table 1) such as ALDH2 (Fig. 5A), thiolase (Fig. 5D), ATP synthase (Fig. 6A), SOD2 (Fig. 7A) and Gpx (Fig. 7D) by immunoblot analysis. We observed moderate reductions in the amounts of ALDH2, thiolase, and SOD2 at 2 h after APAP treatment. The reductions of these proteins were prevented by the NAC co-treatment. However, the levels of ATP synthase and Gpx were unchanged regardless of APAP and/or NAC co-treatment.
Fig. 5.
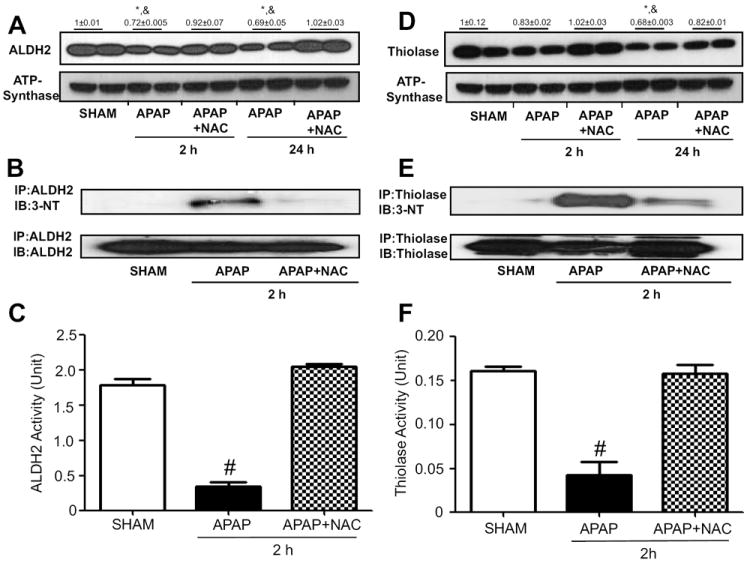
Reversible changes of nitration and activity of ALDH2 or thiolase. The levels of mitochondrial ALDH2 (A) or thiolase (D) in mice exposed to APAP ± NAC for 2 or 24 h were determined by immunoblot analysis (top panels) while the level of ATP synthase was used to demonstrate similar protein loading (lower panels). Numbers above the panels A and D represent densitometric values compared to those of sham group, which was set at 1. (B) Mitochondrial proteins from the indicated groups were immunoprecipitated with anti-ALDH2 (B) or anti-thiolase (E) antibody. The immunoprecipitated proteins were subjected to immunoblot analysis with anti-3-NT antibody (upper panels in B and E) and anti-ALDH2 (B) or anti-thiolase (E) antibody (bottom). ALDH2 (C) or thiolase (F) activity in mitochondrial extracts was determined for the indicated samples. *, significantly different from sham group; &significantly different from corresponding APAP+NAC; #significantly different from other groups (n=4-6/group), (*p<0.05).
Fig. 6.
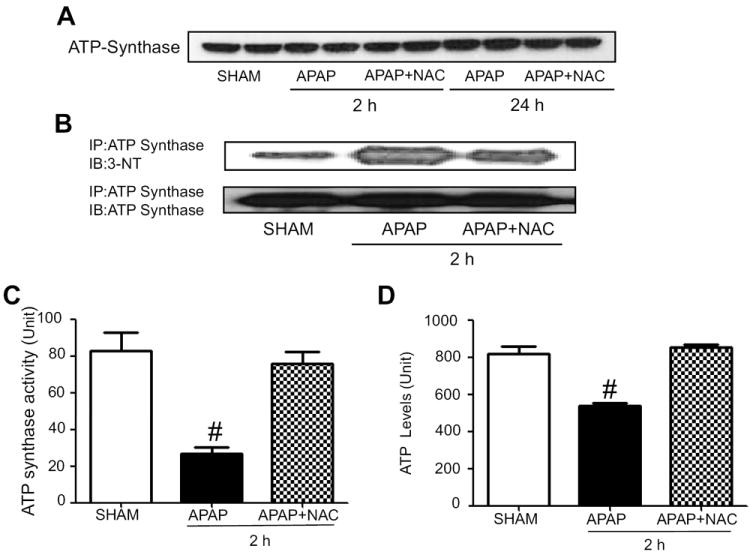
Reversible changes of nitration and activity of ATP synthase. (A) The levels of mitochondrial ATP synthase in APAP-exposed mice ± NAC for 2 or 24 h were determined by immunoblot analysis. (B) Mitochondrial proteins from the indicated groups were immunoprecipitated using the specific anti-ATP synthase antibody. The immunoprecipitated proteins were then subjected to immunoblot analysis with anti-3-NT antibody (upper panel) or anti-ATP synthase antibody (bottom). (C and D) Mitochondrial ATP synthase activity and levels were determined for the indicated samples, respectively. #, significantly different (*p<0.05) from other groups (n=4-6/group).
Fig. 7.
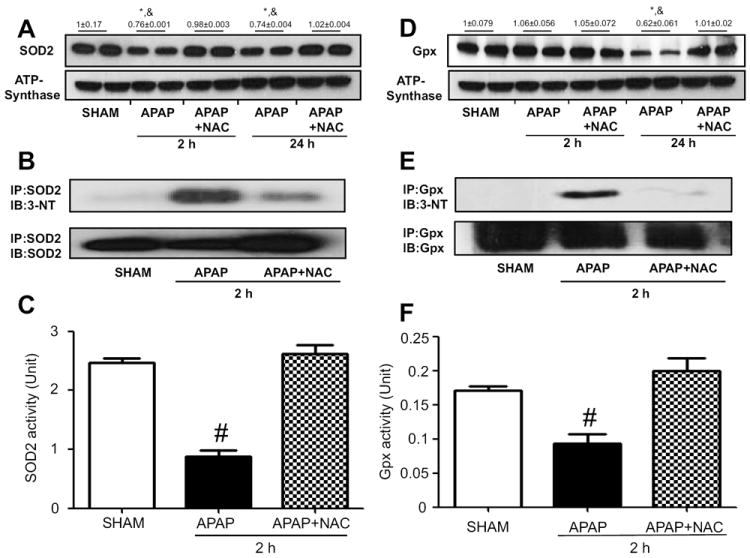
Reversible changes of nitration and activity of SOD2 or Gpx. The levels of mitochondrial SOD2 (A) or Gpx (D) in APAP-exposed mice ± NAC for 2 or 24 h were determined by immunoblot analysis (top panels). ATP synthase levels were used to show similar protein loading (lower panels). Numbers above the panels A and D respresent densitometric values compared to those of sham group which was set at 1. (B) Mitochondrial proteins from the indicated groups were immunoprecipitated with anti-SOD2 (B) or anti-Gpx (E) antibody. The immunoprecipitated proteins were then subjected to immunoblot analysis with anti-3-NT antibody (upper panels in B and E) and anti-SOD2 (B) or anti-Gpx (E) antibody (bottom). SOD2 (C) or Gpx (F) activity in mitochondrial extracts was determined for the indicated samples. *, significantly different from sham group; &significantly different from corresponding APAP+NAC; # significantly different from other groups (n=4-6/group), (*p<0.05).
We further studied direct relationship between nitration and activity changes of the aforementioned five proteins. Each protein in the mitochondrial fraction of saline control or APAP-exposed mice for 2 h in the absence or presence of NAC co-treatment was immunoprecipitated with the specific antibody to each target protein. The immunoprecipitated protein was then immunobloted with either the specific antibody to 3-NT or each target protein. We found that nitration of immunoprecipitated ALDH2 or thiolase protein in control group was extremely low (Fig. 5B or 5E, upper panel, first lane) while the level of nitrated ALDH2 or thiolase was markedly increased in APAP-exposed mice (second lane). The nitrated ALDH2 or thiolase protein was markedly decreased in NAC-co-treated mice (third lane) despite the similar levels of immunoprecipitated ALDH2 or thiolase in the three groups (Fig. 5B or 5E, bottom panel).
To study the functional implications of nitrated mitochondrial proteins in APAP-exposed mice, the catalytic activities of the five selected mitochondrial proteins in APAP-exposed mice in the absence or presence of NAC co-treatment were measured and compared to those of saline control. Fig. 5C and 5F show the results of ALDH2 and thiolase activity measurement, respectively. Hepatic ALDH2 and thiolase activities were markedly suppressed in mice exposed to APAP alone compared to that of saline control but their suppressed activities were restored in the NAC-co-treated samples.
Nitration of ATP synthase (Fig. 6B), SOD2 (Fig. 7B), and Gpx (Fig. 7E) were also observed in mice exposed to APAP alone for 2 h while the nitrated proteins disappeared or markedly decreased in the NAC-co-treated mice (Figs. 6B, 7B, and 7E, respectively). Consistently, we observed reversible inactivation and restoration of the activities of ATP synthase (Fig. 6C), SOD2 (Fig. 7C), and Gpx (Fig. 7F), respectively, in APAP-exposed mice depending on the absence or presence of NAC co-treatment. In addition, we observed significantly reduced levels of ATP in APAP-exposed mice compared to saline control group while the decreased ATP level was restored in the NAC co-treated mice (Fig. 6D), suggesting a sign of mitochondrial dysfunction in APAP-exposed mice. In addition, the activity of recombinant mouse SOD2 was suppressed in a dose-dependent manner after incubation with the nitrating agent tetranitromethane (TNM) (Suppl. Fig. 1). The SOD2 with the lowest activity (TNM/SOD molar ratios of 60:1) was further subjected to MS/MS analysis to identify the nitrated tyrosine (Tyr) residues. As illustrated (Suppl. Fig. 3A-C), MS/MS analysis confirmed that at least 4 Tyr residues of the mature SOD2 were nitrated (Tyr34, Tyr45, and Tyr176) and one of Tyr165, Tyr166, and Tyr1169 (not shown) from the examined peptide coverage map (Suppl. Fig. 2). These data collectively suggest that suppressed activities of some mitochondrial enzymes likely result from post-translational modifications including nitration, as shown in this study. Nitration of some proteins was shown to be conjugated with ubiquitin, leading to the degradation of the proteins with shorter half-lives [44, 45]. These events may explain the reduced levels of some proteins such as ALDH2, thiolase, and SOD2 in APAP-exposed mice.
Discussion
Drug-induced liver injury is a major problem worldwide. Many abused substances as cocaine and amphetamine can damage many organs in the presence of alcohol or other agents [6, 7, 12-15]. Despite numerous incidences of organ injury in response to APAP, alcohol, and other toxic substances in humans [47-49], the mechanisms of tissue injury are still poorly understood. In case of APAP or troglitazone-induced liver injury, several mechanisms have been proposed: metabolic activation of toxic compounds, depletion of cellular antioxidant GSH, the formation of reactive quinine metabolite protein-adducts, increased oxidative/nitrative stress with increased peroxynitrite and protein nitration, activation of the cell death pathway, alteration of calcium homeostasis, mitochondrial dysfunction with mitochondrial permeability transition (MPT) change and energy depletion, induction of hypoxia-inducible factor, activation of innate immune responses, etc. [3, 4] Although each of these mechanisms has its own merit, it is highly likely that these contributing factors work together in concert toward actual tissue damage, contributing to acute liver failure [3, 4].
The formation of quinone-metabolite-protein adducts following APAP and troglitazone rapidly decreases GSH levels, contributing to increased oxidative/nitrative stress. However, the activities of some adduct proteins did not change [22, 23] and oxidative modification of thiol molecules was shown to be minimal in response to APAP [50]. In addition, the generation of similar amounts of liver APAP-protein adducts in the WT mice, the very susceptible IL-10-null mice, and the resistant IL-10/iNOS double knockout mice implies another route for APAP-mediated hepatotoxicity [51]. Furthermore, pharmacokinetics and metabolite profiling studies of urines and sera from both WT and the hepatotoxicity-resistant Cyp2e1-null mice exposed to high doses of APAP revealed minimal differences in NAPQI, thiol conjugates, and detoxification products were minimal [52]. Additionally, pretreatment with gadolinium chloride did not eradicate nor prevent the formation of NAPQI protein adducts although APAP-induced hepatotoxicity was prevented [36]. Lastly, a non-toxic analog of APAP, AMAP produced similar amounts of protein adducts [53]. Taken together, these studies further support the notion that NAPQI production may not be the main/sole cause of APAP-induced liver necrosis and that CYP2E1 in higher APAP doses may have a limited role in the formation of NAPQI.
Therefore, we and others reported the critical role of JNK and mitochondria-dependent cell death pathways contributing to hepatotoxicity by acetaminophen and troglitazone [2, 4, 54-56]. However, recent data observed with CypD-deficient mice or mice treated with Debio 025, an inhibitor of MPT, showed that the effects of JNK seem controversial since it does not work in the absence of MPT change [57], suggesting an potentially important role of nitration in APAP-mediated hepatotoxicity. Formation of 3-NT, a footprint of peroxynitrite, is mediated by reactive nitrogen species as peroxynitrite anion (ONOO.-) and nitrogen dioxide (NO2.-), which can result from NO.- interaction with other oxidants such as superoxide radicals (O2.-), H2O2, and transition metal centers [1 and references within]. Best evidence for the involvement of nitrated proteins in organ damage are from the animal models that iNOS-null mice were resistant to hepatotoxicity by APAP [51] or ethanol [29, 31] and that increased 3-NT immunoreactive proteins and the necrotic areas were co-localized in the central vein areas [28, 58]. In fact, nitrated proteins in mitochondria were critically important in promoting APAP-induced mitochondrial dysfunction and hepatotoxicity [59, 60], although the identities of nitrated proteins were not studied.
Despite the well-established roles of nitrated proteins in APAP-induced acute hepatotoxicity, it is still poorly understood which proteins are nitrated and whether their functions are altered following exposure to APAP. A recent report showed that one protein SOD2 was nitrated and subsequently contributed to APAP-induced liver injury [34]. However, other investigators reported nitration of a few additional proteins in different pathophysiological conditions [37, 61, 62]. Furthermore, our recent data shown on 2-D gels strongly suggest that many more proteins could be nitrated after APAP exposure [1]. Therefore, we aimed to systematically identify the nitrated proteins and study their functional roles in APAP-mediated mitochondrial dysfunction and liver injury by determining the activities of some nitrated mitochondrial proteins and analyzing nitrated proteins at 2-h exposure where hepatotoxicity is still minimal. Herein, we isolated nitrated proteins by antibody-based affinity purification method and determined their protein identities. Mass-spectral analyses revealed that at least 100 mitochondrial (Table 1) and cytosolic proteins (Supplemental Table 1), respectively, were nitrated following APAP exposure. Because of limitation of mass-spectral analysis of detecting low abundant proteins, we believe that the actual number of nitrated proteins could be higher than the current results. In addition, although not performed in the current study, we expect that there are many proteins in cell membranes and nuclear fractions could be nitrated after APAP exposure, contributing to tissue injury. These results suggest that the extent of protein nitration is far greater than that previously reported [34, 37].
Although several methods to identify nitrated proteins have been described [63, 64 and references within], each method has some advantages as well as technical limitations. In this study, we simply used an antibody-based affinity purification method by using a commercially available material. Our results showed that this simple affinity-purification method is highly efficient to purify nitrated proteins, five of which were confirmed by immunoblot analysis of the immunoprecipitated target proteins [Figs. 5-7].
The effects of nitration of certain proteins on their functions are poorly understood. Our results show that nitration of some proteins such as ATP synthase might lead to a decreased activity since this enzyme has Tyr residues on its active site [30]. In addition, Tyr34 of SOD2 is critical in its catalytic activity [65]. In vitro nitration of Tyr34 of recombinant SOD2 and its inactivation (Supplemental data) suggest that SOD2 in APAP-exposed mice was likely inactivated by nitration (Fig 7). On the other hand, the activities of ALDH2, thiolase, and Gpx were suppressed in APAP-intoxicated mice although they do not have Tyr residues on their active sites. This could be explained as nitration of structurally critical Tyr residue(s) in these enzymes may cause conformational change in each protein, leading to modulation of its active site and subsequent inactivation, as elegantly suggested [66].
The inhibition of the β-oxidation of fatty acids was suggested to play a role in the development of APAP-induced hepatotoxicity [67]. Indeed, we monitored oxidative/nitrative modification and activity inhibition of some mitochondrial enzymes in the β-oxidation of fatty acids such as 3-ketoacyl-CoA thiolase (Fig. 3). The inactivation of thiolase, in the fat oxidation pathway which could provide an alternative energy during fasting and other pathological states, and ATP synthase may explain the decreased level of ATP (Fig. 6D) and the dramatic necrosis in APAP-exposed mice. Based on the inactivation of the five critical enzymes (e.g., ALDH2, ATP synthase, 3-ketoacyl-CoA thiolase, SOD2, and Gpx) and potentially many others that are listed in Table 1, we believe that nitrative modifications of these proteins at 2 h likely contribute to mitochondrial dysfunction, resulting in APAP-related liver injury at 24 h. In addition, ROS leakage is likely to be increased due to oxidative/nitrative modification of the respiratory chain complex (complex II in our study) with reduced efficiency of oxidative phosphorylation and decreased ATP synthesis (Fig. 6).
In this study, we used a toxic dose (350 mg/kg) of APAP administered into mice, which are relatively resistant to APAP-induced toxicity compared to humans, where 150 mg/kg APAP is considered toxic [68]. However, based on the temporal changes and positive correlation between the pattern of nitrated proteins and liver injury (including the enzyme activities, ALT values, histology, and 3-NT levels) and on the beneficial effects of NAC, which replenishes GSH leading to prevention of the nitration of many proteins and restoration of their activities, it is likely that increased production of peroxynitrite and consequent protein nitration play a critical role in causing APAP-mediated hepatic toxicity. Our conclusion is in agreement with previous reports about the critical role of peroxynitrite in APAP-mediated liver damage, which can be markedly attenuated by the anti-oxidant GSH [20] or NAC [21]. Similar to the previous works [46], we also observed that some protein levels were decreased at the time of activity measurements, suggesting that decreased protein contents might account for the suppressed enzyme activities. However, the magnitudes of enzyme inhibition (e.g., ALDH2 and thiolase in Fig. 5) were far greater than those of decrement in protein contents, suggesting an alternative mechanism of inactivation such as protein nitration. The inactivation of ATP synthase and Gpx, despite similar protein levels in both APAP-exposed mice and control groups, and the in vitro experiments with recombinant mouse SOD2 with or without nitration also support that nitration plays at a least a partial role in their inactivation, as reported [60].
In summary, based on our results of increased nitrated proteins in APAP-exposed mice, we have affinity-purified nitrated proteins from cytosol and mitochondrial and verified their identities by mass-spectral analysis. To our surprise, many mitochondrial and cytosolic proteins were nitrated at 2 h after APAP exposure where serum ALT levels and histological liver damage were still very low. In contrast, severe hepatotoxicity with markedly elevated ALT levels and histological necrosis were usually observed at later time points such as 24 h. Nitration of five selected proteins at 2 h post-APAP injection was further confirmed by immunoprecipitation of each protein followed by immunoblot analysis with anti-3-NT antibody. It is also worthy to mention that all proteins we randomly selected for further confirmation were found to be prominently nitrated following APAP-exposure compared with the control group, validating our approach of affinity purification of nitrated proteins followed by protein characterization. The levels of nitrated proteins were very low in mice cotreated with APAP and NAC as similar to those in the control groups. The activities of the five nitrated proteins shown in this study were suppressed in APAP-exposed mice but restored by NAC co-treatment although other types of oxidative modifications such as nitrosation and S-nitrosylation might also contribute to inactivation of some enzymes such as ALDH2, as recently reviewed [69 and references within]. Additional in vivo experiments are warranted to confirm these results. All these results likely indicate that nitration of many mitochondrial proteins lead to their inactivation and thus robust mitochondrial dysfunction, ultimately contributing to APAP-induced acute liver injury.
Supplementary Material
Highlights.
-
►
Many mitochondrial proteins were nitrated after acetaminophen (APAP) exposure.
-
►
Nitrated proteins were purified and their identities determined by mass spectrometry.
-
►
Nitrated mitochondrial proteins exhibited decreased activities.
-
►
N-acetylcysteine (NAC) prevented APAP-induced mitochondrial protein nitration.
-
►
Protein nitration correlated with decreased activities and hepatotoxicity.
Acknowledgments
This research was supported by the Intramural Research Program of National Institute on Alcohol Abuse and Alcoholism. We are thankful to Dr. Klaus Gawrisch for supporting this study. We are grateful to ProtTech, Inc and Protea Biosciences, Inc. for determining the protein sequences by mass-spectral analyses.
Footnotes
The abbreviations used are: ALDH2, mitochondrial aldehyde dehydrogenase; ALT, alanine aminotransferase; AMAP, 3-hydroxyacetanilide; APAP, acetaminophen; CHAPS, 3-[(3-cholamidoprpyl)-1dimethylammonio]-propanesulfonic acid; CYP2E1, ethanol-inducible cytochrome P450 2E1; Gpx, glutathione peroxidase; GSH, glutathione; H&E, hematoxylin & eosin; JNK, c-Jun N-terminal protein kinase; iNOS, inducible nitric oxide synthase; MDMA, 3,4-methylenedioxymethamphetamine; MPT, mitochondrial permeability transition; NAC, N-acetylcysteine; NAPQI, N-acetyl-p-benzoquinoneimine; 3-NT, 3-nitrotyrosine; PBS, phosphate buffered saline; RNS, reactive nitrogen species; SOD-1, superoxide dismutase-1; SOD-2, superoxide dismutase-2; WT, wild-type.
AUTHOR DISCLOSURE STATEMENT
The authors declare that they do not have any competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abdelmegeed MA, Moon KH, Chen C, Gonzalez FJ, Song BJ. Role of cytochrome P450 2E1 in protein nitration and ubiquitin-mediated degradation during acetaminophen toxicity. Biochem Pharmacol. 2010;79:57–66. doi: 10.1016/j.bcp.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bae MA, Song BJ. Critical role of c-Jun N-terminal protein kinase activation in troglitazone-induced apoptosis of human HepG2 hepatoma cells. Mol Pharmacol. 2003;63:401–408. doi: 10.1124/mol.63.2.401. [DOI] [PubMed] [Google Scholar]
- 3.Chung RT, Stravitz RT, Fontana RJ, Schiodt FV, Mehal WZ, Reddy KR, Lee WM. Pathogenesis of liver injury in acute liver failure. Gastroenterology. 2012;143:e1–7. doi: 10.1053/j.gastro.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131:165–178. doi: 10.1053/j.gastro.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 5.Carmiel-Haggai M, Cederbaum AI, Nieto N. Binge ethanol exposure increases liver injury in obese rats. Gastroenterology. 2003;125:1818–1833. doi: 10.1053/j.gastro.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 6.Song BJ, Moon KH, Upreti VV, Eddington ND, Lee IJ. Mechanisms of MDMA (ecstasy)-induced oxidative stress, mitochondrial dysfunction, and organ damage. Curr Pharm Biotechnol. 2010;11:434–443. doi: 10.2174/138920110791591436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valente MJ, Carvalho F, Bastos MD, de Pinho PG, Carvalho M. Contribution of Oxidative Metabolism to Cocaine-Induced Liver and Kidney Damage. Curr Med Chem. 2012 Aug 1; doi: 10.2174/092986712803988938. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 8.Ben Ari Z, Avlas O, Pappo O, Zilbermints V, Cheporko Y, Bachmetov L, Zemel R, Shainberg A, Sharon E, Grief F, Hochhauser E. Reduced hepatic injury in Toll-like receptor 4-deficient mice following D-galactosamine/lipopolysaccharide-induced fulminant hepatic failure. Cell Physiol Biochem. 2012;29:41–50. doi: 10.1159/000337585. [DOI] [PubMed] [Google Scholar]
- 9.Harstad EB, Klaassen CD. iNOS-null mice are not resistant to cadmium chloride-induced hepatotoxicity. Toxicology. 2002;175:83–90. doi: 10.1016/s0300-483x(02)00068-9. [DOI] [PubMed] [Google Scholar]
- 10.Laskin JD, Heck DE, Gardner CR, Laskin DL. Prooxidant and antioxidant functions of nitric oxide in liver toxicity. Antioxid Redox Signal. 2001;3:261–271. doi: 10.1089/152308601300185214. [DOI] [PubMed] [Google Scholar]
- 11.Wong FW, Chan WY, Lee SS. Resistance to carbon tetrachloride-induced hepatotoxicity in mice which lack CYP2E1 expression. Toxicol Appl Pharmacol. 1998;153:109–118. doi: 10.1006/taap.1998.8547. [DOI] [PubMed] [Google Scholar]
- 12.McClain CJ, Kromhout JP, Peterson FJ, Holtzman JL. Potentiation of acetaminophen hepatotoxicity by alcohol. JAMA. 1980;244:251–253. [PubMed] [Google Scholar]
- 13.Schiodt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med. 1997;337:1112–1117. doi: 10.1056/NEJM199710163371602. [DOI] [PubMed] [Google Scholar]
- 14.Seeff LB, Cuccherini BA, Zimmerman HJ, Adler E, Benjamin SB. Acetaminophen hepatotoxicity in alcoholics. A therapeutic misadventure. Ann Intern Med. 1986;104:399–404. doi: 10.7326/0003-4819-104-3-399. [DOI] [PubMed] [Google Scholar]
- 15.Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA. 1994;272:1845–1850. doi: 10.1001/jama.1994.03520230055038. [DOI] [PubMed] [Google Scholar]
- 16.Caro AA, Cederbaum AI. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu Rev Pharmacol Toxicol. 2004;44:27–42. doi: 10.1146/annurev.pharmtox.44.101802.121704. [DOI] [PubMed] [Google Scholar]
- 17.Curhan GC, Knight EL, Rosner B, Hankinson SE, Stampfer MJ. Lifetime nonnarcotic analgesic use and decline in renal function in women. Arch Intern Med. 2004;164:1519–1524. doi: 10.1001/archinte.164.14.1519. [DOI] [PubMed] [Google Scholar]
- 18.Lorz C, Justo P, Sanz A, Subira D, Egido J, Ortiz A. Paracetamol-induced renal tubular injury: a role for ER stress. J Am Soc Nephrol. 2004;15:380–389. doi: 10.1097/01.asn.0000111289.91206.b0. [DOI] [PubMed] [Google Scholar]
- 19.Meister A, Anderson ME, Hwang O. Intracellular cysteine and glutathione delivery systems. J Am Coll Nutr. 1986;5:137–151. doi: 10.1080/07315724.1986.10720121. [DOI] [PubMed] [Google Scholar]
- 20.Saito C, Zwingmann C, Jaeschke H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology. 2010;51:246–254. doi: 10.1002/hep.23267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.James LP, McCullough SS, Lamps LW, Hinson JA. Effect of N-acetylcysteine on acetaminophen toxicity in mice: relationship to reactive nitrogen and cytokine formation. Toxicol Sci. 2003;75:458–467. doi: 10.1093/toxsci/kfg181. [DOI] [PubMed] [Google Scholar]
- 22.Hart SG, Cartun RW, Wyand DS, Khairallah EA, Cohen SD. Immunohistochemical localization of acetaminophen in target tissues of the CD-1 mouse: correspondence of covalent binding with toxicity. Fundam Appl Toxicol. 1995;24:260–274. doi: 10.1006/faat.1995.1029. [DOI] [PubMed] [Google Scholar]
- 23.Roberts DW, Bucci TJ, Benson RW, Warbritton AR, McRae TA, Pumford NR, Hinson JA. Immunohistochemical localization and quantification of the 3-(cystein-S-yl)-acetaminophen protein adduct in acetaminophen hepatotoxicity. Am J Pathol. 1991;138:359–371. [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen SD, Khairallah EA. Selective protein arylation and acetaminophen-induced hepatotoxicity. Drug Metab Rev. 1997;29:59–77. doi: 10.3109/03602539709037573. [DOI] [PubMed] [Google Scholar]
- 25.Knight TR, Kurtz A, Bajt ML, Hinson JA, Jaeschke H. Vascular and hepatocellular peroxynitrite formation during acetaminophen toxicity: role of mitochondrial oxidant stress. Toxicol Sci. 2001;62:212–220. doi: 10.1093/toxsci/62.2.212. [DOI] [PubMed] [Google Scholar]
- 26.Pumford NR, Halmes NC, Hinson JA. Covalent binding of xenobiotics to specific proteins in the liver. Drug Metab Rev. 1997;29:39–57. doi: 10.3109/03602539709037572. [DOI] [PubMed] [Google Scholar]
- 27.Qiu Y, Benet LZ, Burlingame AL. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J Biol Chem. 1998;273:17940–17953. doi: 10.1074/jbc.273.28.17940. [DOI] [PubMed] [Google Scholar]
- 28.Hinson JA, Pike SL, Pumford NR, Mayeux PR. Nitrotyrosine-protein adducts in hepatic centrilobular areas following toxic doses of acetaminophen in mice. Chem Res Toxicol. 1998;11:604–607. doi: 10.1021/tx9800349. [DOI] [PubMed] [Google Scholar]
- 29.McKim SE, Gabele E, Isayama F, Lambert JC, Tucker LM, Wheeler MD, Connor HD, Mason RP, Doll MA, Hein DW, Arteel GE. Inducible nitric oxide synthase is required in alcohol-induced liver injury: studies with knockout mice. Gastroenterology. 2003;125:1834–1844. doi: 10.1053/j.gastro.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 30.Moon KH, Hood BL, Kim BJ, Hardwick JP, Conrads TP, Veenstra TD, Song BJ. Inactivation of oxidized and S-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology. 2006;44:1218–1230. doi: 10.1002/hep.21372. [DOI] [PubMed] [Google Scholar]
- 31.Venkatraman A, Shiva S, Davis AJ, Bailey SM, Brookes PS, Darley-Usmar VM. Chronic alcohol consumption increases the sensitivity of rat liver mitochondrial respiration to inhibition by nitric oxide. Hepatology. 2003;38:141–147. doi: 10.1053/jhep.2003.50293. [DOI] [PubMed] [Google Scholar]
- 32.Moon KH, Hood BL, Mukhopadhyay P, Rajesh M, Abdelmegeed MA, Kwon YI, Conrads TP, Veenstra TD, Song BJ, Pacher P. Oxidative inactivation of key mitochondrial proteins leads to dysfunction and injury in hepatic ischemia reperfusion. Gastroenterology. 2008;135:1344–1357. doi: 10.1053/j.gastro.2008.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agarwal R, Hennings L, Rafferty TM, Letzig LG, McCullough S, James LP, MacMillan-Crow LA, Hinson JA. Acetaminophen-induced hepatotoxicity and protein nitration in neuronal nitric-oxide synthase knockout mice. J Pharmacol Exp Ther. 2012;340:134–142. doi: 10.1124/jpet.111.184192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agarwal R, MacMillan-Crow LA, Rafferty TM, Saba H, Roberts DW, Fifer EK, James LP, Hinson JA. Acetaminophen-induced hepatotoxicity in mice occurs with inhibition of activity and nitration of mitochondrial manganese superoxide dismutase. J Pharmacol Exp Ther. 2011;337:110–116. doi: 10.1124/jpet.110.176321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinson JA, Michael SL, Ault SG, Pumford NR. Western blot analysis for nitrotyrosine protein adducts in livers of saline-treated and acetaminophen-treated mice. Toxicol Sci. 2000;53:467–473. doi: 10.1093/toxsci/53.2.467. [DOI] [PubMed] [Google Scholar]
- 36.Michael SL, Pumford NR, Mayeux PR, Niesman MR, Hinson JA. Pretreatment of mice with macrophage inactivators decreases acetaminophen hepatotoxicity and the formation of reactive oxygen and nitrogen species. Hepatology. 1999;30:186–195. doi: 10.1002/hep.510300104. [DOI] [PubMed] [Google Scholar]
- 37.Zhu JH, Zhang X, Roneker CA, McClung JP, Zhang S, Thannhauser TW, Ripoll DR, Sun Q, Lei XG. Role of copper,zinc-superoxide dismutase in catalyzing nitrotyrosine formation in murine liver. Free Radic Biol Med. 2008;45:611–618. doi: 10.1016/j.freeradbiomed.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoo SH, Park O, Henderson LE, Abdelmegeed MA, Moon KH, Song BJ. Lack of PPARalpha exacerbates lipopolysaccharide-induced liver toxicity through STAT1 inflammatory signaling and increased oxidative/nitrosative stress. Toxicol Lett. 2011;202:23–29. doi: 10.1016/j.toxlet.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moon KH, Upreti VV, Yu LR, Lee IJ, Ye X, Eddington ND, Veenstra TD, Song BJ. Mechanism of 3,4-methylenedioxymethamphetamine (MDMA, ecstasy)-mediated mitochondrial dysfunction in rat liver. Proteomics. 2008;8:3906–3918. doi: 10.1002/pmic.200800215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abdelmegeed MA, Banerjee A, Yoo SH, Jang S, Gonzalez FJ, Song BJ. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. J Hepatol. 2012;57:860–866. doi: 10.1016/j.jhep.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abdelmegeed MA, Moon KH, Hardwick JP, Gonzalez FJ, Song BJ. Role of peroxisome proliferator-activated receptor-alpha in fasting-mediated oxidative stress. Free Radic Biol Med. 2009;47:767–778. doi: 10.1016/j.freeradbiomed.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abdelmegeed MA, Yoo SH, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song BJ. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J Nutr. 2011;141:603–610. doi: 10.3945/jn.110.135210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song BJ, Veech RL, Park SS, Gelboin HV, Gonzalez FJ. Induction of rat hepatic N-nitrosodimethylamine demethylase by acetone is due to protein stabilization. J Biol Chem. 1989;264:3568–3572. [PubMed] [Google Scholar]
- 44.Gow AJ, Duran D, Malcolm S, Ischiropoulos H. Effects of peroxynitrite-induced protein modifications on tyrosine phosphorylation and degradation. FEBS Lett. 1996;385:63–66. doi: 10.1016/0014-5793(96)00347-x. [DOI] [PubMed] [Google Scholar]
- 45.Souza JM, Choi I, Chen Q, Weisse M, Daikhin E, Yudkoff M, Obin M, Ara J, Horwitz J, Ischiropoulos H. Proteolytic degradation of tyrosine nitrated proteins. Arch Biochem Biophys. 2000;380:360–366. doi: 10.1006/abbi.2000.1940. [DOI] [PubMed] [Google Scholar]
- 46.Ruepp SU, Tonge RP, Shaw J, Wallis N, Pognan F. Genomics and proteomics analysis of acetaminophen toxicity in mouse liver. Toxicol Sci. 2002;65:135–150. doi: 10.1093/toxsci/65.1.135. [DOI] [PubMed] [Google Scholar]
- 47.Bjornsson E, Olsson R. Suspected drug-induced liver fatalities reported to the WHO database. Dig Liver Dis. 2006;38:33–38. doi: 10.1016/j.dld.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 48.Cholongitas E, Theocharidou E, Vasianopoulou P, Betrosian A, Shaw S, Patch D, O’Beirne J, Agarwal B, Burroughs AK. Comparison of the sequential organ failure assessment score with the King’s College Hospital criteria and the model for end-stage liver disease score for the prognosis of acetaminophen-induced acute liver failure. Liver Transpl. 2012;18:405–412. doi: 10.1002/lt.23370. [DOI] [PubMed] [Google Scholar]
- 49.Testino G, Borro P, Ancarani O, Sumberaz A. Human carcinogenesis and alcohol in hepato-gastroenterology. Eur Rev Med Pharmacol Sci. 2012;16:512–518. [PubMed] [Google Scholar]
- 50.Andringa KK, Bajt ML, Jaeschke H, Bailey SM. Mitochondrial protein thiol modifications in acetaminophen hepatotoxicity: effect on HMG-CoA synthase. Toxicol Lett. 2008;177:188–197. doi: 10.1016/j.toxlet.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bourdi M, Masubuchi Y, Reilly TP, Amouzadeh HR, Martin JL, George JW, Shah AG, Pohl LR. Protection against acetaminophen-induced liver injury and lethality by interleukin 10: role of inducible nitric oxide synthase. Hepatology. 2002;35:289–298. doi: 10.1053/jhep.2002.30956. [DOI] [PubMed] [Google Scholar]
- 52.Chen C, Krausz KW, Idle JR, Gonzalez FJ. Identification of novel toxicity-associated metabolites by metabolomics and mass isotopomer analysis of acetaminophen metabolism in wild-type and Cyp2e1-null mice. J Biol Chem. 2008;283:4543–4559. doi: 10.1074/jbc.M706299200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matthews AM, Hinson JA, Roberts DW, Pumford NR. Comparison of covalent binding of acetaminophen and the regioisomer 3’-hydroxyacetanilide to mouse liver protein. Toxicol Lett. 1997;90:77–82. doi: 10.1016/s0378-4274(96)03831-3. [DOI] [PubMed] [Google Scholar]
- 54.Bae MA, Pie JE, Song BJ. Acetaminophen induces apoptosis of C6 glioma cells by activating the c-Jun NH(2)-terminal protein kinase-related cell death pathway. Mol Pharmacol. 2001;60:847–856. [PubMed] [Google Scholar]
- 55.Henderson NC, Pollock KJ, Frew J, Mackinnon AC, Flavell RA, Davis RJ, Sethi T, Simpson KJ. Critical role of c-jun (NH2) terminal kinase in paracetamol-induced acute liver failure. Gut. 2007;56:982–990. doi: 10.1136/gut.2006.104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim BJ, Ryu SW, Song BJ. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem. 2006;281:21256–21265. doi: 10.1074/jbc.M510644200. [DOI] [PubMed] [Google Scholar]
- 57.LoGuidice A, Boelsterli UA. Acetaminophen overdose-induced liver injury in mice is mediated by peroxynitrite independently of the cyclophilin D-regulated permeability transition. Hepatology. 2011;54:969–978. doi: 10.1002/hep.24464. [DOI] [PubMed] [Google Scholar]
- 58.Knight TR, Ho YS, Farhood A, Jaeschke H. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. J Pharmacol Exp Ther. 2002;303:468–475. doi: 10.1124/jpet.102.038968. [DOI] [PubMed] [Google Scholar]
- 59.Burke AS, MacMillan-Crow LA, Hinson JA. Reactive nitrogen species in acetaminophen-induced mitochondrial damage and toxicity in mouse hepatocytes. Chem Res Toxicol. 2010;23:1286–1292. doi: 10.1021/tx1001755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicol Sci. 2002;65:166–176. doi: 10.1093/toxsci/65.2.166. [DOI] [PubMed] [Google Scholar]
- 61.Turko IV, Murad F. Quantitative protein profiling in heart mitochondria from diabetic rats. J Biol Chem. 2003;278:35844–35849. doi: 10.1074/jbc.M303139200. [DOI] [PubMed] [Google Scholar]
- 62.Vattemi G, Mechref Y, Marini M, Tonin P, Minuz P, Grigoli L, Guglielmi V, Klouckova I, Chiamulera C, Meneguzzi A, Di Chio M, Tedesco V, Lovato L, Degan M, Arcaro G, Lechi A, Novotny MV, Tomelleri G. Increased protein nitration in mitochondrial diseases: evidence for vessel wall involvement. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.002964. M110 002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amoresano A, Chiappetta G, Pucci P, Marino G. A rapid and selective mass spectrometric method for the identification of nitrated proteins. Methods Mol Biol. 2008;477:15–29. doi: 10.1007/978-1-60327-517-0_2. [DOI] [PubMed] [Google Scholar]
- 64.Nardo G, Pozzi S, Mantovani S, Garbelli S, Marinou K, Basso M, Mora G, Bendotti C, Bonetto V. Nitroproteomics of peripheral blood mononuclear cells from patients and a rat model of ALS. Antioxid Redox Signal. 2009;11:1559–1567. doi: 10.1089/ars.2009.2548. [DOI] [PubMed] [Google Scholar]
- 65.Perry JJ, Hearn AS, Cabelli DE, Nick HS, Tainer JA, Silverman DN. Contribution of human manganese superoxide dismutase tyrosine 34 to structure and catalysis. Biochemistry. 2009;48:3417–3424. doi: 10.1021/bi8023288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Savvides SN, Scheiwein M, Bohme CC, Arteel GE, Karplus PA, Becker K, Schirmer RH. Crystal structure of the antioxidant enzyme glutathione reductase inactivated by peroxynitrite. J Biol Chem. 2002;277:2779–2784. doi: 10.1074/jbc.M108190200. [DOI] [PubMed] [Google Scholar]
- 67.Chen C, Krausz KW, Shah YM, Idle JR, Gonzalez FJ. Serum metabolomics reveals irreversible inhibition of fatty acid beta-oxidation through the suppression of PPARalpha activation as a contributing mechanism of acetaminophen-induced hepatotoxicity. Chem Res Toxicol. 2009;22:699–707. doi: 10.1021/tx800464q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rowden AK, Norvell J, Eldridge DL, Kirk MA. Updates on acetaminophen toxicity. Med Clin North Am. 2005;89:1145–1159. doi: 10.1016/j.mcna.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 69.Song BJ, Abdelmegeed MA, Yoo SH, Kim BJ, Jo SA, Jo I, Moon KH. Post-translational modifications of mitochondrial aldehyde dehydrogenase and biomedical implications. J Proteomics. 2011;74:2691–2702. doi: 10.1016/j.jprot.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.