

INTRODUCTION
Complex inflammatory processes underlie the progression of atherosclerosis towards its complications 1 such as coronary plaque thrombosis 2 and abdominal aortic aneurysms (AAA) 3. Several antigens become immune targets in atherosclerotic patients 4, 5 possibly because the regulation of immune responses is defective 6, 7. The trans-homophilic 8 inhibitory immunoreceptor CD31 (PECAM-1) 9 is expressed exclusively and constitutively by the cells of the blood-vessel interface and may therefore play a major role in vascular homeostasis 10. Notably, experimental studies have shown that CD31 signaling is necessary to prevent blood leukocyte cell-cell adhesion 11, chronic inflammatory diseases 12, and platelet thrombosis 13.
We previously observed that a reinforcement of the physiologic CD31 T-cell regulatory pathway, operating before the development of the disease, prevents plaque development in atherosclerosis-prone mice 14. However, this approach requires the presence of the trans-homophilic CD31 extracellular domains 14 which are typically lost on peripheral T-cells of mice 15 and patients 16 that have already developed atherosclerotic complications. Recently, a new therapeutic option has arisen from our latest data showing that a truncated extracellular CD31 fragment is indeed expressed by T-cells that apparently lack CD31 17 and that a CD31-derived peptide is able to engage this fragment. In particular this peptide showed an immunosuppressive effect in vivo through restoration of the CD31 inhibitory pathway 17. The aim of this study was therefore to evaluate whether restoring the CD31-mediated regulatory pathway with this peptide could harness the inflammatory responses underlying atherosclerosis progression and aneurysmal complication in an experimental model. We chose to test this hypothesis in aged apolipoprotein E knockout (ApoE KO) mice submitted to chronic infusion of angiotensin II because, as in patients 18, angiotensin II promotes atherosclerotic progression and AAA formation via its pro-inflammatory effect in this model 19, 20.
METHODS
Peptides
The murine synthetic CD31 peptide (aa 551-574, MW 2606.0) (purified or 5,6-FAM-conjugated, >95% pure) was synthesized by Genosphere (France) or Mimotopes (Australia) and dissolved at 1 mg/ml in sterile PBS (DMSO 0.5%). Endotoxin levels were consistently < 0.01 ng/μg of peptide as determined by the LAL test.
Mice
Male, 28-week old ApoE KO mice (B6.129P2-Apoetm1Unc/Crl, Charles River France) were maintained on a regular chow diet under standard conditions. The experiments were repeated four times and included 2 groups (n =8–10 mice/group) assigned to the administration of 50 μl of either the peptide solution (“peptide” group) or of vehicle alone (“control” group). The dose of the murine CD31 peptide (1.5 mg/Kg/d) was chosen on the basis of our previous in vivo studies 17. The treatment was administered subcutaneously, for 28 days during which Angiotensin II (Sigma, #A9525) was infused (1 mg/kg/d) using osmotic mini-pumps (Alzet, #2004) as previously described 21. At the end of the study, mice were euthanized by exsanguination under anesthesia (i.p. injection of Ketamine-HCl 100mg/Kg and Xylazine 20 mg/kg). Blood was withdrawn from the right heart ventricle and collected in heparinized and EDTA tubes for blood cell and plasma analysis. The heart and the aorta were dissected for AAA assessment, measurement of plaque size and phenotypic analysis. Four additional mice of the “control” group received a single injection of 1.5 mg/Kg fluorescent (5,6-FAM-conjugated) peptide, 30 minutes before euthanasia (day 28). All the investigations conformed to the Directive 2010/63/EU of the European Parliament and formal approval was granted by the Local Animal Ethics Committee (Comité d’éthique Bichat – Debré).
AAA and atherosclerotic lesions
The presence of an AAA was blindly assessed by two investigators (A-T. G. and A. N.). Plaque size was measured on oil-red-O stained frozen cross-sections of the aortic root, as previously described 15. Morphometric analysis was performed on Masson’s trichrome stained slides. Adventitial cell infiltrate was calculated as the fraction of the total surface area of the tissue outside the external elastic lamina occupied by nuclei (black); plaque extracellular matrix density was calculated as the ratio between the green-stained surface area and the surface of the plaque. All computer-assisted image analyses were performed using the Leica Qwin® software.
Blood cells and plasma analysis
Blood was centrifuged at 900 g for 30 minutes to separate plasma. The Th1/Th2/Th17 CBA kit (BD Biosciences) was used to measure cytokines in the plasma samples. Cholesterol was measured using Infinity® Cholesterol-Liquid reagents and an Olympus AU-400 multiparametric analyzer. Leukocyte pellets were stained with CD3-Alexa Fluor®700, CD4-PerCP, CD8-Pacific Blue, CD19-APC, CD69-PE-Cy7, CD115-PE, Ly-6G-APC-Cy7, CD31-FITC, (all from BD Biosciences) or with anti-mouse CD4-PerCP, CD25-APC (clone PC61, BD) and intracellular FoxP3-PE (clone PE-FJK-16s, eBiosciences). Analysis was performed using a LSRII® flow cytometer and BD FACSDiva® Software 6.0.
T-lymphocyte function
TCR stimulation
CD4+ splenocytes (CD4 T Lymphocyte Enrichment Set, BD Biosciences) were plated at 0.2×106 cells/well in U bottom 96-well plates (Costar®) pre-coated with anti-mouse CD3ε antibody (BD Biosciences) and cultured for 3 days in the presence of 1 μg/ml anti-mouse CD28 antibody (BD Biosciences) and different concentrations of the CD31 peptide. Cytokines were measured in the supernatants using the Th1/Th2/Th17 CBA kit (BD, Biosciences) and T-cell proliferation was assessed using the CellTrace™ CFSE Cell Proliferation Kit (Invitrogen). Phospho-SHP2 was evaluated by flow cytometry on TCR-stimulated splenocytes as detailed in the supplementary method section.
Antigen-specific stimulation
Four individual ApoE KO male mice aged 24 weeks were immunized (4 footpads) with 100 μg autologous oxidized (ox) LDL (prepared as previously described 22) in complete Freund’s adjuvant. Ten days later, the animals were euthanized and the popliteal and axillary lymph nodes draining the immunization sites were collected. Single cell suspensions obtained from pooled lymph nodes from each mouse were stained with CFSE (5 μM) and plated in complete medium in 96-well plates (round bottom, 2×105 cells/well) in the presence of oxLDL (1 μg/ml) and in the presence of increasing doses of CD31 peptide (0, 12.5, 25, 50 and 100 μg/ml). Culture supernatants were harvested four days later for soluble CD31 analysis and cells were submitted to cytometry to analyze the CFSE-based proliferation index within the CD4+ lymphocyte gate. The mean fluorescent intensity of surface CD31 was analyzed in the same wells and soluble CD31 concentration was assessed in the supernatants (supplementary methods).
Macrophage function
Bone marrow-derived macrophages were differentiated and IFNγ-primed as previously described 23. Cells were then cultured in complete medium and stimulated with 100 ng/ml angiotensin II in the presence of increasing doses (0, 25, 50μg/ml) of CD31 peptide. Macrophage intracellular MMP-2/9 activity was quantified by flow cytometric detection of a fluorescent enzyme substrate, according to the manufacturer’s instructions (Invitrogen). Briefly, 10 μg/ml fluorescein-conjugated DQ™ type IV collagen, analog of the MMP-2/9 natural substrate, were added to stimulated macrophages for six hours and the fluorescence derived from the enzyme-driven hydrolysis of the fluorescein-conjugated DQ™ was quantified by flow cytometry. IL-1α, IL-1β, IL-12/IL-23p40, RANTES, IL-6, MCP-1 and MIP-1α and MIP-1β (CBA flex sets, BD Biosciences) were measured in the culture supernatants from each condition. Individual mouse data (n =4), expressed as Median Fluorescence Intensity (MFI) have been used for statistical analysis.
Statistical Analysis
Data are expressed as means ± SD unless otherwise indicated in the text. Differences between groups were analyzed by Student t-test or chi-squared test, as appropriate. Differences were considered statistically significant when the p value was ≤0.05.
RESULTS
Loss of T-cell extracellular CD31 and development of AAA
Plaque-infiltrating leukocyte analysis showed that CD4+ cells were all positive for intracellular CD31 but not all of them co-expressed extracellular CD31 (supplementary Figure 1a). The lack of extracellular CD31 was also specifically observed on peripheral blood CD4+ T lymphocytes and associated with the development of an AAA (supplementary Figure 1b and 1c).
CD31 peptide treatment reduces AAA formation and plaque growth
Since AAA development was associated with the loss of extracellular CD31 on CD4+ T-cells we reasoned that this receptor could be shed from mouse leucocytes as we have previously described for human cells 17. Accordingly, we evaluated the therapeutic potential of a CD31-derived peptide, which is able to restore the lost inhibitory function in vitro and in vivo 17 in this model. CD31 peptide treatment significantly reduced the incidence of AAA formation (Figure 1a) and the extent of atherosclerotic lesions (Figure 1b and 1c). Aortic root adventitial cell infiltrate was associated with the presence of an AAA (Figure 1d and 1e) and was also reduced in peptide-treated mice. The peptide stabilized the phenotype of the plaques as determined by higher extracellular matrix density (44.52 ± 5.5 vs 31.61 ±4.9 a.u, p=0.05), lower expression of VCAM-1 and higher αSMA content (supplementary Figure 2). Fibrillar collagen was more abundant in the aortic root plaques and significantly higher at the site of the abdominal aorta that is prone to dissection in this model, suggesting that the peptide treatment confers a higher resistance against rupture to the arterial wall (supplementary Figure 3). Furthermore, the presence of T-cells (CD4+), macrophages (Mac3+) and MMP9 within the plaques was also blunted by the treatment (supplementary Figure 2). CD31-treatement did not affect body weight (34.8 ± 0.8 vs 36.4 ± 0.9 g), lipoprotein profile (supplementary Figure 4) or plasma levels of total cholesterol (11.9 ± 0.75 vs 10.9 ± 0.6 mmol/l), HDL (1.2 ± 0.1 vs 1.1± 0.1 mmol/l), and triglycerides (1.1 ± 0.1 vs 1.2 ± 0.1 mmol/l). No differences could be found between the two groups in terms of total IgG and IgM and of specific anti-oxLDL IgG and IgM serum levels (supplementary data, Figure 5) or circulating blood cell count (supplementary Table 1).
Figure 1. CD31 peptide-treatment prevents plaque growth and aneurysm formation.
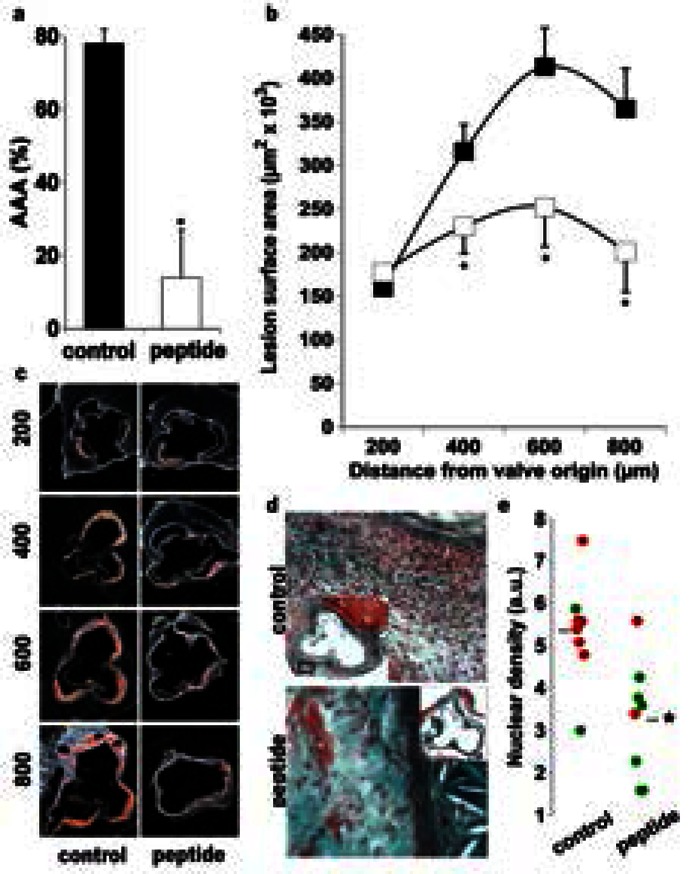
a. The incidence of AAA (% of mice/group) was significantly reduced by CD31 treatment (14.5 ± 3 vs 75 ± 7, *p<0.001). b. Quantification of atherosclerotic lesion surface area in serial cross-sections of the aortic root (200, 400, 600, 800 μm from the appearance of the first cusp) in control (■, n=10) and CD31 peptide-treated (□, n=8) mice. *p<0.05 vs control. c. Representative micrographs of oil-red-O stained sections. d. Representative images showing Masson’s trichrome staining of aortic root cross-sections. Control mice showed increased adventitial cell infiltration (nuclear staining) as compared to peptide-treated mice. e. Quantitative analysis of the adventitial cell infiltration. Peptide-treated mice (n=8) showed significantly reduced (*p<0.01) cell infiltration as compared to control mice (n=8). Interestingly, a higher adventitial nuclear density was correlated with the presence of abdominal aortic aneurysms (AAA, red dots; no AAA, green dots).
CD31 peptide modulates the peripheral T-cell compartment in vivo and directly inhibits T-cell responses in vitro
Peptide-treated mice showed increased CD4+CD25+FoxP3+ (Tregs) and reduced CD69+ (recently activated) T-cell % in the peripheral blood (Figure 2a). The treatment decreased plasma levels of IL-2, IFNγ, IL-4 and IL-6 (Figure 2b) but not those of TNFα, IL-10 or IL-17A (data not shown).
Figure 2. Immunoregulatory effects of the CD31 peptide in vivo.
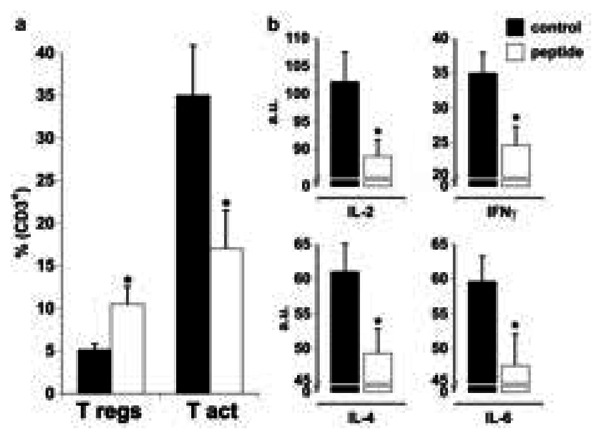
a. The percentage of Tregs (CD25+ FoxP3+) was increased while the relative fraction of activated T-cells (CD69+) CD4+ T-cells among total blood CD3+ T-cells was diminished by the peptide treatment (*p<0.05). b. Plasma IL-2, IFNγ, IL-4 and IL-6 (a.u.) were significantly decreased by the peptide treatment in vivo (*p<0.05).
We therefore assessed if the peptide exerts direct T-cell suppressive effects in vitro. Our data show that, as for human cells 17, the peptide reduces mouse T-cell proliferation in response to TCR engagement (Figure 3a). In addition, it inhibited the production of IL-2, IFNγ, IL-4, IL-6, TNF and IL-10 (Figure 3b). To assess whether the peptide triggered the physiological T-cell CD31 inhibitory signaling, we evaluated the levels of the SHP2 phosphorylation [tyrosine (Y) 542] in TCR-stimulated splenocytes. Similar to the specific control (antibody-mediated cross-linking of surface CD31) a significant increase in intracellular SHP2 pY542 was induced by the peptide, in a dose-dependent manner (Figure 3c).
Figure 3. The CD31 peptide inhibits TCR-stimulated T cell responses in vitro and drives SHP2 phosphorylation.
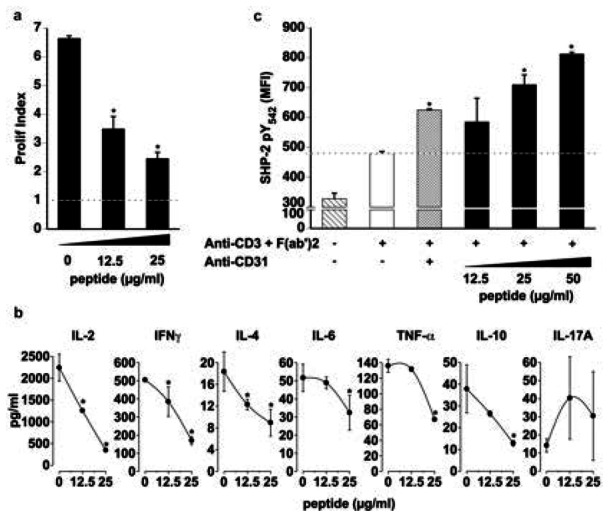
a. Proliferation in response to TCR engagement is inhibited as a function of dose by the CD31 peptide (*p<0.05 vs dose “0”). Proliferation was analyzed by the Modfit® software and expressed as “Prolif Index”. The dotted line represents baseline proliferation (Prolif Index for unstimulated CD4+ cells). b. Cytokine concentrations (pg/ml) in the supernatants of stimulated CD4+ T-cells were significantly reduced by the peptide. (*p<0.05 vs dose “0”). c. Flow cytometric quantification of SHP2 pY542 in stimulated splenocytes. Dotted line (baseline) represents SHP2 phosphorylation induced by TCR engagement alone. Crosslinking of the TCR with surface CD31 molecules induced SHP2 phosphorylation (*p<0.01 vs baseline); Increasing doses of CD31 peptide induced further increments of SHP2 phosphorylation (*p<0.01 vs baseline). Data are expressed as Median Fluorescent Intensity (MFI).
The peptide was also able to suppress the CD4+-specific immune response directed against ox-LDL, a major atherosclerosis-related antigen (Figure 4a). Interestingly, the expression of CD31 extracellular domains at the cell surface was reduced after antigenic T-cell stimulation, concomitantly with an increase in soluble CD31 concentration in the supernatants, further adding to the evidence that CD31 shedding is driven by (antigen-specific) TCR-mediated T-cell activation (Figure 4b and 4c).
Figure 4. oxLDL stimulation drives the cleavage and shedding of CD31 and the CD31 peptide inhibits oxLDL-specific T-cell responses.

a The peptide inhibits the proliferation of oxLDL-specific CD4+ splenocytes derived from oxLDL-immunized apoE KO mice (*p<0.05 vs dose “0”). Proliferation was analyzed at 4 days after a challenge with oxLDL in vitro by the Modfit® software and expressed as “Prolif Index”. b. Representative density plot and c. quantitative analysis of CD31 by flow cytometry on a CD4+ splenocyte surface (MFI) and by ELISA in matched culture supernatants (ng/ml). Upon oxLDL stimulation in vitro, the expression of CD31 decreases at the surface of CD4+ cells while its detection in the soluble form increases in parallel (*p<0.01).
CD31 peptide prevents angiotensin II-induced macrophage activity in vitro
While T cells are certainly involved in the acceleration of plaque formation in the angiotensin II infusion model, macrophages may play a more important role in AAA formation 24, 25. We therefore assessed whether the peptide is able to bind to macrophages in vivo and to exert a direct effect on angiotensin II-induced macrophage activation, in vitro. Fluorescent peptide tracking showed that plaque-infiltrating and peri-aneurysmal macrophages could indeed be targeted by the peptide, in vivo (Figure 5). In vitro, the CD31 peptide reduced intracellular MMP-2/9 activity (Figure 6a and 6b) and release of IL-6, MCP-1, MIP-1α and MIP-1β in the culture medium (Figure 6c). The concentrations of IL-1α, IL-1β, IL-12 and RANTES were not affected by the peptide (data not shown).
Figure 5. The CD31 peptide binds to plaque-infiltrating and peri-aneurysmal adventitial macrophages.

Representative in vivo tracking immunofluorescent cross-section micrographs. The fluorescent peptide (green) bound to cells in clusters (blue = nuclei) including CD68+ cells (red, macrophages) within and surrounding abdominal aortic aneurysm (left panel) as well as within aortic root plaques (right panel). Numbered white boxes in the top panels indicate the position of the magnified insets. Scale bar=100 μm. Hem=intramural hematoma; Adv=adventitia, EL=elastin, L=lumen.
Figure 6. The CD31 peptide inhibits macrophage response to angiotensin II.

a. Representative phase contrast and fluorescent images of angiotensin II-stimulated macrophages in the absence (left panel) or presence (50 μg/ml) of the CD31 peptide (right panel). Collagenolytic activity (MMP, green) was detected by the presence of the fluorogenic degradation product of DQ collagen. Blue = nuclei. Scale bar = 100 μm. b. Flow cytometric quantification of the collagenolytic activity shown in a. At 50 μg/ml, the peptide significantly inhibits collagen degradation (*p<0.05 vs dose “0”) MFI = median fluorescent intensity of viable macrophages in the fluorescein channel. c. The concentration of IL-6, MCP-1, MIP-1α and MIP-1β in the culture supernatants was significantly reduced by the peptide. (*p<0.05 vs dose “0”).
DISCUSSION
The present study provides further evidence for a protective role of T-cell CD31 against atherosclerotic complications, such as AAA. Indeed, as in patients 16, the presence of an AAA was linked to the loss of CD31 on both plaque-infiltrating and peripheral T-cells in angiotensin II-infused apoE KO mice. We also found that the incidence of AAA correlated with adventitial cell infiltration at the level of the aortic root, distant from the abdominal aorta. While such a perivascular leukocyte infiltration had previously been described at the site of aneurysm formation 24, our findings suggest that the adventitial inflammatory process induced by angiotensin II extends to the whole arterial tree and contributes to both the acceleration of plaque-growth (aortic root) and the occurrence of wall dissection/aneurysm formation (abdominal aorta) depending on site-specific hemodynamics and leukocyte environment. In support of this hypothesis, the CD31-peptide treatment effectively reduced perivascular inflammation and equally protected against aneurysmal complications and atherosclerosis progression in this study.
Although the CD31 peptide can potentially act on CD31-positive cells other than leukocytes (platelets and endothelial cells), in this study we focused our attention on its putative effect on the cells of the immune system. Indeed T-cell activation is a key cellular process linking the hypertensive and pro-inflammatory effects of angiotensin II 19, 26, 27. Our data suggest that the inhibition of T-cell activation achieved in vivo 17, 28 by the CD31 peptide prevents the angiotensin II-driven inflammatory vascular damage. However, the T-cell suppression cannot explain, alone, the anti-inflammatory and protective effect of the peptide in this model because angiotensin II receptors are expressed both by T lymphocytes and macrophages 29. The latter are particularly relevant in angiotensin II-driven arterial wall dissection and AAA formation 24, 25. Interestingly, the peptide was found to bind to both plaque and adventitial macrophages, which were both reduced by the treatment. In addition, the peptide inhibited macrophage collagenolytic activity and the release of IL-6 and MCP-1 by angiotensin II-stimulated inflammatory macrophages, possibly explaining the resistance of the aortic wall to dissection in peptide-treated mice 30.
In agreement with our findings in human lymphocytes 17, our in vitro data suggest that restoring CD31-driven intracellular SHP-2 phosphorylation in activated leukocytes constitutes a novel immunomodulatory strategy. Interestingly, it has recently been shown that the CD31-SHP2 pathway simultaneously inhibits tyrosin kinase-dependent activation while promoting T-cell survival via the Erk/MAP kinase pathway 31. The latter is also involved in extrathymic Treg induction 32 and function 33. This may account for the enrichment of the Treg peripheral pool that we have observed in treated mice and represent an additional immunoregulatory mechanism exerted by the peptide.
As compared to other potential immunosuppressive atheroprotective agents, such mycophenolate mofetil 34, anti-CD3 35, 36 and anti-CD20 37 antibodies, the peptide that we used in this study induces a “targeted” immunosuppressor effect because it does not cause lymphocyte depletion and reduces the release of selected disease-related cytokines. Moreover, since disease severity is linked to the extent of CD31 shedding from leukocytes 16, which can be easily measured in plasma samples 17, the dosing of the peptide could be finely adjusted to optimize the benefit/risk ratio of its use as a drug.
Supplementary Material
Acknowledgments
FUNDING
This work was supported in part by the “Fondation de France” (Engt 2008-002724), the “Agence Nationale de la Recherche” (project “RELATE” and project “BROSCI”), the “Fighting Aneurysmal Disease” (FAD, #217) European integrated project (Health-F2-2008-200647). G.F. was the recipient of grants from the “Groupe de Reflexion sur la Recherche Cardio-Vasculaire”, “Féderation Française de Cardiologie” and “Fondation pour la Recherche Médicale”. M.C. is the recipient of a doctoral grant provided by the “CODDIM” from the Region île de France.
We thank Devy Diallo (Inserm U698) for his help with the lipoprotein profile analysis and Mary Osborne-Pellegrin (Inserm U698) for her help in editing the manuscript.
Footnotes
CONFLICT OF INTEREST
None declared.
References
- 1.Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis: from pathophysiology to practice. Journal of the American College of Cardiology. 2009;54:2129–2138. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 3.Hellenthal FA, Buurman WA, Wodzig WK, Schurink GW. Biomarkers of abdominal aortic aneurysm progression. Part 2: inflammation. Nature reviews. 2009;6:543–552. doi: 10.1038/nrcardio.2009.102. [DOI] [PubMed] [Google Scholar]
- 4.Caligiuri G, Paulsson G, Nicoletti A, Maseri A, Hansson GK. Evidence for antigen-driven T-cell response in unstable angina. Circulation. 2000;102:1114–1119. doi: 10.1161/01.cir.102.10.1114. [DOI] [PubMed] [Google Scholar]
- 5.Caligiuri G, Stahl D, Kaveri S, Irinopoulous T, Savoie F, Mandet C, et al. Autoreactive antibody repertoire is perturbed in atherosclerotic patients. Laboratory investigation; a journal of technical methods and pathology. 2003;83:939–947. doi: 10.1097/01.lab.0000077010.90550.ff. [DOI] [PubMed] [Google Scholar]
- 6.Caligiuri G, Nicoletti A. Tregs and human atherothrombotic diseases: toward a clinical application? Arteriosclerosis, thrombosis, and vascular biology. 2010;30:1679–1681. doi: 10.1161/ATVBAHA.110.209668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mallat Z, Taleb S, Ait-Oufella H, Tedgui A. The role of adaptive T cell immunity in atherosclerosis. Journal of lipid research. 2009;50 (Suppl):S364–369. doi: 10.1194/jlr.R800092-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newton JP, Buckley CD, Jones EY, Simmons DL. Residues on both faces of the first immunoglobulin fold contribute to homophilic binding sites of PECAM-1/CD31. The Journal of biological chemistry. 1997;272:20555–20563. doi: 10.1074/jbc.272.33.20555. [DOI] [PubMed] [Google Scholar]
- 9.Newman PJ. Switched at birth: a new family for PECAM-1. The Journal of clinical investigation. 1999;103:5–9. doi: 10.1172/JCI5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newman PJ, Newman DK. Signal transduction pathways mediated by PECAM-1: new roles for an old molecule in platelet and vascular cell biology. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:953–964. doi: 10.1161/01.ATV.0000071347.69358.D9. [DOI] [PubMed] [Google Scholar]
- 11.Brown S, Heinisch I, Ross E, Shaw K, Buckley CD, Savill J. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature. 2002;418:200–203. doi: 10.1038/nature00811. [DOI] [PubMed] [Google Scholar]
- 12.Wong MX, Hayball JD, Hogarth PM, Jackson DE. The inhibitory co-receptor, PECAM-1 provides a protective effect in suppression of collagen-induced arthritis. J Clin Immunol. 2005;25:19–28. doi: 10.1007/s10875-005-0354-7. [DOI] [PubMed] [Google Scholar]
- 13.Falati S, Patil S, Gross PL, Stapleton M, Merrill-Skoloff G, Barrett NE, et al. Platelet PECAM-1 inhibits thrombus formation in vivo. Blood. 2006;107:535–541. doi: 10.1182/blood-2005-04-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Groyer E, Nicoletti A, Ait-Oufella H, Khallou-Laschet J, Varthaman A, Gaston AT, et al. Atheroprotective effect of CD31 receptor globulin through enrichment of circulating regulatory T-cells. Journal of the American College of Cardiology. 2007;50:344–350. doi: 10.1016/j.jacc.2007.04.040. [DOI] [PubMed] [Google Scholar]
- 15.Caligiuri G, Groyer E, Khallou-Laschet J, Al Haj Zen A, Sainz J, Urbain D, et al. Reduced immunoregulatory CD31+ T cells in the blood of atherosclerotic mice with plaque thrombosis. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:1659–1664. doi: 10.1161/01.ATV.0000172660.24580.b4. [DOI] [PubMed] [Google Scholar]
- 16.Caligiuri G, Rossignol P, Julia P, Groyer E, Mouradian D, Urbain D, et al. Reduced immunoregulatory CD31+ T cells in patients with atherosclerotic abdominal aortic aneurysm. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:618–623. doi: 10.1161/01.ATV.0000200380.73876.d9. [DOI] [PubMed] [Google Scholar]
- 17.Fornasa G, Groyer E, Clement M, Dimitrov J, Compain C, Gaston AT, et al. TCR stimulation drives cleavage and shedding of the ITIM receptor CD31. J Immunol. 2010;184:5485–5492. doi: 10.4049/jimmunol.0902219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Montecucco F, Pende A, Mach F. The renin-angiotensin system modulates inflammatory processes in atherosclerosis: evidence from basic research and clinical studies. Mediators of inflammation. 2009;2009:752406. doi: 10.1155/2009/752406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. The Journal of experimental medicine. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Ait-Oufella H, Herbin O, Bonnin P, Ramkhelawon B, Taleb S, et al. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. The Journal of clinical investigation. 2010;120:422–432. doi: 10.1172/JCI38136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. The Journal of clinical investigation. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caligiuri G, Khallou-Laschet J, Vandaele M, Gaston AT, Delignat S, Mandet C, et al. Phosphorylcholine-targeting immunization reduces atherosclerosis. Journal of the American College of Cardiology. 2007;50:540–546. doi: 10.1016/j.jacc.2006.11.054. [DOI] [PubMed] [Google Scholar]
- 23.Khallou-Laschet J, Varthaman A, Fornasa G, Compain C, Gaston AT, Clement M, et al. Macrophage plasticity in experimental atherosclerosis. PLoS One. 2010;5:e8852. doi: 10.1371/journal.pone.0008852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saraff K, Babamusta F, Cassis LA, Daugherty A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:1621–1626. doi: 10.1161/01.ATV.0000085631.76095.64. [DOI] [PubMed] [Google Scholar]
- 25.Uchida HA, Kristo F, Rateri DL, Lu H, Charnigo R, Cassis LA, et al. Total lymphocyte deficiency attenuates AngII-induced atherosclerosis in males but not abdominal aortic aneurysms in apoE deficient mice. Atherosclerosis. 2010;211:399–403. doi: 10.1016/j.atherosclerosis.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1089–1097. doi: 10.1152/ajpregu.00373.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, et al. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ Res. 2010;107:263–270. doi: 10.1161/CIRCRESAHA.110.217299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y, Schlegel PG, Tran N, Thompson D, Zehnder JL, Chao NJ. Administration of a CD31-derived peptide delays the onset and significantly increases survival from lethal graft-versus-host disease. Blood. 1997;89:1452–1459. [PubMed] [Google Scholar]
- 29.Shimada K, Yazaki Y. Binding sites for angiotensin II in human mononuclear leucocytes. J Biochem. 1978;84:1013–1015. doi: 10.1093/oxfordjournals.jbchem.a132183. [DOI] [PubMed] [Google Scholar]
- 30.Tieu BC, Lee C, Sun H, Lejeune W, Recinos A, 3rd, Ju X, et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. The Journal of clinical investigation. 2009;119:3637–3651. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma L, Mauro C, Cornish GH, Chai JG, Coe D, Fu H, et al. Ig gene-like molecule CD31 plays a nonredundant role in the regulation of T-cell immunity and tolerance. Proc Natl Acad Sci U S A. 2010;107:19461–19466. doi: 10.1073/pnas.1011748107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishihara M, Ogura H, Ueda N, Tsuruoka M, Kitabayashi C, Tsuji F, et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int Immunol. 2007;19:695–702. doi: 10.1093/intimm/dxm045. [DOI] [PubMed] [Google Scholar]
- 33.Adler HS, Kubsch S, Graulich E, Ludwig S, Knop J, Steinbrink K. Activation of MAP kinase p38 is critical for the cell-cycle-controlled suppressor function of regulatory T cells. Blood. 2007;109:4351–4359. doi: 10.1182/blood-2006-09-047563. [DOI] [PubMed] [Google Scholar]
- 34.van Leuven SI, Kastelein JJ, Allison AC, Hayden MR, Stroes ES. Mycophenolate mofetil (MMF): firing at the atherosclerotic plaque from different angles? Cardiovasc Res. 2006;69:341–347. doi: 10.1016/j.cardiores.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 35.Sasaki N, Yamashita T, Takeda M, Shinohara M, Nakajima K, Tawa H, et al. Oral anti-CD3 antibody treatment induces regulatory T cells and inhibits the development of atherosclerosis in mice. Circulation. 2009;120:1996–2005. doi: 10.1161/CIRCULATIONAHA.109.863431. [DOI] [PubMed] [Google Scholar]
- 36.Steffens S, Burger F, Pelli G, Dean Y, Elson G, Kosco-Vilbois M, et al. Short-term treatment with anti-CD3 antibody reduces the development and progression of atherosclerosis in mice. Circulation. 2006;114:1977–1984. doi: 10.1161/CIRCULATIONAHA.106.627430. [DOI] [PubMed] [Google Scholar]
- 37.Ait-Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L, et al. B cell depletion reduces the development of atherosclerosis in mice. The Journal of experimental medicine. 2010;207:1579–1587. doi: 10.1084/jem.20100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.