

Abstract
BACKGROUND
Sonic hedgehog (Shh) signaling plays a pivotal role in stromal-epithelial interaction during normal development but its role in tumor-stromal interaction during carcinogenic progression is less well defined. Since hormone refractory prostate cancer with bone metastasis is difficult to treat, it is crucial to investigate how androgen independent (AI) human prostate cancer cells communicate with their associated stroma.
METHODS
Shh and its target transcription factor, Gli1 mRNA, were assessed by RT-PCR and/or quantitative RT-PCR in co-cultured cell recombinants comprised of AI C4-2 either with NPF (prostate fibroblasts from normal/benign prostate gland) or CPF cancer-associated stromal fibroblasts) under Shh/cyclopamine (a hedgehog signaling inhibitor) treatment. Human bone marrow stromal (HS27A) cells were used as controls. In vivo investigation was performed by checking serum PSA and immunohistochemical staining for the apoptosis-associated M30 gene in mice bearing chimeric C4-2/NPF tumors.
RESULTS
CONCLUSIONS
Based on co-culture and chimeric tumor models, active Shh-mediated signaling was demonstrated between AI prostate cancer and NPF in a paracrine- and tumor progression-dependent manner. Our study suggests that drugs like cyclopamine that interfere with Shh signaling could be beneficial in preventing AI progression in prostate cancer cells.
Keywords: Sonic hedgehog, androgen independent prostate cancer, prostate stroma, bone marrow stroma, tumor-stromal interaction
INTRODUCTION
Sonic hedgehog (Shh) signaling relates to stromal-epithelial interaction during normal development. How it works in tumor-stromal interaction during carcinogenic progression is not well known. The general schema of activation involves the binding of hedgehog (Hh) ligand to its receptor, Patched (Ptch), which has an inhibitory function on the protein Smoothened (Smo), a co-receptor for Hh signaling. This allows activation of downstream Gli transcriptional factors to activate target genes.1-3 Coordinated regulation of Gli target genes could enhance cell survival by promoting G1/S cell cycle progression and providing anti-apoptotic cues, and accelerate cancer cell growth by the induction/recruitment of “reactive” stroma, increasing the ability of prostate cancer cells to respond to Hh-signaling and undergo stem cell renewal.3
Hh signaling may contribute to the development of solid tumors in humans through gene alterations of Ptch/Smo resulting in constitutive activated Hh signaling.4 Although early reports indicated that Shh signaling is activated in advanced metastatic prostate cancers1, the activation of these signaling pathways could occur via various mechanisms including ligand-independent Hh signaling activation. Recently, Shh expression was shown to be markedly elevated in human prostate cancer cells during androgen deprivation.5 These findings could be highly significant since ligand-dependent Shh paracrine signaling between human prostate cancer cells and mouse osteoblasts occurs in co-culture.6 These results, taken together, suggest the potential value of targeting Hh signaling in human primary and bone metastatic prostate cancer.5 The purposes of the present study are to determine if Hh paracrine signaling is activated between androgen independent (AI) human prostate cancer cells and human prostate stromal fibroblasts and to identify potential Gli1 target genes in human prostate stromal fibroblasts.
Stromal fibroblasts have recently been recognized as more than just silent partners of cancer epithelial cells.7 They have growth- and differentiation-inductive potential, recruit endothelial, inflammatory, and mesenchymal stem cells, and confer survival benefits for cancer epithelium through secreted soluble growth-modulating and insoluble matrix factors. Stromal fibroblasts are also moving targets which respond to inductive cues from the cancer epithelium, transitioning to become “reactive” stroma.8, 9 The status of “reactive” stroma, including growth inductive potential and gene expression profiles, has been shown to predict recurrence in prostate cancer patients.10 Reciprocal tumor-stroma interactions were demonstrated in 3-D co-culture conditions9 and in chimeric tumors in mice.11 Remarkably, non-random co-evolution of the genetic constituents of tumor-stroma interactions can increase tumor growth in mice. Molecular signatures responsible for tumor-stroma interactions in these chimeric tumor models were found to be expressed in patient serum specimens and to have prognostic value for cancer bone metastases.8, 9
Sanchez et al. demonstrated that activated Shh-Gli signaling occurs in an autocrine/paracrine manner in prostate cancer cells.12, 13 Several Shh-Gli1 target genes are reported.14 Osteopontin supports osteoblastogenesis and cell adhesion14, and γ-catenin regulates cell growth and provides a molecular link between pathways regulating cell adherence and the balance of cell growth and cell death.14 In addition, the Ptch and Wnt genes are considered as downstream targets of Gli115, and biochemical evidence has established HNF-3 (Hepatocyte Nuclear Factor-3) as a target of Gli1 during development.16
We compared the effects of prostate stromal fibroblasts isolated from either prostate carcinoma or normal/benign prostate glandular epithelium-associated areas on the growth of human prostate carcinoma cells in culture and as coinoculates in mice, and defined the roles of Shh signaling under these growth conditions. We constructed cell recombinants consisting of androgen-independent human prostate cancer cells (C4-2) and normal human prostate stromal fibroblasts (NPF), and investigated the roles of Shh in these cell recombinants. Our work expanded on information gathered from a previous study where the roles of Hh signaling were investigated in an androgen-resistant CWR22rv model. 2
Our results suggest that within a given prostate carcinoma cell, Shh signaling activation appears to occur between AI human prostate cancer and NPF at an early stage, but not in advanced stages of disease, i.e. between the progressing AI prostate carcinoma cell and CPF. We also identified a new Shh-Gli1 target gene, osteonectin (ON), in normal human prostate stromal cells responding to prostate cancer cells and Shh protein. Activation of Shh-Gli1 signaling via ON expression in NPF is coordinated with osteomimicry by AI human prostate cancer cells, which may help explain the growth, survival, invasion and metastasis of prostate cancer to the skeleton.17
MATERIALS AND METHODS
Cells, cell cultures, reagents, and conditioned medium (CM) collection
The androgen independent (AI) human prostate cancer C4-2 cell line was established by our laboratory in 1994 and commercialized and fully characterized by ViroMed Laboratories (Minnetonka, MN). The HS27A human bone stromal cell line was obtained from ATCC (CRL 2496; Manassas, VA). These cells were cultured in T-medium (Invitrogen corporation, Carlsbad, CA) supplemented with 5% fetal bovine serum (FBS) (Sigma, St. Louis, MO) and 1% penicillin/streptomycin as described previously.18 C4-2 cells, a lineage-derived subline from LNCaP, is an androgen receptor (AR)-positive tumorigenic and bone metastatic human prostate cancer cell line. The cells were maintained at 37°C in 5% CO2. For conditioned medium (CM) collection, cells were cultured in T-medium with 5% FBS until 80% confluent. The cells were then washed twice in phosphate-buffered saline (PBS) and incubated in T-medium without serum or in serum-free and phenol red-free RPMI 1640 (Invitrogen). After 2 days of incubation, CM was collected, centrifuged, and stored at -20°C until use. Cyclopamine (Toronto Chemical Inc., Toronto, ON, Canada) was dissolved in DMSO at 10 mM of the stock solution and stored at -20 °C until use.
Prostate stromal cells
Tissues for primary prostate stromal culture were obtained from consenting patients with prostate cancer undergoing radical prostatectomy at the Emory Clinic, Emory University School of Medicine. The use of fresh prostate tissue specimens for research was approved by the IRB Committee at the Emory University School of Medicine. Prostate cancer specimens diagnosed by a pathologist as Gleason score 6 were obtained, minced with surgical scissors and subcultured as tissue fragments using previously described procedures.9 Outgrowths of stromal fibroblasts were established as cell strains through a subculture protocol using the established differential trypsinization method. Stromal fibroblasts were cultured in T-medium with 10% FBS and used within 6 passages. The origins of stromal fibroblasts were: 1) normal prostate fibroblast (NPF) from normal/benign lesions of the human prostate gland and 2) cancer-associated prostate fibroblasts (CPF) from cancerous lesions of the prostate gland.
Determining cell growth in co-cultured prostate stromal fibroblasts and C4-2-Luc cells
AR-positive and AI C4-2-Luc cells (C4-2 stably expressing luciferase gene)19 and NPF or CPF were co-cultured in 6-well plastic dishes seeded at 2 x 105 cells/well in T-medium containing 5 % FBS cultured for 24 hrs, then changed to serum free T-media containing 2% TCM (MP Biomedicals LLC, Solon, OH). TCM™ is a fortified, multipurpose serum replacement medium for long-term culturing of many types of The concentration of TCM used in our study was based on the manufacturer’s recommendations.
One μg/ml human Shh protein (or 51.0 nM of Shh, Neuromics, Edina, MN) was added in the presence or absence of 5 μM cyclopamine, a Shh-Gli signaling inhibitor, for 48 hrs. Human Shh protein was diluted with PBS to 50 μg/ml and served as stock solution. The growth of prostate cancer cells (C4-2 Luc cells) was monitored by the measurement of luciferase activity. Twenty μl of the lysate supernatant was mixed with 100 μl of Luciferin (or 2.62mM, Promega, Madison, WI) according to the manufacturer’s protocol, and light emission was measured by a luminometer (Monolight 2010, Analytical Luminescence Laboratory, Sparks, MD).19
Cell proliferation assay
Cell proliferation was determined with the CellTiler 96 Aqueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI) using the manufacturer-recommended protocol. Cells were seeded in 96-well plates at 5 × 103 cells/well in T-medium containing 5 % FBS for 24 hrs, then changed to serum free T-medium containing 2 % TCM. Cells were then recovered for another 48 hrs with or without the C4-2 CM (50% of the culture volume). MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfonyl)-2H- tetrazolium, was added to each well and the incubation was extended at 37°C for another 1 hr, followed by recording the absorbance of the converted dye at a wavelength of 570 nm. All experiments were carried out in triplicate and repeated two times. Data were expressed as mean ± S.D. and evaluated for statistical significance.
Reverse transcription-PCR analysis and quantitative real-time RT-PCR (qRT-PCR)
Total RNA was isolated from confluent monolayers of cells using an RNA mini Kit (Qiagen, Valencia, CA). Total RNA was used as a template and random hexanucleotide primers (0.15 μg) were added for reverse transcription and amplification in a reaction volume of 20 μl according to the manufacturer’s instructions (Invitrogen). After a reverse transcription reaction, first-strand cDNA (3-5 μl) was used for PCR with a PTC-100 programmable thermal controller (MJ Research, Inc., Waltham, MA). The oligonucleotide primer sets used for PCR analysis of cDNA are human Gli1, 5′-CAGAGAATGGAGCATCCTCC-3′ (forward) and 5′-TTCTGGCTCTTCCTGTAGCC-3′ (reverse), human osteonectin (ON), 5′-ATGAGGGCCTGGATCTTC-3′ (forward) and 5′-CTCCAGGCGCTTCTCATT-3′ (reverse)., human Shh, 5′-ACTGGGTGTACTACGAGTCCAAGG-3′ (forward) and 5′-AAACTGAGGAAGTCGCTGTAGAGC-3′ (reverse). For human Gli1 amplification, the PCR condition was 95°C, 15 min followed by 40 cycles of 94°C, 30 sec; 60°C, 30 sec; 72°C, 60 sec; and 72°C, 10 min final extension. For human ON amplification, the PCR condition was 94°C, 5 min followed by 40 cycles of 94°C, 30 sec; 60°C, 30 sec; 72°C, 45 sec; and 72°C, 10 min final extension. For human Shh amplification, the PCR condition was 94°C, 3 min followed by 40 cycles of 94°C, 45 sec; 63°C, 45 sec; 72°C, 45 sec; and 72°C, 10 min final extension. The reverse transcription PCR products were analyzed by 1% agarose gel electrophoresis. The intensity of the RT-PCR products was quantified by Image J (http://rsb.info.nih/ij/) after scanning the images and converted to TIFF files. Relative Gli1 expression was semi-quantified after normalization to endogenous β-actin.
For qRT-PCR, First-strand cDNA were prepared as describe above. First-strand cDNA (5 μl) was used for qPCR with a ABI 7500 Fast Real-Time PCR System (Applied Biosystem., Carlsbad, CA). The oligonucleotide primer sets used for qPCR analysis of cDNA are human Gli1, 5′-AGGGAGTGCAGCCAATACAG-3′ (forward) and 5′-ATTGGCCGGAGTTGATGTAG-3′ (reverse); human ON,5′-GAGACCTGTGACCTGGACAATG-3′ (forward) and 5′-GGAAGGAGTGGATTTAGATCACAAGA-3′ (reverse); and β-actin, 5′-CAAGGCCAACCGCGAGAAGATGAC-3′ (forward) and 5′-GCCAGAGGCGTACAGGGATAGCACA-3′ (reverse). A hot start at 95°C for 5 minutes was followed by 40 cycles at denaturation at 95°C for 15 s, annealing of the primers at 65°C for 30 s and elongation at 72°C for 30 s. Data were normalized to β-actin and represented as the average ratio of duplicates.
In vivo chimeric tumor growth studies
Chimeric tumors, consisting of NPF and C4-2 cells were established by co-inoculation at a ratio of 1:1 (1×106 cells each) in both flanks of mice. Three weeks post tumor cell inoculation, tumor appearance and serum PSA were evaluated, and treatment with cyclopamine or vehicle control was initiated. Blood specimens (70 μl) were obtained from the retroorbital sinus vein at the initiation of treatment and at sacrifice for serum PSA determination. Serum PSA was determined by microparticle ELISA using an Abbott IMx instrument (Abbott Laboratories, Abbott Park, IL). A total of 8 athymic male nude mice (BALB/c nu/nu; National Cancer Institute, Bethesda, MD) were divided into 2 groups, a vehicle-treated control group (n=4) and a cyclopamine treatment group (n=4). C4-2 cells mixed with NPF, at 1 x106 cells each, were inoculated in mouse flanks and shoulder areas with Matrigel (BD bioscience, San Jose, CA) containing T-medium at a ratio of 1:1 as described previously.11 Cyclopamine (5 μM) or T-medium (serving as a vehicle control) was delivered as Gelform (Johnson & Johnson, New Brunswick, NJ) adsorbent inserted inside of the growing tumor for 5 days before removal for histomorphologic and immunohistochemical examination.20, 21 Mice were sacrificed and serum PSA was measured as described previously.21
Immunohistochemical analysis
Histopathology of the mouse flank tumor specimens was assessed on H&E stained slides. Immunohistochemical (IHC) staining of tissue specimens was performed by a Dako Autostainer Plus System (Dako Corporation, Carpinteria, CA). The respective antibodies used in IHC analyses were mouse monoclonal antibody against M30 CytoDeath (1:60, DiaPharma Group, Inc., Wester Chester, OH), rabbit Gli1 polyclonal antibody against Gli1 (1:100, Santa Cruz Biotechnology, Santa Cruz, CA), mouse monoclonal antibody against androgen receptor (AR) (1:300, Santa Cruz Biotechnology), and mouse monoclonal antibody against PSA (1:100, Santa Cruz Biotechnology). Tissues were deparaffinized, rehydrated and subjected to 5-minute pressure-cooking antigen retrieval, 10-minute double endogenous enzyme block, and 30-minute primary antibody reaction followed by 30-minute EnVision plus Dual Link System (Peroxidase) incubation at room temperature. Antigenic signals were detected by adding diaminobenzidine as a chromogen of horseradish peroxidase, and slides were counterstained by hematoxylin. All reagents were obtained from Dako Corporation unless otherwise indicated. For quantification, 200 cells at 3 randomly selected areas were assessed at ×100 magnification and positive AR, PSA, Gli1 and M30 stained cells were recorded.
Statistical analysis
Statistical analysis was conducted with the JSTAT - Java Virtual Machine Statistics Monitoring Tool (Sun Microsystems, Inc., Santa Clara, CA). Statistical significance was established at p<0.05.
RESULTS
Reciprocal cellular interaction between prostate cancer and normal prostate fibroblasts mediated by soluble factors in conditioned medium
The growth-promoting effects of the conditioned medium (CM) isolated from androgen-independent (AI) human prostate cancer C4-2 cells, normal prostate stromal fibroblasts (NPF) and cancer-associated prostate stromal fibroblasts (CPF) were compared. CM and/or cyclopamine were added for 48 hrs to cells once the cell confluence reached 70%. Figure 1 shows that C4-2 CM induced NPF cell growth two fold over the control, whereas the same CM did not affect the growth of CPF. The effect of CM appears to be prostate cancer epithelial cell-specific since similar CM isolated from ARCaP cells, an advanced and highly metastatic human prostate cancer cell line isolated from the ascites fluid of a patient with bone metastatic disease22, failed to show a growth stimulatory effect on NPF and CPF in vitro (data not shown).
Figure 1. C4-2 conditioned media (CM) stimulated cell growth of normal prostate stroma (NPF) but not cancer-associated prostate stroma (CPF) and cyclopamine (Cyc) blocked this stimulated cell growth.
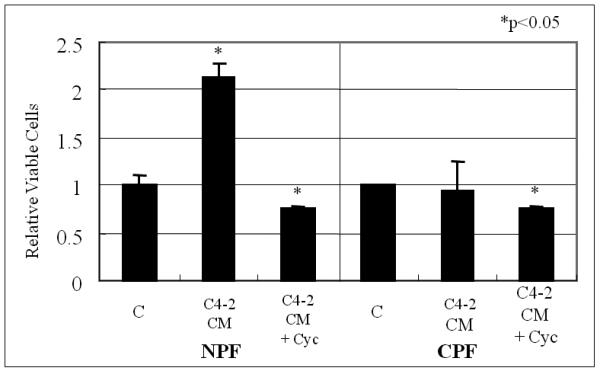
Prostate stroma cells (NPF and CPF) were treated with C4-2 CM. Culturing with C4-2 CM stimulated the growth of NPF but not CPF. The induced cell growth was blocked by Cyc (5 μM Cyc for 48 hrs in the absence of serum). Vehicle-treated control (V) lanes were set as 1.0. Asterisks (*) show significant change compare with vehicle-treated control (V). Data represent relative cell survival +/− S.D. NPF growth stimulated by C4-2 CM and its inhibition by Cyc were both statistically significant compared to vehicle-treated control (V) (p<0.05).
The growth-promoting effects of CM harvested from prostate cancer cells on prostate stroma appears to be reciprocal in nature, as revealed in two in vitro studies. First, CM harvested from NPF, but not CPF, induced C4-2 cell growth (Fig. 2A). Second, C4-2-Luc growth was stimulated when co-cultured with NPF, but not CPF (Fig. 2B). Note that the addition of Shh alone or the combination of Shh plus cyclopamine did not affect the growth of C4-2-Luc cells. These results suggest that other factors in the CM may modify Shh and confer the growth stimulatory effects Shh. No growth stimulatory effects were detected in ARCaP cells when exposed to CM harvested either from NPF or CPF cells (data not shown).
Figure 2. Prostate cancer cell growth induced by normal/benign prostate stroma CM or co-culture in the presence or absence of Shh was blocked by cyclopamine.
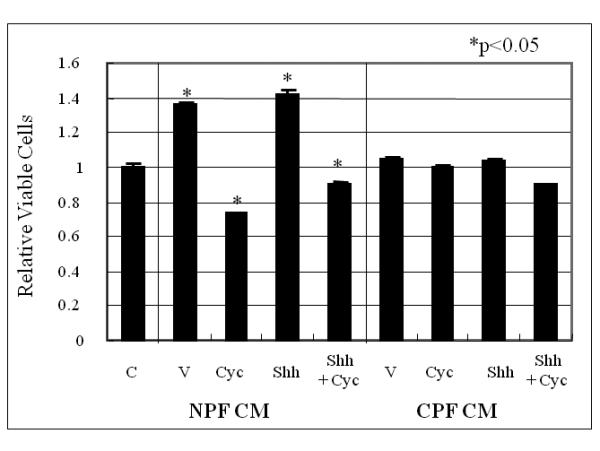
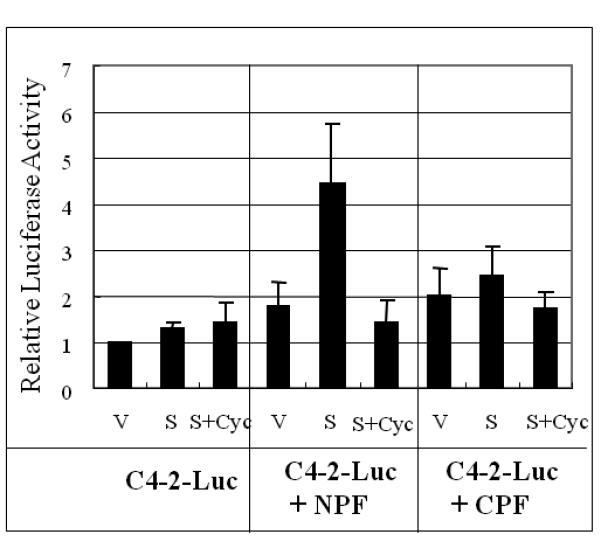
Panel A: CM from NPF but not CPF induced C4-2 cell growth and this induction was abrogated by cyclopamine. C = control (vehicle controls); V = CM-treated (either NPF- or CPF-CM treated); Shh = Shh added CM (NPF or CPF collected from cultured media in the presence of 1 μg/ml recombinant human Shh protein); Shh + Cyc (NPF or CPF collected in the presence of 1 μg/ml recombinant human Shh protein plus Cyc). Treatments were performed for 48 hrs and C4-2 cell survival was evaluated by MTS assay. The vehicle control lane (C) was set as 1.0. Asterisks (*) show significant change compared to vehicle-treated control (V). Data were tabulated as relative viable cells compared to vehicle controls expressed as average +/− S.D. with a statistical significance detected in NPF CM treated cells and the effects of Cyc. No statistically significant difference was observed in Shh treated samples. Panel B; Coculture of NPF but not CPF induced growth of C4-2-Luc cells (C4-2 stably luciferase transfected cells) in a Shh-dependent manner. Experiments were performed in duplicate. In the presence of 1 μg/ml recombinant human Shh protein and NPF, C4-2-Luc cell growth increased by 4.3 fold in a Shh-dependent manner. Cyc inhibited this growth stimulation. In contrast, the growth of C4-2-Luc was not increased when co-cultured with CPF either in the presence or absence of added recombinant Shh protein. The growth of C4-2-Luc cells in the absence of NPF or CPF was not affected by exogeneously added recombinant Shh protein and/or Cyc. V= vehicle control (assigned as 1.0). Treatments were performed for 48 hrs. C4-2-Luc cell growth was evaluated by luciferase activity which was found to correlate lineally with cell number. Cyc (5 μM for 48 h) was used in the absence of serum.
Shh is a potential mediator of the reciprocal cellular interaction between C4-2 and NPF
We sought to determine if Shh may be the soluble mediator responsible for the reciprocal communication between C4-2 and NPF. Cyclopamine was chosen to block Shh mediated downstream signaling between C4-2 and NPF. Figures 1 and 2 show that cyclopamine inhibited the growth of both C4-2 and NPF in vitro, with their respective growth stimulated by the addition of CM collected from the reciprocal cell types under co-culture conditions. These results were also confirmed by RT-PCR data where Gli1 expression in NPF but not CPF was upregulated by Shh in a ligand-dependent manner, and this upregulation was completely inhibited by cyclopamine (Fig. 3A). Unlike NPF and CPF, the steady-state levels of endogenous Shh and Gli1 in C4-2 cells were not regulated by Shh (Fig. 3B). These results were confirmed by qRT-PCR (Figs. 3C and D). These data are consistent with several previously published studies showing that Shh produced by cultured LNCaP cells23 and recombinant Shh protein failed to stimulate the growth of LNCaP24 and C4-2 cells in culture (data not shown). These results collectively emphasize the importance of paracrine rather than autocrine roles of Shh in the induction of prostate cancer cell growth by prostate stromal fibroblasts.
Figure 3. Activated Shh signaling in normal but not cancer-associated prostate stromal fibroblasts.
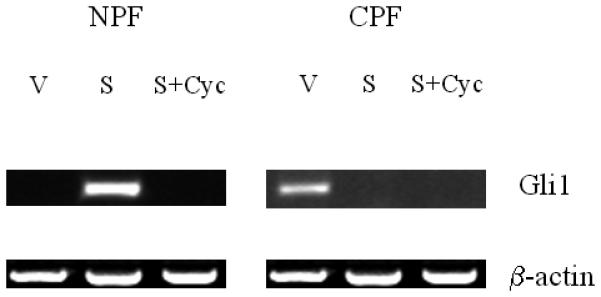
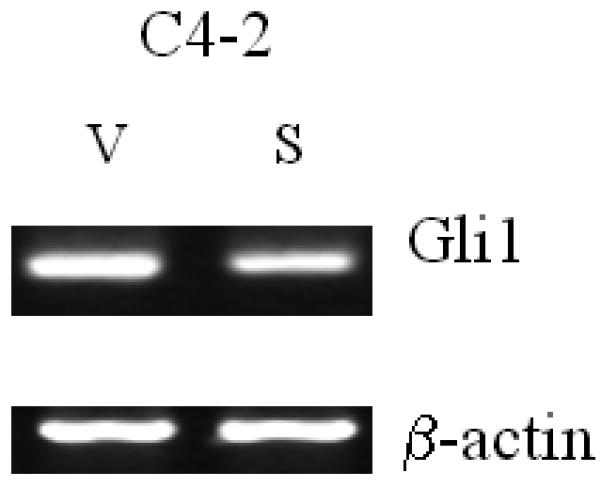
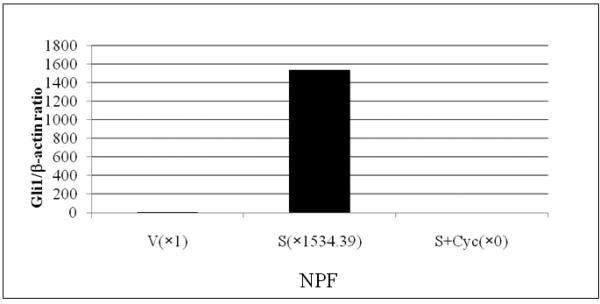

Panel A: Shh induced Gli1 expression in NPF and this induction was completely abrogated by cyclopamine (Cyc). In contrast to NPF, CPF did not reveal any consistent expression of Gli1 (data not shown). Panel B: In contrast to NPF, C4-2 did not show any consistent response to Cyc (data not shown). Exogenously added Shh did not induce Shh and Gli1 expression in C4-2 cells. V: vehicle added controls; S: recombinant human Shh protein (1 μg/ml) treated; Cyc: cyclopamine (5 μM). The treatment was performed for 48 hrs. Panel C: qRT-PCR data also revealed recombinant Shh (1μg/ml) (S) induced Gli1 expression in NPF compared to vehicle control (V) and this induction was completely abrogated by cyclopamine (5 μM) (S+Cyc). Panel D: qRT-PCR data showed that Gli1 mRNA was expressed in C4-2 cells and the levels of Gli1 mRNA was suppressed by Shh (1μg/ml) in a ligand-dependent manner (i.e. reversal in part by Cyc).
Shh induced Gli1 expression in NPF, but not CPF
Because the induction of NPF growth in vitro by the presence of either CM or C4-2 cells can be blocked by cyclopamine, we tested the possibility that Shh may be the active inducer for downstream signaling, accounting for the Shh-induced NPF response. Figure 4 shows that the addition of Shh to two pairs of prostate stromal fibroblasts (denoted as 003004 and 002004), NPF and CPF, induced Gli1 expression only in NPF but not CPF according to our quantification analyses [Gli1 band intensity calculated as Shh-treated (S) / vehicle-treated (V)]. The specificity of Gli1 induction in NPF was demonstrated by the addition of cyclopamine (Fig. 3A). This result was confirmed by qRT-PCR (Fig. 3C).
Figure 4. Comparison of the responsiveness of Gli1 in prostate stromal fibroblasts isolated from either normal/benign or cancerous areas of prostate specimens.
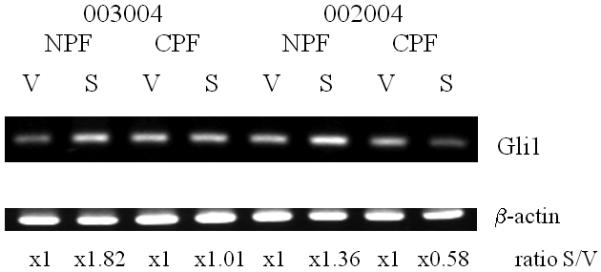
Two pairs of NPF and CPF (denoted as 003004 and 002004) were isolated from human prostate cancer tissue specimens. These cells were then treated with recombinant Shh (1 μg/ml). Gli1 expression was analyzed by RT-PCR. V: vehicle-treated controls; S: recombinant human Shh protein (1 μg/ml) treated stromal fibroblasts. The numbers below the bands represent relative ratios of Gli1 from Shh-treated (S) / vehicle-treated (V).
Osteonectin (ON) was identified as one of the Shh-Gli1 signaling targets in NPF, but not HS27A human marrow stromal cells,
We evaluated the potential target gene of the Shh-Gli1 signaling pathway in NPF cells upon the addition of Shh. Upon the addition of Shh to NPF, we observed the induction of ON mRNA as detected by RT-PCR in NPF and not in HS27A cells (Fig. 5A). This induction in NPF cells can be completely abrogated by the addition of cyclopamine (Fig. 5B). These results were confirmed by qRT-PCR in NPF cells (Fig. 5C).
Figure 5. Induction of Gli1 and osteonectin (ON) expression by Shh in NPF but not a normal human bone marrow stromal cell line, HS27A.

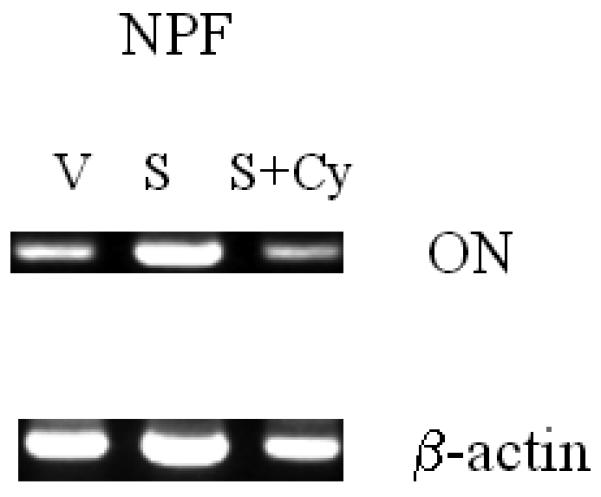
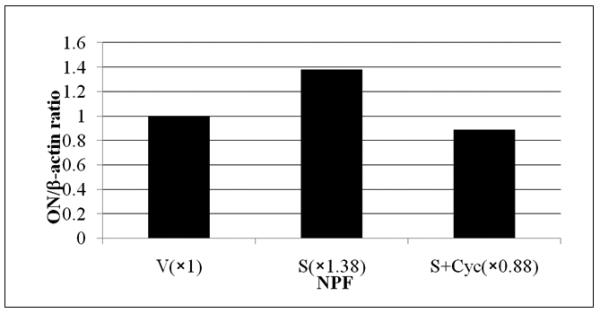
To identify the possible Shh target genes in NPF cells, we compared the expression of osteomimetic-related genes in NPF and in a normal human marrow stromal cell line, HS-27A. Panel A shows the addition of recombinant Shh at 1 μg/ml induced Gli1 and ON mRNA expression, as assessed by RT-PCR, in NPF but not HS27A cells. Panel B shows that recombinant Shh-induced ON expression in NPF cells was abrogated by 5 μM cyclopamine (Cyc). Panel C shows that addition of Shh (1μg/ml) induced ON mRNA expression and the Shh-induced ON expression was abrogated by cyclopamine (Cyc, 5 μM) in NPF cells confirmed by qRT-PCR.
Cyclopamine inhibited the growth of chimeric tumors comprised of C4-2 and NPF in mice
To see if reciprocal cellular communication between C4-2 and NPF occurs in vivo, we established subcutaneous chimeric C4-2 and NPF tumors in mice by co-inoculating these cells using well-established protocols.11 After tumors reached approximately 100 mm3, the mice were treated intratumorally with cyclopamine embedded in Gelform as described previously.20 After five days of cyclopamine treatment, serum PSA in the cyclopamine-treated group decreased by about 30% compared to vehicle-treated tumors (Fig. 6). Tumor specimens from vehicle and cyclopamine-treated mice were subsequently isolated and compared by both immunohistochemical and histopathologic protocols. Figure 7 shows significantly decreased AR and PSA, and enhanced cell death (M30) (p<0.05) in representative tissue specimens. These results suggest that the blockade of Shh-Gli signaling between C4-2 and NPF could abrogate the growth and survival signaling mediated by AR and cause significant apoptosis in prostate cancer cells.
Figure 6. Cyclopamine lowered serum PSA in mice implanted subcutaneously with chimeric prostate cancer tumors comprised of C4-2 androgen independent human prostate cancer and NPF cells.
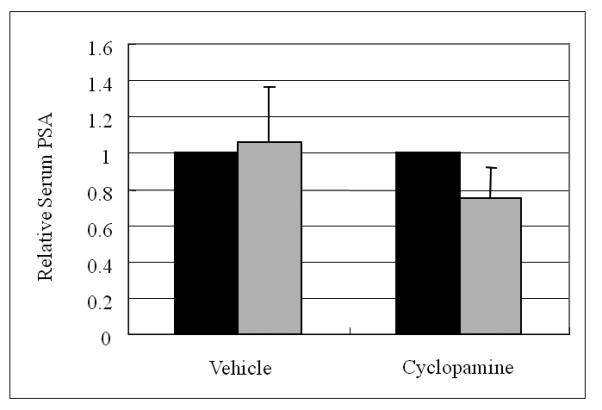
Cyclopamine depressed serum PSA in mice bearing chimeric C4-2 tumors. Cyclopamine (5 μM) was delivered as Gelform adsorbent inserted inside the growing tumor for 5 days. Left lane (black) is the serum PSA value at the time of the initiation of the cyclopamine treatment and right lane (gray) is the level of serum PSA in mice on the 5th day of cyclopamine treatment. V = vehicle-treated controls; Cyc = cyclopamine treatment. Treatments were evaluated at the end of day 5. Data represent the relative serum PSA value (with vehicle control = 1.0) +/− S.D. of 4 samples per group at the time of termination of the animal study.
Figure 7. Cyclopamine inhibited the expression of AR, PSA, and Gli1 and induced apoptosis in chimeric prostate tumors.
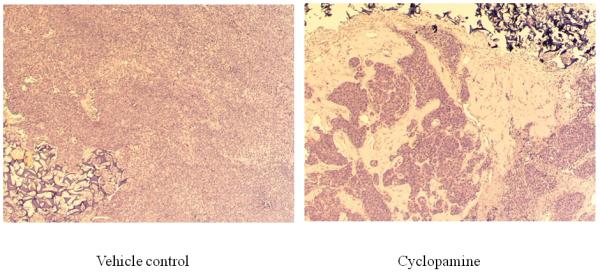
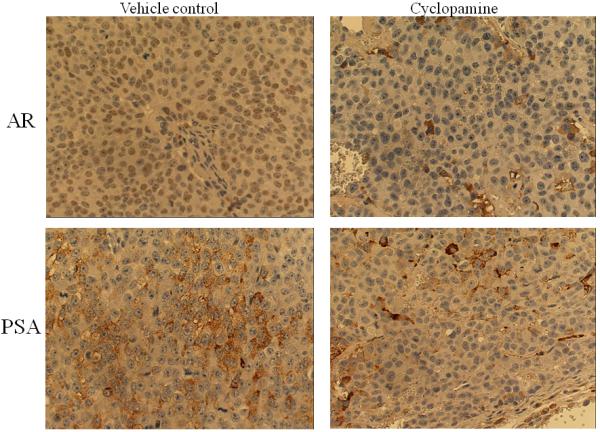
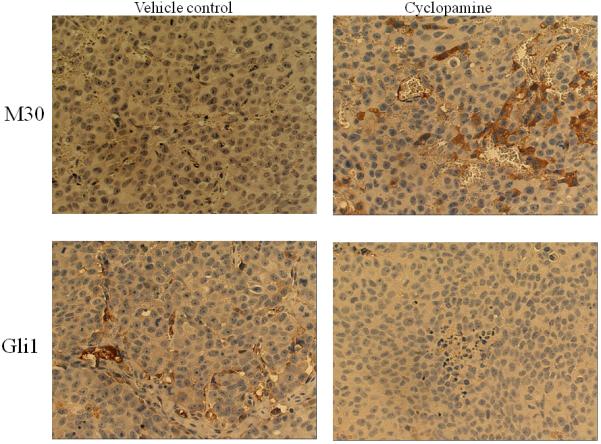
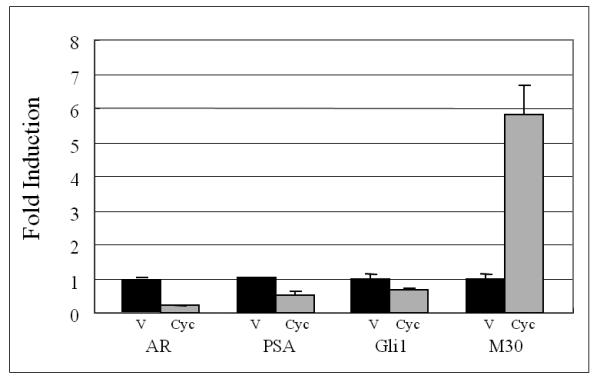
Panel A: hematoxylin & eosin stained slides from chimeric tumors treated with vehicle control or cyclopamine. Cyc imbedded in Gelform delivered to chimeric tumors induced cell death. As shown, very few viable cells were detected in cyclopamine-treated tumors compared to vehicle controls (Arrows indicate Gelform.) (x100). Panel B: cyclopamine inhibited AR, PSA, and Gli1 expression and induced apoptosis (M30) more than vehicle controls in vivo as analyzed by immunohistochemistry (x200). Panel C: detailed comparative quantification of cyclopamine (Cyc) therapy compared to vehicle controls on the expression of markers indicative of AR, PSA, Gli1, and apoptosis (M30) (p<0.05). Relative immunostaining of AR, PSA, Gli1 and M30 was set as 1.0 in control vehicle-treated mouse tumors. V: Vehicle-treated control; Cyc: cyclopamine treatment.
DISCUSSION
Prostate cancer progression has been shown to be determined by genetic as well as epigenetic factors in which reciprocal interactions of cancer cells with host stromal fibroblasts, either at the primary or at metastatic sites, could play key roles.25 A complex cell signaling network, mediated by soluble growth factors and extracellular matrices contributed by both prostate cancer cells and host factors from stromal fibroblasts, has been proposed to account for the growth, progression, and metastasis of prostate cancer.26, 27 Such investigations have resulted in the development of novel concepts and molecular-based therapeutic agents for the treatment of localized and disseminated liquid and solid tumors, including prostate cancer in man.28 The purposes of the present investigation are: 1) to explore the potential functional roles of Shh as a mediator between co-cultured AI human prostate cancer and prostate stromal fibroblasts and in chimeric prostate tumors in mice; 2) to compare Shh signaling between human AI prostate cancer and prostate stromal fibroblasts derived either from normal/benign or malignant regions of the prostate gland; 3) to examine potential Shh target genes in prostate stromal fibroblasts; and 4) to test the potential co-targeting of activated reciprocal Shh signaling between AI prostate cancer and prostate stromal fibroblasts in a chimeric prostate tumor model. Our results revealed the following findings: 1) Shh is a paracrine mediator that reciprocally promotes growth in AI human prostate cancer and human prostate stromal fibroblasts obtained from normal/benign prostate, but not advanced malignant prostate glands. While ligand-dependent autocrine Shh signaling activation may not be prevalent in AI human prostate cancer or prostate stromal fibroblasts, constitutive activated ligand-independent and prostate cancer cell-specific Shh-Gli1 signaling has been demonstrated in co-culture models as shown here (see Fig. 2 A and B, and Ref. 24). 2) ON, an osteomimicry marker and cell adhesion molecule capable of conferring prostate cancer cell adhesion to bone matrix proteins via integrin isotypes and prostate cancer cell migratory and invasive behaviors29, 30, was found to be one of the Shh target genes in prostate stromal fibroblasts. Further work is warranted to determine if ON is a target gene in marrow stromal cells exposed to human prostate cancer cells. 3) Though Shh and Gli1 were found to be co-expressed by all prostate cancer and stromal cell lines studied, ligand-dependent and stromal fibroblast-restricted Shh signaling activation appears in AI human prostate cancer and stromal fibroblasts isolated from normal/benign but not from cancer-associated prostate human stromal fibroblasts. These results suggest the importance of this signaling and its downstream components as determinants of early stages of human AI prostate cancer progression. 4) Targeting Shh signaling activation resulted in the inhibition of the chimeric prostate tumor growth and induction of apoptosis in a mouse model.
Ligand-dependent Shh signal activation in prostate cancer cells has been of considerable interest and a diverse array of data appears in the literature. Zhang et al. recently showed that there is no evidence of ligand-dependent autocrine signaling in three human prostate cancer cell lines (LNCaP, PC3, and 22RV1). They found, however, that cyclopamine inhibited the growth of these cells in vitro, suggesting that ligand-independent Shh activation may play a role.24 These observations were confirmed by Fan et al. who investigated signaling between cancer epithelial and the surrounding stromal fibroblasts in sections of prostate tumors by in situ hybridization.23 They showed that LNCaP cells overexpressing Shh after implantation in mice did not activate human but did activate mouse Ptch1 or Gli1, indicating a lack of autocrine (within human prostate cancer cells) but an activation of paracrine Shh signaling (between mouse and human cells) in human prostate cancer cells and mouse host stromal cells grown in vivo.
These results, however, contrasted with data obtained by Karhadkar et al.2 who demonstrated expression of Shh, Ptch1, and Gli1 in LNCaP cells and, upon cyclopamine treatment, decreased cell proliferation with a concomitant decreased expression of Gli1 expression.31 Datta et al. reported in situ hybridization results showing expression of Shh, Ptch1, and Gli1 by primary human prostate epithelial tumor cells.1 By blocking Shh signaling with a Shh antibody, they observed a 75 % decrease in cell proliferation in primary prostate cell cultures. Thus, it should be emphasized that despite the importance of Shh signaling activation in human prostate cancer, the status of ligand-dependent autocrine and paracrine Shh signaling activation is still controversial and dependent on the cell lines chosen and the experimental conditions employed.
Our studies revealed a ligand-dependent paracrine role of Shh signaling between AI human prostate cancer cells and stromal fibroblasts from normal/benign but not from malignant prostate stromal fibroblasts. This signaling pathway can be targeted by a Shh inhibitor in mice. We showed the upregulation of an apoptotic marker (M30) by treatment with cyclopamine, indicating that a Shh-Gli signaling inhibitor might be useful for treating AI prostate cancer. Our data are consistent with several published reports in which Shh signaling was shown to be an attractive molecular target in solid tumor progression. Ma et al. reported that elevated expression of hedgehog target genes, Gli1 and Ptch1, occurs in primary esophageal cancers and stroma with the expression of Shh transcript localized primarily to the tumor tissue. They showed that treatment with cyclopamine or the neutralizing antibodies of Shh reduced esophageal cancer growth and induced apoptosis.32 Hwang et al. revealed that cathepsin B, which is known to contribute to cancer invasiveness, is a Shh-Gli1 downstream target gene in the Shh-positive pancreatic cancer cell line PANC-1 .33
In our model, we identified ON as a new Shh-Gli1 signaling target gene in prostate stroma. ON was shown to regulate cell adhesion, proliferation, migration, and tissue remodeling, and is expressed during development and in processes requiring extracellular matrix turnover such as wound healing and tumor progression.34 In addition, ON is important for osteoblast formation, maturation, and survival.35-39 Elevated ON levels occur in a multitude of malignant tumors, including breast, brain, esophageal, and prostate carcinomas, as well as gliomas and melanomas40, 41, further suggesting that Shh-Gli signaling between tumor-stroma could be associated with osteomimicry and the malignant progression of solid tumors.
ON can be expressed by prostate tumor cells. The Shh-Gli pathway is constitutively upregulated in prostate cancer cells as the result of enhanced osteomimicry during prostate cancer progression.6 ON expression in prostate stroma was shown to be upregulated by the addition of exogenous Shh in AI prostate cancer cells, and this elevation can be blocked by cyclopamine (Fig. 6). Our model of elevated ON expression in benign/normal prostate stromal cells and AI prostate cancer cells but not human bone marrow stromal cells adds to our understanding of the initiation of bone metastasis in AI prostate cancer. ON expression by prostate cancer cells could lead to the activation of VEGF-VEGFR2 and αvβ3 and αvβ5 integrins in prostate cancer cells and bone stroma, which could prepare and facilitate the migration, invasion, and metastasis of prostate cancer cells and their subsequent adhesion to bone.42 It is conceivable that Shh-induced ON in normal/benign prostate stroma could help maintain the constitutive activated ON expression in prostate cancer cells which have an activated Shh-Gli1 pathway under either ligand-dependent or independent regulation.
The differential Shh-Gli1 signaling between C4-2 cells and NPF but not between C4-2 cells and CPF suggests that this signaling network might be associated with the early stage of AI prostate carcinogenesis before stromal fibroblasts become morphologically and functionally activated as “reactive” stroma.43
This interpretation, however, requires caution, considering the well-established fact that prostate cancer is heterogenous44 and Shh-Gli1 signal activation could occur regionally. However, an expanded role for Shh should be considered since Chen and associates showed that Shh expression by LNCaP cells is markedly enhanced upon androgen deprivation5 and Shh plays an important mediatory role between prostate cancer cells and osteoblasts.6 Since cancer-associated stromal fibroblasts are known to undergo marked morphologic and molecular transition and become highly inductive of cancer growth45, our results suggest that activated Shh-Gli1 signaling in NPF could lead to a subsequent metastatic cascade of AI prostate cancer cells and their adhesion to cells in the metastatic bone microenvironment. Additional studies are warranted to dissect the differential interaction between prostate cancer cells and stromal fibroblasts of different origins, including prostate and bone, and to define their pathophysiologic significance.
CONCLUSIONS
Shh-Gli1 signaling was demonstrated to occur specifically between androgen independent human prostate cancer cells and stromal fibroblasts originating from the normal/benign area of prostate glands. Activation of the Shh-Gli1 signaling network between hormone androgen independent prostate cancer cells and specific (benign/normal) prostate stromal fibroblasts could facilitate subsequent androgen-independent prostate cancer bone metastasis via the expression of ON, a new Shh-Gli1 downstream target. In a chimeric prostate cancer model composed of androgen-independent human prostate cancer cells and inductive stromal fibroblasts, we demonstrated that cyclopamine, an inhibitor of Shh signaling, inhibited prostate tumor growth in mice in vivo.
Acknowledgments
Grant support (in part): NIH/NCI P01 CA98912, R01 CA122602, and Department of Defense PC060866.
Abbreviations
- Shh
Sonic hedgehog
- Hh
hedgehog
- Ptch
Patched
- NPF
Normal prostate stroma
- CPF
Cancer associated prostate stroma
- CM
Conditioned media
- AR
Androgen receptor
- PSA
Prostate specific antigen
- AD
Androgen dependent
- AI
Androgen independent
- Smo
Smoothened
- VEGF
Vascular endothelial growth factor
- VEGFR2
Vascular endothelial growth factor receptor 2
- OPG
Osteoprotegerin
- RANKL
Receptor activator of nuclear factor- B ligand
- ON
Osteonectin
- FBS
Fetal bovine serum
- PBS
phosphate-buffered saline
- quantitative real time RT-PCR
qRT-PCR
REFERENCES
- 1.Datta S, Datta MW. Sonic Hedgehog signaling in advanced prostate cancer. Cell Mol Life Sci. 2006;63:435–48. doi: 10.1007/s00018-005-5389-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, Isaacs JT, Berman DM, Beachy PA. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–12. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 3.Lamm ML, Catbagan WS, Laciak RJ, Barnett DH, Hebner CM, Gaffield W, Walterhouse D, Iannaccone P, Bushman W. Sonic hedgehog activates mesenchymal Gli1 expression during prostate ductal bud formation. Dev Biol. 2002;249:349–66. doi: 10.1006/dbio.2002.0774. [DOI] [PubMed] [Google Scholar]
- 4.Sun LS, Li XF, Li TJ. PTCH1 and SMO gene alterations in keratocystic odontogenic tumors. J Dent Res. 2008;87:575–9. doi: 10.1177/154405910808700616. [DOI] [PubMed] [Google Scholar]
- 5.Chen M, Tanner M, Levine AC, Levina E, Ohouo P, Buttyan R. Androgenic regulation of hedgehog signaling pathway components in prostate cancer cells. Cell Cycle. 2009;8:149–57. doi: 10.4161/cc.8.1.7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zunich SM, Douglas T, Valdovinos M, Chang T, Bushman W, Walterhouse D, Iannaccone P, Lamm ML. Paracrine sonic hedgehog signalling by prostate cancer cells induces osteoblast differentiation. Mol Cancer. 2009;8:12. doi: 10.1186/1476-4598-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–7. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shigemura K, Isotani S, Wang R, Fujisawa M, Gotoh A, Marshall FF, Zhau HE, Chung LW. Soluble factors derived from stroma activated androgen receptor phosphorylation in human prostate LNCaP cells: Roles of ERK/MAP kinase. Prostate. 2009;69:949–55. doi: 10.1002/pros.20944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sung SY, Hsieh CL, Law A, Zhau HE, Pathak S, Multani AS, Lim S, Coleman IM, Wu LC, Figg WD, Dahut WL, Nelson P, Lee JK, Amin MB, Lyles R, Johnstone PA, Marshall FF, Chung LW. Coevolution of prostate cancer and bone stroma in three-dimensional coculture: implications for cancer growth and metastasis. Cancer Res. 2008;68:9996–10003. doi: 10.1158/0008-5472.CAN-08-2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edlund M, Sung SY, Chung LW. Modulation of prostate cancer growth in bone microenvironments. J Cell Biochem. 2004;91:686–705. doi: 10.1002/jcb.10702. [DOI] [PubMed] [Google Scholar]
- 11.Hsieh CL, Gardner TA, Miao L, Balian G, Chung LW. Cotargeting tumor and stroma in a novel chimeric tumor model involving the growth of both human prostate cancer and bone stromal cells. Cancer Gene Ther. 2004;11:148–55. doi: 10.1038/sj.cgt.7700665. [DOI] [PubMed] [Google Scholar]
- 12.Sanchez P, Clement V, Ruiz i Altaba A. Therapeutic targeting of the Hedgehog-GLI pathway in prostate cancer. Cancer Res. 2005;65:2990–2. doi: 10.1158/0008-5472.CAN-05-0439. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez P, Hernandez AM, Stecca B, Kahler AJ, DeGueme AM, Barrett A, Beyna M, Datta MW, Datta S, Ruiz i Altaba A. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc Natl Acad Sci U S A. 2004;101:12561–6. doi: 10.1073/pnas.0404956101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoon JW, Kita Y, Frank DJ, Majewski RR, Konicek BA, Nobrega MA, Jacob H, Walterhouse D, Iannaccone P. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J Biol Chem. 2002;277:5548–55. doi: 10.1074/jbc.M105708200. [DOI] [PubMed] [Google Scholar]
- 15.Mullor JL, Dahmane N, Sun T, Ruiz i Altaba A. Wnt signals are targets and mediators of Gli function. Curr Biol. 2001;11:769–73. doi: 10.1016/s0960-9822(01)00229-9. [DOI] [PubMed] [Google Scholar]
- 16.Sasaki H, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1997;124:1313–22. doi: 10.1242/dev.126.17.3915. [DOI] [PubMed] [Google Scholar]
- 17.Koeneman KS, Yeung F, Chung LW. Osteomimetic properties of prostate cancer cells: a hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate. 1999;39:246–61. doi: 10.1002/(sici)1097-0045(19990601)39:4<246::aid-pros5>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 18.Wang R, Xu J, Juliette L, Castilleja A, Love J, Sung SY, Zhau HE, Goodw TJ, Chung LW. Three-dimensional co-culture models to study prostate cancer growth, progression, and metastasis to bone. Semin Cancer Biol. 2005;15:353–64. doi: 10.1016/j.semcancer.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Huang WC, Xie Z, Konaka H, Sodek J, Zhau HE, Chung LW. Human osteocalcin and bone sialoprotein mediating osteomimicry of prostate cancer cells: role of cAMP-dependent protein kinase A signaling pathway. Cancer Res. 2005;65:2303–13. doi: 10.1158/0008-5472.CAN-04-3448. [DOI] [PubMed] [Google Scholar]
- 20.Gleave ME, Hsieh JT, von Eschenbach AC, Chung LW. Prostate and bonefibroblasts induce human prostate cancer growth in vivo: implications for bidirectional tumor-stromal cell interaction in prostate carcinoma growth and metastasis. J Urol. 1992;147:1151–9. doi: 10.1016/s0022-5347(17)37506-7. [DOI] [PubMed] [Google Scholar]
- 21.Shigemura K, Arbiser JL, Sun SY, Zayzafoon M, Johnstone PA, Fujisawa M, Gotoh A, Weksler B, Zhau HE, Chung LW. Honokiol, a natural plant product, inhibits the bone metastatic growth of human prostate cancer cells. Cancer. 2007;109:1279–89. doi: 10.1002/cncr.22551. [DOI] [PubMed] [Google Scholar]
- 22.Zhau HE, Odero-Marah V, Lue HW, Nomura T, Wang R, Chu G, Liu ZR, Zhou BP, Huang WC, Chung LW. Epithelial to mesenchymal transition (EMT) in human prostate cancer: lessons learned from ARCaP model. Clin Exp Metastasis. 2008;25:601–10. doi: 10.1007/s10585-008-9183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan L, Pepicelli CV, Dibble CC, Catbagan W, Zarycki JL, Laciak R, Gipp J, Shaw A, Lamm ML, Munoz A, Lipinski R, Thrasher JB, Bushman W. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology. 2004;145:3961–70. doi: 10.1210/en.2004-0079. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, Lipinski R, Shaw A, Gipp J, Bushman W. Lack of demonstrable autocrine hedgehog signaling in human prostate cancer cell lines. J Urol. 2007;177:1179–85. doi: 10.1016/j.juro.2006.10.032. [DOI] [PubMed] [Google Scholar]
- 25.Niu YN, Xia SJ. Stroma-epithelium crosstalk in prostate cancer. Asian J Androl. 2009;11:28–35. doi: 10.1038/aja.2008.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaminski A, Hahne JC, Haddouti el-M, Florin A, Wellmann A, Wernert N. Tumour-stroma interactions between metastatic prostate cancer cells and fibroblasts. Int J Mol Med. 2006;18:941–50. [PubMed] [Google Scholar]
- 27.Wernert N, Kaminski A, Haddouti el-M, Hahne JC. Tumor-stroma interactions of metastatic prostate cancer cell lines: analyses using microarrays. Methods Mol Biol. 2007;382:223–37. doi: 10.1007/978-1-59745-304-2_14. [DOI] [PubMed] [Google Scholar]
- 28.Chung LW, Hsieh CL, Law A, Sung SY, Gardner TA, Egawa M, Matsubara S, Zhau HE. New targets for therapy in prostate cancer: modulation of stromal-epithelial interactions. Urology. 2003;62:44–54. doi: 10.1016/s0090-4295(03)00796-9. [DOI] [PubMed] [Google Scholar]
- 29.Jacob K, Webber M, Benayahu D, Kleinman HK. Osteonectin promotes prostate cancer cell migration and invasion: a possible mechanism for metastasis to bone. Cancer Res. 1999;59:4453–7. [PubMed] [Google Scholar]
- 30.De S, Chen J, Narizhneva NV, Heston W, Brainard J, Sage EH, Brainard J, Sage EH, Byzova TV. Molecular pathway for cancer metastasis to bone. J Biol Chem. 2003;278:39044–50. doi: 10.1074/jbc.M304494200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berman DM, Desai N, Wang X, Karhadkar SS, Reynon M, Abate-Shen C, Beachy PA, Shen MM. Roles for Hedgehog signaling in androgen production and prostate ductal morphogenesis. Dev Biol. 2004;267:387–98. doi: 10.1016/j.ydbio.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 32.Ma X, Sheng T, Zhang Y, Zhang X, He J, Huang S, Chen K, Sultz J, Adegboyega PA, Zhang H, Xie J. Hedgehog signaling is activated in subsets of esophageal cancers. Int J Cancer. 2006;118:139–48. doi: 10.1002/ijc.21295. [DOI] [PubMed] [Google Scholar]
- 33.Hwang JH, Lee SH, Lee KH, Lee KY, Kim H, Ryu JK, Yoon YB, Kim YT. Cathepsin B is a target of Hedgehog signaling in pancreatic cancer. Cancer Lett. 2009;273:266–72. doi: 10.1016/j.canlet.2008.08.028. [DOI] [PubMed] [Google Scholar]
- 34.Koblinski JE, Kaplan-Singer BR, VanOsdol SJ, Wu M, Engbring JA, Wang S, Goldsmith CM, Piper JT, Vostal JG, Harms JF, Welch DR, Kleinman HK. Endogenous osteonectin/SPARC/BM-40 expression inhibits MDA-MB-231 breast cancer cell metastasis. Cancer Res. 2005;65:7370–7. doi: 10.1158/0008-5472.CAN-05-0807. [DOI] [PubMed] [Google Scholar]
- 35.Sage H, Vernon RB, Funk SE, Everitt EA, Angello J. SPARC, a secreted protein associated with cellular proliferation, inhibits cell spreading in vitro and exhibits Ca+2-dependent binding to the extracellular matrix. J Cell Biol. 1989;109:341–56. doi: 10.1083/jcb.109.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenblatt S, Bassuk JA, Alpers CE, Sage EH, Timpl R, Preissner KT. Differential modulation of cell adhesion by interaction between adhesive and counter-adhesive proteins: characterization of the binding of vitronectin to osteonectin (BM40, SPARC) Biochem J. 1997;324:311–9. doi: 10.1042/bj3240311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Funk SE, Sage EH. The Ca2(+)-binding glycoprotein SPARC modulates cell cycle progression in bovine aortic endothelial cells. Proc Natl Acad Sci U S A. 1991;88:2648–52. doi: 10.1073/pnas.88.7.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Francki A, Bradshaw AD, Bassuk JA, Howe CC, Couser WG, Sage EH. SPARC regulates the expression of collagen type I and transforming growth factor-ß1 in mesangial cells. J Biol Chem. 1999;274:32145–52. doi: 10.1074/jbc.274.45.32145. [DOI] [PubMed] [Google Scholar]
- 39.Delany AM, Kalajzic I, Bradshaw AD, Sage EH, Canalis E. Osteonectin-null mutation compromises osteoblast formation, maturation, and survival. Endocrinology. 2003;144:2588–96. doi: 10.1210/en.2002-221044. [DOI] [PubMed] [Google Scholar]
- 40.Framson PE, Sage EH. SPARC and tumor growth: where the seed meets the soil? J Cell Biochem. 2004;92:679–90. doi: 10.1002/jcb.20091. [DOI] [PubMed] [Google Scholar]
- 41.Bos TJ, Cohn SL, Kleinman HK, Murphy-Ulrich JE, Podhajcer OL, Rempel SA. International Hermelin brain tumor symposium on matricellular proteins in normal and cancer cell-matrix interactions. Matrix Biol. 2004;23:63–9. doi: 10.1016/j.matbio.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 42.De S, Chen J, Narizhneva NV, Heston W, Brainard J, Sage EH, Byzova TV. Molecular pathway for cancer metastasis to bone. J Biol Chem. 2003;278:39044–50. doi: 10.1074/jbc.M304494200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cunha GR, Hayward SW, Wang YZ, Ricke WA. Role of the stromal microenvironment in carcinogenesis of the prostate. Int J Cancer. 2003;107:1–10. doi: 10.1002/ijc.11335. [DOI] [PubMed] [Google Scholar]
- 44.Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, Macvicar GR, Varambally S, Harwood J, Bismar TA, Kim R, Rubin MA, Pienta KJ. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64:9209–16. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 45.Nakagawa H, Liyanarachchi S, Davuluri RV, Auer H, Martin EW, Jr, de la Chapelle A, Frankel WL. Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene. 2004;23:7366–77. doi: 10.1038/sj.onc.1208013. [DOI] [PubMed] [Google Scholar]