

Abstract
The homologous Rho kinases, ROCK1 and ROCK2, are involved in stress fiber assembly and cell adhesion and are assumed to be functionally redundant. Using mouse embryonic fibroblasts (MEFs) derived from ROCK1−/− and ROCK2−/− mice, we have recently reported that they play different roles in regulating doxorubicin-induced stress fiber disassembly and cell detachment: ROCK1 is involved in destabilizing the actin cytoskeleton and cell detachment, whereas ROCK2 is required for stabilizing the actin cytoskeleton and cell adhesion. Here, we present additional insights into the roles of ROCK1 and ROCK2 in regulating stress-induced impairment of cell-matrix and cell-cell adhesion. In response to doxorubicin, ROCK1−/− MEFs showed significant preservation of both focal adhesions and adherens junctions, while ROCK2−/− MEFs exhibited impaired focal adhesions but preserved adherens junctions compared with the wild-type MEFs. Additionally, inhibition of focal adhesion or adherens junction formations by chemical inhibitors abolished the anti-detachment effects of ROCK1 deletion. Finally, ROCK1−/− MEFs, but not ROCK2−/− MEFs, also exhibited preserved central stress fibers and reduced cell detachment in response to serum starvation. These results add new insights into a novel mechanism underlying the anti-detachment effects of ROCK1 deletion mediated by reduced peripheral actomyosin contraction and increased actin stabilization to promote cell-cell and cell-matrix adhesion. Our studies further support the differential roles of ROCK isoforms in regulating stress-induced loss of central stress fibers and focal adhesions as well as cell detachment.
Keywords: Rho kinase, isoform, actin cytoskeleton, detachment, cell-matrix and cell-cell adhesion, stress fibers, doxorubicin, serum starvation
Introduction
Rho-associated coiled coil-containing protein kinase (ROCK) is one of the best-characterized effectors of small GTPase RhoA and belongs to the AGC (protein kinase A/protein kinase G/protein kinase C) family of serine/threonine kinases.1-4 The ROCK family contains two members, ROCK1 and ROCK2, that share 65% overall identity and 92% identity in the kinase domain. Due to the high degree of sequence homology, ROCK1 and ROCK2 are believed to share more than 30 immediate downstream substrates and are assumed to be functionally redundant and involved in modulating actin cytoskeleton organization, stress fiber formation and cell adhesion.5-13 ROCK can increase myosin light chain (MLC) phosphorylation through direct effect on MLC or indirectly by inactivating MLC phosphatase (MYPT1), resulting in the stimulation of actomyosin contractility.14,15 ROCK stabilizes actin filaments through LIM kinases activation, resulting in cofilin phosphorylation and thereby inhibiting its actin-depolymerization activity.16,17 Both ROCK/MYPT1/MLC and ROCK/LIM kinase/cofilin pathways are heavily involved in stress fiber assembly and cell adhesion.
Regardless of structural similarities and possible functional redundancy of the two ROCK isoforms, a growing body of evidence supports that they possess unique functions, particularly in pathological processes (reviewed in refs. 11 and 18). Using mouse embryonic fibroblasts (MEFs) derived from ROCK1−/− and ROCK2−/− mice, we recently demonstrated that ROCK1 is involved in destabilizing actin cytoskeleton and cell detachment through regulating MLC2 phosphorylation and peripheral actomyosin contraction, whereas ROCK2 is required for stabilizing actin cytoskeleton and cell adhesion through regulating cofilin phosphorylation in response to cytotoxic stress induced by doxorubicin, a chemotherapeutic drug.19 These results support a novel concept that ROCK1 and ROCK2 can differently regulate stress fiber disassembly and cell adhesion under stress conditions.
Here, we further showed ROCK isoform functions in regulating the stress-induced impairment of cell-cell and cell-matrix adhesion. ROCK1−/− MEFs displayed significant preservation of F-actin to free G-actin ratio, focal adhesions and cadherin junctions in response to doxorubicin, while ROCK2−/−MEFs exhibited impaired focal adhesions but preserved adherens junctions compared with the wild-type (WT) MEFs. In addition, the inhibition of cell-matrix or cell-cell interactions by focal adhesion kinase (FAK) inhibitor 14 (F14), or EGTA abolished the anti-detachment effects of ROCK1 deletion. Finally, ROCK1−/− MEFs, but not ROCK2−/− MEFs, exhibited preserved central stress fibers and reduced cell detachment under another cell stress condition, serum starvation. These results add new insights into a novel mechanism underlying the anti-detachment effects of ROCK1 deletion, which are mediated by reduced peripheral actomyosin contraction, resulting in enhanced cell-cell adhesion, and also by increased actin stabilization leading to enhanced cell-matrix adhesion.
Results and Discussion
ROCK1 or ROCK2 deletion has no significant effect on cell adhesion rate under normal condition
Stress fibers are prominent bundles of actin and myosin filaments seen in many cell types and play important roles in numerous cell activities such as contraction, adhesion, movement, survival, etc. They can be broadly divided into thick and dense stress fibers, which are located in the peripheral portion of the cell (“cortical actin”), and stress fibers, which are located in the central portion of the cell (“central stress fibers”). Stress fibers terminate directly on focal adhesions where several adhesion-related proteins that connect the cell membrane to the underlying substrate are accumulated. Previous studies using ROCK inhibitors and/or ROCK dominant active/negative mutants support that ROCK activity is required for stress fiber organization and focal adhesion formation through promoting both myosin activity and actin polymerization.14-17,20
We recently reported that the architecture of the actin cytoskeleton in ROCK1−/− or ROCK2−/− MEF cells was largely similar compared with the WT MEFs at baseline condition, absent of cytotoxic stress, although we detected a significant reduction in the number of cells with cortical ring formation in ROCK1−/− MEFs (1.89% vs. 4.62% in WT cells) and a significant increase in the number of cells with periphery membrane folding in ROCK2−/− MEFs (10.2% vs. 4.32% in WT cells).19 To further characterize ROCK isoform function in regulating cell adhesion, we examined cell adhesion rates of WT, ROCK1−/− and ROCK2−/− MEFs under baseline condition (Fig. 1). MEFs were plated onto substratum from 5–120 min followed by gentle wash to remove unbound cells. The remaining adherent and viable cells were measured by methylthiazole tetrazolium (MTT) assay. The cell adhesion rate reached 90% after plating for 60 min for WT, ROCK1−/− and ROCK2−/− MEFs, and no significant increase was observed beyond the 120 min time point, which was considered to be 100%. ROCK1−/− MEFs showed a trend toward increased cell adhesion rate compared with WT MEFs, but this difference was not significant (Fig. 1A). A trend toward reduced cell adhesion rate was noticed in ROCK2−/− MEFs (Fig. 1B). These results indicate that ROCK1−/− and ROCK2−/− MEFs exhibit subtle differences in cell adhesive tendency at baseline, which is consistent with the mild differences in the architecture of actin cytoskeleton among ROCK1−/−, ROCK2−/− and WT MEFs.19
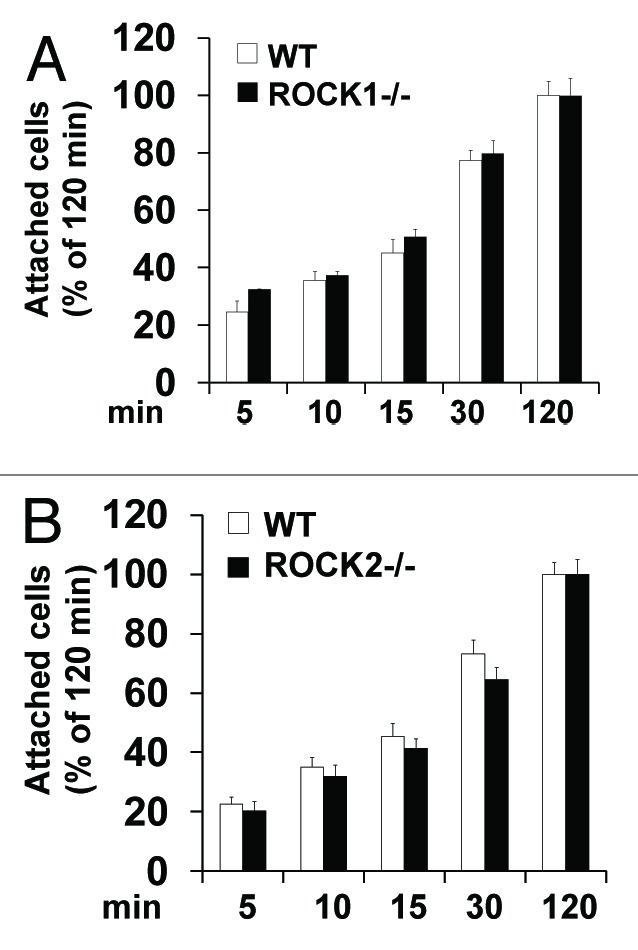
Figure 1. Effects of ROCK1 or ROCK2 deletion on cell adhesion rate. WT and ROCK1−/− cells (A) and WT and ROCK2−/− cells (B) were seeded at a density of 1 × 105/well in 10% FBS DMEM and left to adhere for the indicated times. After removal of unattached cells, attached cell viability was measured by MTT assay. Assays were performed in triplicate. Attached cell viability was expressed as percentage of the cells attaching for 120 min. No significant difference on cell adhesion rate was detected among WT, ROCK1−/− and ROCK2−/− cells. Error bars represent standard deviation *p < 0.05 vs. WT under the same condition.
These observations on the architecture of actin cytoskeleton at baseline condition are in agreement with previous studies characterizing ROCK1−/− or ROCK2−/− MEFs.21 It is worth noting that small interfering ribonucleic acids (siRNA)-based gene silencing studies may induce more prominent changes in the actin cytoskeleton than those observed in ROCK1- or ROCK2-deficient MEFs at baseline. For example, ROCK2 siRNA-transfected cells showed increased periphery membrane folding and disruption of central stress fibers, a phenomenon which is more prominent than that observed in ROCK2−/− cells at baseline,19 and this may be attributed to unknown compensatory mechanisms in ROCK2−/− cells. Previous siRNA-based studies have also shown that ROCK1 and ROCK2 have functional differences in regulating actin cytoskeleton in a variety of cell types.21-29 Future studies with conditional deletion of ROCK1 or ROCK2 in MEFs are needed to determine immediate vs. long-term consequences of isoform deletion on actin cytoskeleton organization.
ROCK1 and ROCK2 play different roles in cell detachment induced by doxorubicin
The subtle differences between WT cells and ROCK1- or ROCK2-deficient MEFs observed at baseline can be amplified under stress conditions.19 ROCK1−/− MEFs, under cytotoxic stress induced by doxorubicin, exhibited improved actin cytoskeleton stability characterized by attenuated periphery actomyosin ring formation, and preserved central stress fibers, resulting in reduced cell detachment compared with the WT MEFs. In contrast, ROCK2−/− MEFs showed increased periphery membrane folding and a trend toward increased cell detachment. In addition, ROCK1−/− MEFs exhibited reduced MLC phosphorylation but preserved cofilin phosphorylation, whereas ROCK2−/− MEFs showed reduced phosphorylation of both MLC and cofilin, suggesting that reduced actomyosin contraction and preserved actin polymerization contribute to the anti-detachment effects of ROCK1 deletion. To further support this concept, we investigated the effect of ROCK1 deletion on F-actin to free G-actin ratio (Fig. 2). Cells were treated with 3 μM doxorubicin for 4 h and fractionated cell extracts containing F-actin and nonpolymerized G-actin were prepared and analyzed for G- and F-actin contents. As shown in Figure 2B, ROCK1−/− MEFs exhibited a trend toward increased F to G-actin ratio at baseline condition. Doxorubicin treatment induced a decrease in F to G-actin ratio in WT MEFs but not in ROCK1−/− MEFs. These results further highlight that ROCK1 deficiency can preserve F-actin stability.
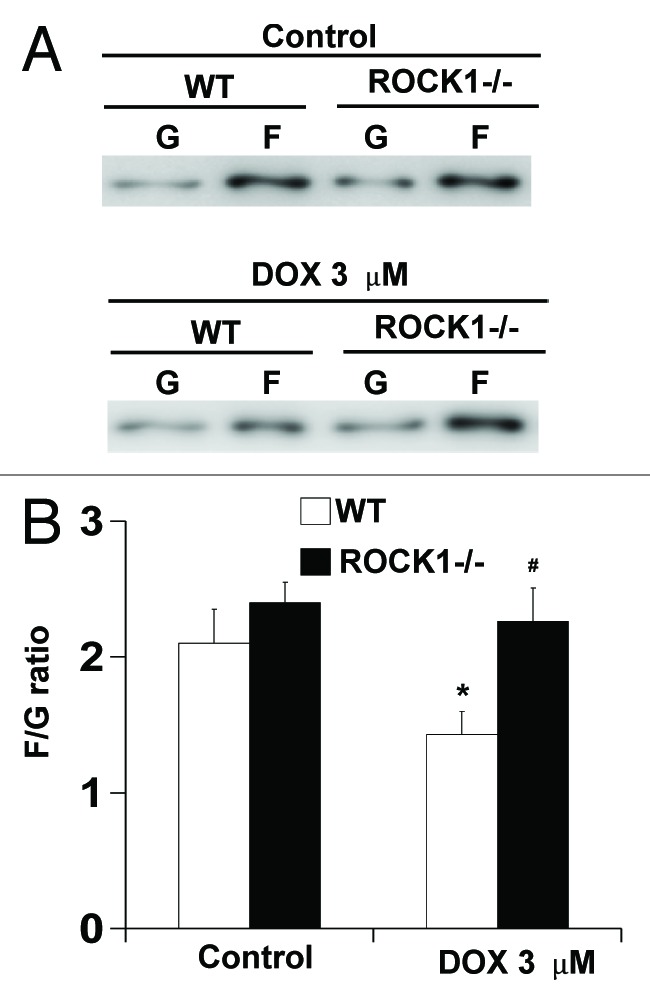
Figure 2. ROCK1 deletion preserves cell F-actin content in response to doxorubicin. WT and ROCK1−/− MEFs with or without 3 μM doxorubicin for 4 h. G- and F-actin were separated by ultracentrifuge followed by western blot of supernatants (G-actin) and pellets (F-actin). Representative western blots (A) were analyzed for F/G-actin ratios (B). *p < 0.05 vs. control of the same genotype; #p < 0.05 vs. WT under the same treatment condition.
Although our results suggest that reduced MLC phosphorylation and increased cofilin phosphorylation contribute to the increased actin cytoskeleton stability due to ROCK1 deletion,19 additional proximal ROCK effectors may also be involved, as more than 30 immediate ROCK downstream substrates have been identified, and most of them are involved in regulating actin cytoskeleton organization.5-13,30 Future studies are needed to dissect the mechanisms underlying the isoform specificity in the regulation of stress-induced central stress fiber disassembly.
ROCK1 and ROCK2 play different roles in regulating focal adhesions under normal and stress conditions
To further evaluate individual role of ROCK1 and ROCK2 in regulating actin cytoskeleton organization, we examined focal adhesion formations, which are located along and at both ends of the central stress fibers and also at the leading edge of the cell. Immunofluorescence staining was performed for activated FAK (p-FAK-Tyr925), a key component of focal adhesions, under baseline condition and after doxorubicin treatment (Fig. 3). At baseline, both ROCK1−/− and WT MEFs showed comparable p-FAK staining. In contrast, substantially reduced p-FAK staining was noticed in ROCK2−/− MEFs, especially at the ends of the stress fibers, consistent with a mild increase in periphery membrane folding19 and a trend of reduction of adhesion rate (Fig. 1B). This is likely due to the reduced actomyosin contraction and actin polymerization mediated by reduced phosphorylation of both MLC and cofilin in ROCK2−/− MEFs under baseline condition.19

Figure 3. ROCK1 deletion, but not ROCK2 deletion, reduces doxorubicin-induced impairment of focal adhesion formations. Representative images of focal adhesion formation revealed by p-FAK staining (green) of individual WT, ROCK1−/− and ROCK2−/− cells at baseline and after treatment with 3 μM doxorubicin for 16 h. Rhodamine-phalloidin staining for F-actin (red), and DAPI staining for nuclei (blue) are shown in merged images. Typical focal adhesions are indicated with white arrows. Bar, 50 μm.
After 16 h treatment with 3 μM doxorubicin, p-FAK staining was significantly reduced in the WT MEFs, consistent with reduced central stress fibers and increased formation of cortical rings (Fig. 3). However, ROCK1−/− MEFs showed preserved p-FAK staining at focal adhesions associated with preserved central stress fibers (Fig. 3). These results support preserved F-actin/G-actin ratio in ROCK1−/− MEFs compared with WT MEFs (Fig. 2). Together, these results reveal a non-redundant role for ROCK1 and ROCK2 in the regulation of focal adhesions: ROCK1 is involved in destabilizing focal adhesions under stress conditions, whereas ROCK2 is required for focal adhesion formation under both normal and stress conditions, consistent with their differential roles in the regulation of central stress fiber disassembly.
Both ROCK1 and ROCK2 are involved in stress-induced disruption of cell-cell adhesion
We have also examined the effects of ROCK1 or ROCK2 deletion on cell-cell adhesion under both baseline and doxorubicin treatment conditions by immunofluorescence staining for β-catenin, a key component of adherens junctions (Fig. 4). As expected, β-catenin was localized at the cell membrane with more intense and punctate staining in the area of cell-cell contacts representing adherens junctions. In untreated cells, β-catenin staining revealed similar patterns with overlapping cell borders among adjacent cells (Fig. 4). In response to doxorubicin treatment, a substantial loss of normal cell-cell contacts associated with cell shrinkage was observed in WT MEFs. In contrast, adherens junctions were well preserved in ROCK1−/− and to a less degree also in ROCK2−/− MEF cells (Fig. 4). The preservation of adherens junctions is likely linked to the reduced cortical ring formation and MLC phosphorylation observed in both ROCK1−/− and ROCK2−/− MEFs.19

Figure 4. ROCK1 or ROCK2 deletion reduces doxorubicin-induced impairment of adherens junction formations. Representative images of adherens junction formations revealed by β-catenin staining (green) of adjacent WT, ROCK1−/− and ROCK2−/− cells. Rhodamine-phalloidin staining for F-actin (red) and DAPI staining for nuclei (blue) are shown in merged images. Overlapping cell-cell contacts are indicated with white arrows. Adherens junctions are indicated with arrow heads. Bar, 50 μm.
Inhibition of cell-cell or cell-matrix adhesion abolishes the anti-detachment effects of ROCK1 deletion
We have validated the contributions of enhanced cell-matrix and cell-cell interactions to the anti-detachment effects of ROCK1 deletion using chemical inhibitors F14 and EGTA (Fig. 5). F14 (IC50 = 1 μM) targets the tyrosine 397 activation domain of FAK and prevents FAK activation.31 EGTA, a calcium chelator, induces disassembly of adherens junctions.32 Treatment with 1 μM F14 treatment did not show significant effect on cell detachment at baseline condition, but significantly increased cell detachment in doxorubicin-treated WT and ROCK1−/− MEFs to a similar degree (Fig. 5A). On the other hand, the treatment with 2 mM EGTA increased cell detachment similarly in WT and ROCK1−/− cells at baseline and after doxorubicin treatment (Fig. 5B). Together, these data indicate that inhibition of cell-matrix or cell-cell interactions abolished the anti-detachment effects of ROCK1 deletion, further supporting the notion that the beneficial effects of ROCK1 deletion are mediated through preserved cell-matrix and cell-cell adhesion.
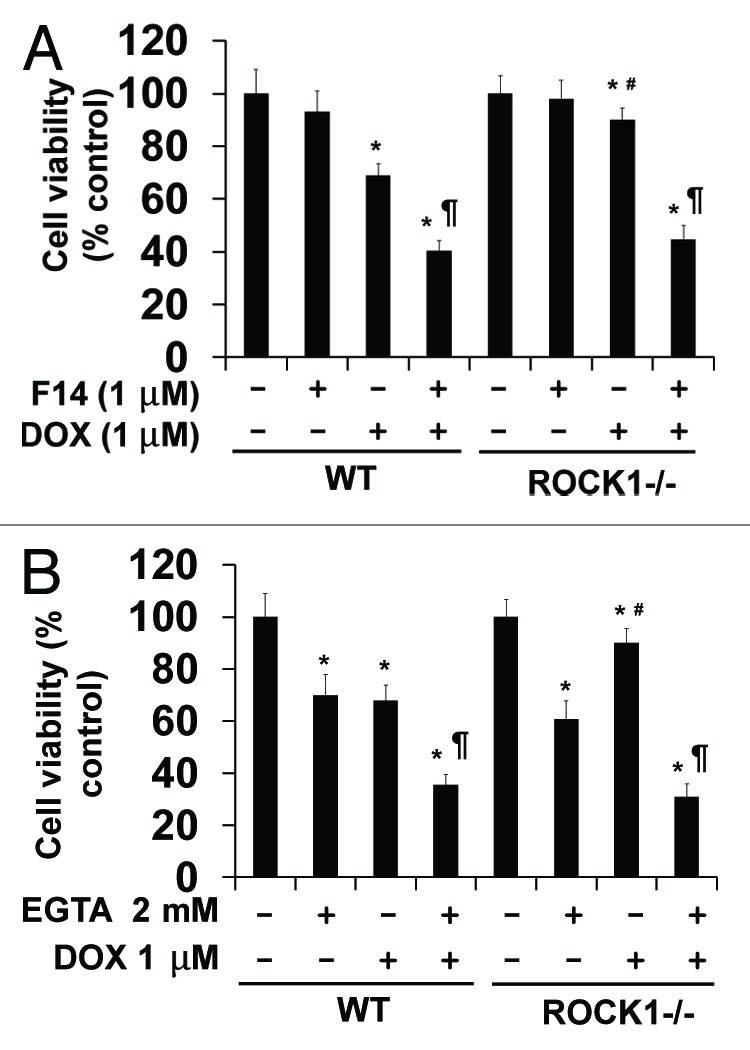
Figure 5. Disruptions of cell-matrix and cell-cell interactions by F14 and EGTA abolish the protective effects of ROCK1 deletion in response to doxorubicin. MTT assay performed with WT and ROCK1−/− MEFs with 1 μM doxorubicin and/or 1 μM F14 (A) or 2 mM EGTA (B) for 16 h. Cell viability was expressed as percentage of control cells without treatment. *p < 0.05 vs. control of the same genotype; #p < 0.05 vs. WT under the same treatment condition. ¶p < 0.05 vs. the same genotype under doxorubicin only condition.
Depending on the cell type and the apoptotic stimulus, inhibition of ROCK activity by ROCK inhibitors can act either as a pro-apoptotic or as a pro-survival regulator. Our findings with ROCK1- or ROCK2-deficient MEFs in the preservation of adherens junctions share some similarity with the pro-survival effects of ROCK inhibitors in preventing apoptosis through enhancing cell-cell interactions in some contexts, including embryonic stem cells grown in suspension culture conditions.33-35 On the other hand, the pro-apoptotic effects of ROCK inhibitors were mainly observed in the adhesion-dependent and differentiated cells,36-38 most likely due to the loss of focal adhesions as observed in ROCK2-deficient MEFs. It is worth noting that enhanced cell-matrix and cell-cell interactions due to ROCK1 deletion observed in MEFs may represent a mechanism underlying the in vivo protections observed in ROCK1-deficient mice in the context of heart failure and stressed erythropoiesis.39-41 Future studies are needed to evaluate the roles of ROCK1 and ROCK 2 in other cell types relevant to the pathogenesis in various disease models, including cardiovascular diseases, metabolic disorders and cancers.
ROCK1 deletion, but not ROCK2 deletion, inhibits cell detachment induced by serum starvation
As ROCK1 deletion inhibited cell detachment and apoptosis induced by doxorubicin, it is important to determine if these protective effects of ROCK1 deletion can be extended to other stress conditions. Serum starvation is one of the most frequently performed environmental stresses in cell biology aimed at dissecting the mechanisms of cell proliferation, differentiation and survival.42,43 It is known that cell detachment can be induced upon serum starvation in many cell types. Serum starvation inhibits cell growth, causes cellular rounding up and finally leads to cell detachment and subsequent death by apoptosis.44 As expected, a significant increase in cell detachment was detected for serum-starved WT MEFs, but it was delayed for ROCK1−/− MEFs (Fig. 6). Bright field photography (Fig. 6A), phalloidin staining for F-actin (Fig. 6B) and cell counting (Fig. 6C) showed a time-dependent increase in cell detachment upon serum starvation of the WT cells, while a significant reduction was observed in ROCK1−/− cells. Again, cell detachment was not reduced in ROCK2−/− cells (Fig. 6D). These results indicate that only ROCK1 deletion, but not ROCK2 deletion has anti-detachment effects after serum starvation.

Figure 6. ROCK1 deletion, but not ROCK2 deletion, reduces serum starvation-induced cell detachment. (A) Representative image of bright field photography of WT and ROCK1−/− cells before and after 8 h of serum starvation showing starvation-induced cell detachment. Bar, 400 μm. (B) Representative images of rhodamine-phalloidin staining for F-actin (red) and DAPI staining (blue) of WT and ROCK1−/− cells before and after 8 h of serum starvation showing starvation-induced disruption of central stress fibers and cell shrinkage. Bar, 100 μm. (C andD) Floating and attached cells were separately collected from WT and ROCK1−/− cells or from WT and ROCK2−/− cells at indicated time points after serum starvation. Floating cell ratio was expressed as percentage of total cells (floating plus attached cells) under indicated condition. *p < 0.05 vs. control of the same genotype; #p < 0.05 vs. WT under the same treatment condition.
In addition to cell detachment, we also examined if ROCK1 deletion inhibits apoptosis of attached cells induced by serum starvation. As expected, a significant increase in caspase-3 activation was detected for serum-starved WT MEFs after 4 h of serum starvation, but it was attenuated in ROCK1−/− MEFs (Fig. 7A). In addition to serum starvation, cell detachment from the extracellular matrix is also a potent apoptotic inducer.45 We observed that the expression levels of cleaved caspase-3 and cleaved poly(ADP-ribose) polymerase (PARP), which is a nuclear caspase-3 substrate, were significantly increased in floating WT cells compared with the attached cells after 4 or 8 h of serum starvation (Fig. 7B). The level of cleaved caspase-3 reached at least 15-fold higher in the floating cells compared with the level in attached cells after 8 h of serum starvation (Fig. 7B), indicating that cell detachment is a more predominant trigger of apoptosis than serum starvation. Like in doxorubicin-induced cell detachment,19 ROCK1 deficiency did not reduce the levels of cleaved caspase-3 in floating cells (data not shown), further supporting that ROCK1 deficiency has no significant inhibitory effect on apoptosis occurring after MEF detachment.

Figure 7. ROCK1 deletion inhibits apoptosis induced by serum starvation in attached cells. (A) Representative image (left panel) of western blot of full-length and cleaved ROCK1 and cleaved caspase-3 in cell lysates from attached WT and ROCK1−/− MEFs before and after 4 or 8 h of serum starvation. Quantitative analysis (right panel) of immunoreactive bands of cleaved capsase-3 (n = 4–6 for each condition), expressed as percent change relative to WT cells after 8 h of serum starvation. *p < 0.05 vs. control of the same genotype; #p < 0.05 vs. WT under the same treatment condition. (B) Representative image (left panel) of western blot of full-length and cleaved PARP, cleaved caspase-3 in cell lysates from attached or floating WT cells after 4 or 8 h of serum starvation. Densitometry analysis (right panel) of immunoreactive bands of cleaved caspase-3. Expression of cleaved caspase-3 was expressed as fold change relative to attached cells after 8 h of serum starvation. *p < 0.05 vs. 8 h attached cells.
Conclusion
Our studies, with isoform-selective deletions of ROCK, support a novel mechanism underlying the anti-detachment effects of ROCK1 deletion, which is mediated through reduced MLC phosphorylation but preserved cofilin phosphorylation, leading to reduced actomyosin contraction and preserved actin polymerization, which, in turn, result in increased central stress fiber stability and cell-matrix/cell-cell interactions under stress conditions. In addition, these studies have shown that ROCK1 vs. ROCK2 deletion produce different effects on actin stress fiber disassembly, leading to the different consequences on cell detachment under stressed conditions. ROCK2 deletion results in reduced MLC phosphorylation, reduced actomyosin contraction and preserved cell-cell interactions, which are shared beneficial effects with ROCK1 deletion. However, ROCK2 deletion also leads to reduced cofilin phosphorylation, reduced central stress fiber stability and focal adhesion formation, which differ from ROCK1 deletion and impair cell adhesion. Consequently, ROCK1 deletion, but not ROCK2 deletion, inhibits cell detachment induced by doxorubicin or serum starvation. A model to summarize all of these findings is schemed in Figure 8. In this model, actomyosin contraction and actin polymerization both promote stress fiber formation and focal adhesion formation resulting in cell adhesion at normal condition, but they play opposite roles in cortical ring formation and central stress fiber disruption under stress conditions, which leads to cell detachment and apoptosis.
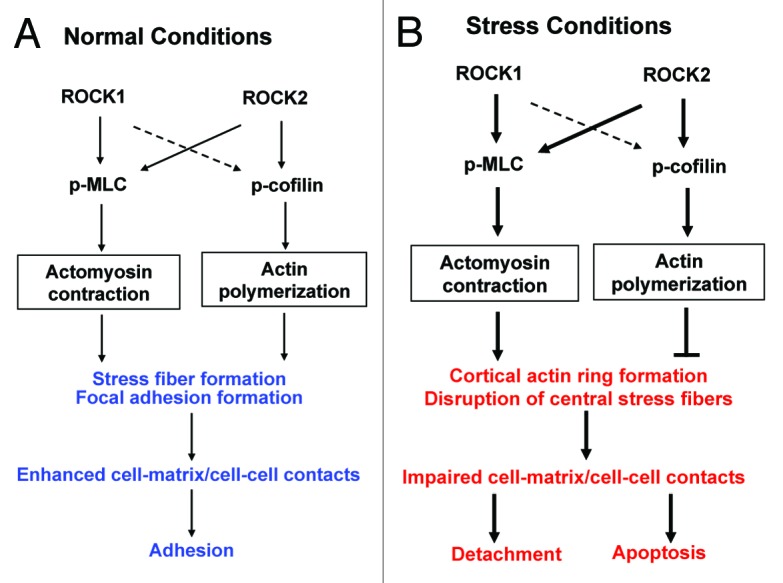
Figure 8. Schematic summary of roles of ROCK1 and ROCK2 in regulating actin cytoskeleton organization under normal (A) or cytotoxic stress (B) conditions (e.g., doxorubicin or serum starvation). Both ROCK1 and ROCK2 are involved in promoting actomyosin contraction via MLC phosphorylation. ROCK2 plays a preferential role in promoting actin polymerization via cofilin phosphorylation. Under normal conditions, both actomyosin contraction and actin polymerization promote stress fiber formation and focal adhesion formation resulting in cell adhesion. Under stress conditions, increased actomyosin contraction and actin polymerization play opposite roles in cortical ring formation and central stress fiber disruption. These stress-induced actin cytoskeleton remodeling events lead to cell detachment and apoptosis. The thin solid lines stand for baseline action. The thick solid lines stand for stimulated actions. The broken lines stand for alternative actions in the absence of ROCK2.
During the last decade, the ROCK family has attracted significant interest as a promising target for the treatment of a wide range of human diseases, including cardiovascular disorders, neurologic disorders, metabolic disorders and cancers.5-12 Most of the studies have been performed with non-isoform selective pharmacological inhibitors, which therefore inhibit ROCK1 and ROCK2 with equal potency and also have non-selective effects.46-48 Since ROCK pan-inhibitors are able to reduce phosphorylation of both MLC and cofilin, treatment with these inhibitors can’t prevent (or could even exaggerate) cell detachment induced by doxorubicin, which is a shared characteristic with ROCK2 deletion in MEFs.19 Future studies are needed, with specific targeting of ROCK1 and ROCK2 via genetic and chemical approaches, to determine whether the beneficial and detrimental effects of ROCK pan-inhibitors in experimental and clinical studies are mediated by inactivation of individual or both ROCK isoforms.
Materials and Methods
Cell culture and treatments
ROCK1- or ROCK2-deficient MEF cells were prepared from ROCK1−/− or ROCK2−/− embryos as previously described.19 All animal experiments were conducted in accordance with the National Institutes of Health “Guide for the Care and Use of Laboratory Animals” (NIH Publication No. 85–23, revised 1996) and were approved by the Institutional Animal Care and Use Committee at Indiana University School of Medicine. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals) and penicillin-streptomycin in a humidified incubator with 5% CO2 at 37°C. Cells at 90% confluency were treated with various drugs at indicated times and dosages. These drugs include pan-ROCK inhibitor Y27632 (Enzo Life Sciences); doxorubicin (Sigma), F14 (Santa Cruz Biotechnology). For serum starvation experiment, cells at 90% confluency were switched to serum-free DMEM and then incubated for indicated times.
Fluorescence imaging
Phalloidin staining of F-actin was performed as previously described.19 For visualizing focal adhesion and cadherin junction formations, MEF cells were seeded on gelatin coated glass coverslips. The cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min and permeabilized with 0.2% Triton X-100 for 1 h. After blocking with 5% goat serum in PBS for 1 h, the cells were incubated with rabbit polyclonal antibodies to p-FAK (#3284; Cell Signaling) or β-catenin (Ab16051, Abcam) overnight followed by incubation with Alexa Fluor 488 goat anti-rabbit IgG (A-11034), and rhodamine phalloidin (R-415, Life Technologies). The coverslips were then mounted with Vectashield mounting media (Vector Laboratories) containing Diamidino-2-phenylindole (DAPI) for counterstaining the nuclei. The fluorescent images were taken with a Leica DM5500B microscope (objectives: HCX PL FUOTAR 20.0 × 0.50, HCX PL FUOTAR 40 × 0.75) equipped with a DFC300FXR2 camera and analyzed with the Leica AF6000 software. For individual cells, focal adhesion formations can be visualized by p-FAK or β-catenin staining. For grouped cells, cadherin junction formations can be visualized by β-catenin staining.
Protein analysis
Following serum starvation at indicated time points, attached cells were harvested and analyzed for caspase activation by western blot analysis as previously described.19 The blots were then probed with primary antibodies to ROCK1 (sc-5560), ROCK2 (sc-5561, Santa Cruz Biotechnology), cleaved caspase-3 (#9661) and PARP (#9542, Cell Signaling). After blotting with corresponding secondary antibodies conjugated with horseradish peroxidase, the membranes were developed with ECL western blotting or SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific), and the blots were visualized by using a Fujifilm LAS-4000 Imager. All blots were normalized to GAPDH (10R-G109a, Fitzgerald Industries International) or to actin (sc-1616; Santa Cruz Biotechnology).
Measurement of F-actin/G-actin ratio
Determination of the amount F-actin content compared with free G-actin content was performed using the G-actin/F-actin assay kit (Cytoskeleton Inc.) according to the manufacturer's instructions. Briefly, after various treatments, MEF cells were homogenized in cell lysis and F-actin stabilization (LAS) buffer (50 mM PIPES, pH6.9, 50 mM NaCl, 5 mM MgCl2, 5 mM EGTA, 5% glyceral, 0.1% Nonidet P-40, 0.1% Triton X-100, 0.1% Tween 20, 0.1% 2-mercaptoethanol, 0.001% Antifoam C plus protease inhibitor cocktail) supplemented with 1 mM ATP followed by centrifugation at 100,000 g for 1 h at 37°C to separate the F-actin from G-actin pool. Supernatants were saved as G-actin fraction. The pellets were resuspended in ice-cold milli-Q H2O plus 1 μM cytochalasin D and then incubated on ice for 1 h to dissociate F-actin. Equal volumes of both the supernatant fraction (G-actin) and the resuspended pellet fraction (F-actin) were subjected to western blot analysis with the use of an actin antibody (Cytoskeleton Inc.).
Cell viability and detachment assays
For the cell viability assay, following treatment with desired drugs at indicated concentrations and time points, an MTT assay was performed as previously described.19 At least three independent experiments were analyzed. For cell detachment assay induced by serum starvation, cells were first cultured in DMEM supplemented with 10% FBS to reach 90% confluency, then switched to serum free DMEM. Detached cells in culture medium (floating cells) were collected at indicated times and counted with a hemacytometer. The attached cells were harvested by trypsinization for counting. Cell viability of floating and attached cells was also determined by assessing cellular uptake of trypan blue dye as previously described.19 At least three independent experiments were analyzed for each condition.
Adhesion assays
In a 24-well plate, 1 × 105 cells/well were seeded with 1 ml of culture media. Cells were left to adhere for the indicated times and unattached cells were then aspirated. The remaining cells were gently washed three times to remove unbound or loosely attached cells. Viability of attached cells was determined by MTT assay as previously described.19 The samples were prepared in triplicates. At least three independent experiments were analyzed.
Statistical analysis
Data are reported as mean ± SE. Comparisons between groups were analyzed by Student’s t-test or ANOVA as appropriate, with p < 0.05 considered as significant.
Acknowledgments
This work was supported by National Institutes of Health grants (HL085098 to L.W.), a Grant-in-Aid award from American Heart Association, Midwest Affiliate (to L.W.) and the Riley Children's Foundation.
Glossary
Abbreviations:
- DAPI
Diamidino-2-phenylindole
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- FAK
focal adhesion kinase
- F14
focal adhesion kinase inhibitor 14
- MEF
mouse embryonic fibroblast
- MLC2
myosin light chain 2
- MYPT1
myosin light chain phosphatase 1
- MTT
methylthiazole tetrazolium
- PARP
poly(ADP-ribose) polymerase
- PBS
phosphate-buffered saline
- ROCK
Rho-associated coiled coil-containing protein kinase
- siRNA
small interfering ribonucleic acid
- WT
wild type
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24699
References
- 1.Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, et al. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 1996;15:2208–16. [PMC free article] [PubMed] [Google Scholar]
- 2.Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, et al. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J. 1996;15:1885–93. [PMC free article] [PubMed] [Google Scholar]
- 3.Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996;392:189–93. doi: 10.1016/0014-5793(96)00811-3. [DOI] [PubMed] [Google Scholar]
- 4.Leung T, Chen XQ, Manser E, Lim L. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol Cell Biol. 1996;16:5313–27. doi: 10.1128/mcb.16.10.5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hahmann C, Schroeter T. Rho-kinase inhibitors as therapeutics: from pan inhibition to isoform selectivity. Cell Mol Life Sci. 2010;67:171–7. doi: 10.1007/s00018-009-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loirand G, Pacaud P. The role of Rho protein signaling in hypertension. Nat Rev Cardiol. 2010;7:637–47. doi: 10.1038/nrcardio.2010.136. [DOI] [PubMed] [Google Scholar]
- 7.Miyamoto S, Del Re DP, Xiang SY, Zhao X, Florholmen G, Brown JH. Revisited and revised: is RhoA always a villain in cardiac pathophysiology? J Cardiovasc Transl Res. 2010;3:330–43. doi: 10.1007/s12265-010-9192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nunes KP, Rigsby CS, Webb RC. RhoA/Rho-kinase and vascular diseases: what is the link? Cell Mol Life Sci. 2010;67:3823–36. doi: 10.1007/s00018-010-0460-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong M, Yan BP, Liao JK, Lam YY, Yip GW, Yu CM. Rho-kinase inhibition: a novel therapeutic target for the treatment of cardiovascular diseases. Drug Discov Today. 2010;15:622–9. doi: 10.1016/j.drudis.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 2010;67:545–54. doi: 10.1002/cm.20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Surma M, Wei L, Shi J. Rho kinase as a therapeutic target in cardiovascular disease. Future Cardiol. 2011;7:657–71. doi: 10.2217/fca.11.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olson MF. Applications for ROCK kinase inhibition. Curr Opin Cell Biol. 2008;20:242–8. doi: 10.1016/j.ceb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rath N, Olson MF. Rho-associated kinases in tumorigenesis: re-considering ROCK inhibition for cancer therapy. EMBO Rep. 2012;13:900–8. doi: 10.1038/embor.2012.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, et al. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase) J Biol Chem. 1996;271:20246–9. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- 15.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–8. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 16.Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–8. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- 17.Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K. Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem. 2000;275:3577–82. doi: 10.1074/jbc.275.5.3577. [DOI] [PubMed] [Google Scholar]
- 18.Shi J, Zhang L, Wei L. Rho-kinase in development and heart failure: insights from genetic models. Pediatr Cardiol. 2011;32:297–304. doi: 10.1007/s00246-011-9920-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi J, Wu X, Surma M, Vemula S, Zhang L, Yang Y, et al. Distinct roles for ROCK1 and ROCK2 in the regulation of cell detachment. Cell Death Dis. 2013;4:e483. doi: 10.1038/cddis.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katoh K, Kano Y, Noda Y. Rho-associated kinase-dependent contraction of stress fibres and the organization of focal adhesions. J R Soc Interface. 2011;8:305–11. doi: 10.1098/rsif.2010.0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noguchi M, Hosoda K, Fujikura J, Fujimoto M, Iwakura H, Tomita T, et al. Genetic and pharmacological inhibition of Rho-associated kinase II enhances adipogenesis. J Biol Chem. 2007;282:29574–83. doi: 10.1074/jbc.M705972200. [DOI] [PubMed] [Google Scholar]
- 22.Yoneda A, Multhaupt HA, Couchman JR. The Rho kinases I and II regulate different aspects of myosin II activity. J Cell Biol. 2005;170:443–53. doi: 10.1083/jcb.200412043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoneda A, Ushakov D, Multhaupt HA, Couchman JR. Fibronectin matrix assembly requires distinct contributions from Rho kinases I and -II. Mol Biol Cell. 2007;18:66–75. doi: 10.1091/mbc.E06-08-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Zheng XR, Riddick N, Bryden M, Baur W, Zhang X, et al. ROCK isoform regulation of myosin phosphatase and contractility in vascular smooth muscle cells. Circ Res. 2009;104:531–40. doi: 10.1161/CIRCRESAHA.108.188524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mong PY, Wang Q. Activation of Rho kinase isoforms in lung endothelial cells during inflammation. J Immunol. 2009;182:2385–94. doi: 10.4049/jimmunol.0802811. [DOI] [PubMed] [Google Scholar]
- 26.Bryan BA, Dennstedt E, Mitchell DC, Walshe TE, Noma K, Loureiro R, et al. RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. FASEB J. 2010;24:3186–95. doi: 10.1096/fj.09-145102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lock FE, Hotchin NA. Distinct roles for ROCK1 and ROCK2 in the regulation of keratinocyte differentiation. PLoS ONE. 2009;4:e8190. doi: 10.1371/journal.pone.0008190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chun KH, Araki K, Jee Y, Lee DH, Oh BC, Huang H, et al. Regulation of glucose transport by ROCK1 differs from that of ROCK2 and is controlled by actin polymerization. Endocrinology. 2012;153:1649–62. doi: 10.1210/en.2011-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darenfed H, Dayanandan B, Zhang T, Hsieh SH, Fournier AE, Mandato CA. Molecular characterization of the effects of Y-27632. Cell Motil Cytoskeleton. 2007;64:97–109. doi: 10.1002/cm.20168. [DOI] [PubMed] [Google Scholar]
- 30.Brown K, Bhowmick NA. Linking TGF-beta-mediated Cdc25A inhibition and cytoskeletal regulation through RhoA/p160(ROCK) signaling. Cell Cycle. 2004;3:408–10. doi: 10.4161/cc.3.4.778. [DOI] [PubMed] [Google Scholar]
- 31.Golubovskaya VM, Nyberg C, Zheng M, Kweh F, Magis A, Ostrov D, et al. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J Med Chem. 2008;51:7405–16. doi: 10.1021/jm800483v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kartenbeck J, Schmid E, Franke WW, Geiger B. Different modes of internalization of proteins associated with adhaerens junctions and desmosomes: experimental separation of lateral contacts induces endocytosis of desmosomal plaque material. EMBO J. 1982;1:725–32. doi: 10.1002/j.1460-2075.1982.tb01237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol. 2007;25:681–6. doi: 10.1038/nbt1310. [DOI] [PubMed] [Google Scholar]
- 34.Koyanagi M, Takahashi J, Arakawa Y, Doi D, Fukuda H, Hayashi H, et al. Inhibition of the Rho/ROCK pathway reduces apoptosis during transplantation of embryonic stem cell-derived neural precursors. J Neurosci Res. 2008;86:270–80. doi: 10.1002/jnr.21502. [DOI] [PubMed] [Google Scholar]
- 35.Braam SR, Nauw R, Ward-van Oostwaard D, Mummery C, Passier R. Inhibition of ROCK improves survival of human embryonic stem cell-derived cardiomyocytes after dissociation. Ann N Y Acad Sci. 2010;1188:52–7. doi: 10.1111/j.1749-6632.2009.05083.x. [DOI] [PubMed] [Google Scholar]
- 36.Svoboda KK, Moessner P, Field T, Acevedo J. ROCK inhibitor (Y27632) increases apoptosis and disrupts the actin cortical mat in embryonic avian corneal epithelium. Dev Dyn. 2004;229:579–90. doi: 10.1002/dvdy.20008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore M, Marroquin BA, Gugliotta W, Tse R, White SR. Rho kinase inhibition initiates apoptosis in human airway epithelial cells. Am J Respir Cell Mol Biol. 2004;30:379–87. doi: 10.1165/rcmb.2003-0019OC. [DOI] [PubMed] [Google Scholar]
- 38.Shi J, Wei L. Rho kinase in the regulation of cell death and survival. Arch Immunol Ther Exp (Warsz) 2007;55:61–75. doi: 10.1007/s00005-007-0009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang J, Xie M, Shah VR, Schneider MD, Entman ML, Wei L, et al. Activation of Rho-associated coiled coil protein kinase 1 (ROCK-1) by caspase-3 cleavage plays an essential role in cardiac myocyte apoptosis. Proc Natl Acad Sci USA. 2006;103:14495–500. doi: 10.1073/pnas.0601911103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi J, Zhang YW, Yang Y, Zhang L, Wei L. ROCK1 plays an essential role in the transition from cardiac hypertrophy to failure in mice. J Mol Cell Cardiol. 2010;49:819–28. doi: 10.1016/j.yjmcc.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vemula S, Shi J, Mali RS, Ma P, Liu Y, Hanneman P, et al. ROCK1 functions as a critical regulator of stress erythropoiesis and survival by regulating p53. Blood. 2012;120:2868–78. doi: 10.1182/blood-2011-10-384172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pirkmajer S, Chibalin AV. Serum starvation: caveat emptor. Am J Physiol Cell Physiol. 2011;301:C272–9. doi: 10.1152/ajpcell.00091.2011. [DOI] [PubMed] [Google Scholar]
- 43.Tavaluc RT, Hart LS, Dicker DT, El-Deiry WS. Effects of low confluency, serum starvation and hypoxia on the side population of cancer cell lines. Cell Cycle. 2007;6:2554–62. doi: 10.4161/cc.6.20.4911. [DOI] [PubMed] [Google Scholar]
- 44.Kulkarni GV, McCulloch CA. Serum deprivation induces apoptotic cell death in a subset of Balb/c 3T3 fibroblasts. J Cell Sci. 1994;107:1169–79. doi: 10.1242/jcs.107.5.1169. [DOI] [PubMed] [Google Scholar]
- 45.Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13:555–62. doi: 10.1016/S0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- 46.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–4. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]