

Abstract
DNA damaging agents, including those used in the clinic, activate cell cycle checkpoints, which blocks entry into mitosis. Given that checkpoint override results in cell death via mitotic catastrophe, inhibitors of the DNA damage checkpoint are actively being pursued as chemosensitization agents. Here we explored the effects of gemcitabine in combination with Chk1 inhibitors in a panel of pancreatic cancer cell lines and found variable abilities to override the S phase checkpoint. In cells that were able to enter mitosis, the chromatin was extensively fragmented, as assessed by metaphase spreads and Comet assay. Notably, electron microscopy and high-resolution light microscopy showed that the kinetochores and centromeres appeared to be detached from the chromatin mass, in a manner reminiscent of mitosis with unreplicated genomes (MUGs). Cell lines that were unable to override the S phase checkpoint were able to override a G2 arrest induced by the alkylator MMS or the topoisomerase II inhibitors doxorubicin or etoposide. Interestingly, checkpoint override from the topoisomerase II inhibitors generated fragmented kinetochores (MUGs) due to unreplicated centromeres. Our studies show that kinetochore and centromere fragmentation is a defining feature of checkpoint override and suggests that loss of cell viability is due in part to acentric genomes. Furthermore, given the greater efficacy of forcing cells into premature mitosis from topoisomerase II-mediated arrest as compared with gemcitabine-mediated arrest, topoisomerase II inhibitors maybe more suitable when used in combination with checkpoint inhibitors.
Keywords: cell cycle checkpoints, MUGs, DNA damage, centromere, mitotic catastrophe
Introduction
Cells possess an evolutionary conserved checkpoint pathway that prevents cells with DNA damage from progressing through the cell cycle. Many chemotherapies induce DNA damage that normally triggers a p53-dependent G1 arrest. As p53 is compromised in approximately 50% of all cancers,1 most tumor cells rely on S phase or G2 checkpoints.2 In this context, DNA damage activates ATM and ATR kinases, which, in turn, phosphorylate and activate effector kinases, Chk1 and Chk2.3 Cell cycle arrest occurs through the inhibitory phosphorylations on Cdc2 and Cdc25.4 If the damage is successfully repaired, cells will re-enter the cell cycle. Thus, cell cycle checkpoints maintain genome stability by ensuring cells enter mitosis with accurately replicated DNA. Based on the notion that cell cycle regulators are required to maintain cell viability, the use of pharmacological inhibitors to disrupt the checkpoint arrest has emerged as an attractive target for therapeutic intervention.5 The idea of using kinase inhibitors to enhance chemotherapeutic efficacy was first shown for caffeine.6 More recent studies have focused on using DNA damaging agents with the concomitant addition of relevant checkpoint inhibitors. Notably, inhibiting Chk1,7 ATR8 and Wee19 sensitizes cancer cells to various DNA damaging agents such as gemcitabine,10 cisplatin, 5-fluorouracil,11 SN3812 and adriamycin.13 The mechanism of sensitization as reported for HCT116 cells appears to be death via mitotic catastrophe.12
Currently, there is a lack of detailed information about which chemotherapeutic agents respond best to checkpoint override, and whether there are cellular determinants that may affect the response of cells to combination treatments with chemotherapy and checkpoint inhibitors. Here we report that cells exhibit variable responses to S phase checkpoint override, but all cells tested were able to override a G2 checkpoint arrest. Checkpoint override induced by replication or topoisomerase II (topoII) inhibitors induced centromere and kinetochore fragmentation, which is a defining feature of mitotic catastrophe. We suggest that inhibitors of the DNA damage checkpoint should work most effectively with agents that inhibit centromere replication, as this results in acentric genomes that cannot be segregated. Our studies provide information that should be taken into consideration when developing protocols for using checkpoint inhibitors as chemosensitizers.
Results
Cells can be forced into premature mitosis following cell cycle arrest and Chk1 inhibition
Inhibitors of the DNA damage checkpoint kinase 1 (Chk1) will cause drug-arrested cells to prematurely enter mitosis.7,12,14 We wanted to understand the nature of the mitotic defect in greater detail. We treated gemcitabine-arrested PANC1 cells stably expressing H2B:gfp with UCN-01, an inhibitor of Chk1, and monitored cell fates by time-lapse microscopy for 24 h (Fig. 1A). 67.8 ± 8.8% of vehicle-treated cells progressed through a normal mitosis, while only 2% of the cells treated with gemcitabine entered mitosis. 98.4 ± 2.7% of the cells were arrested in S phase (also based on FACs, Fig. S4) for up to 48 h. Addition of UCN-01 to gemcitabine-arrested cells forced 58.9 ± 11.1% of cells to prematurely enter mitosis during the 24 h movie. These mitotic cells were highly abnormal, because their chromosomes did not align properly, and we consistently saw mitotic chromatin pushed outside of the mitotic spindle and separated from centromeres (Fig. 1B). These cells were confirmed to be in mitosis, as they were positive for phospho-histone H3 (pH3SER10) staining (Fig. S1A). We noted that between 5 and 12 h after UCN-01 addition, 94% of cells entered mitosis, with < 5% of cells apoptotic (data not shown). Cells with highly condensed chromatin that were consistent with “apoptotic bodies” were negative for pH3SER10 (denoted as “A”). We were therefore able to discern the condensed DNA morphology of mitotic cells from apoptotic cells. Similar observations were made when cells were treated with gemcitabine followed by caffeine, an inhibitor of ATM/ATR (data not shown).
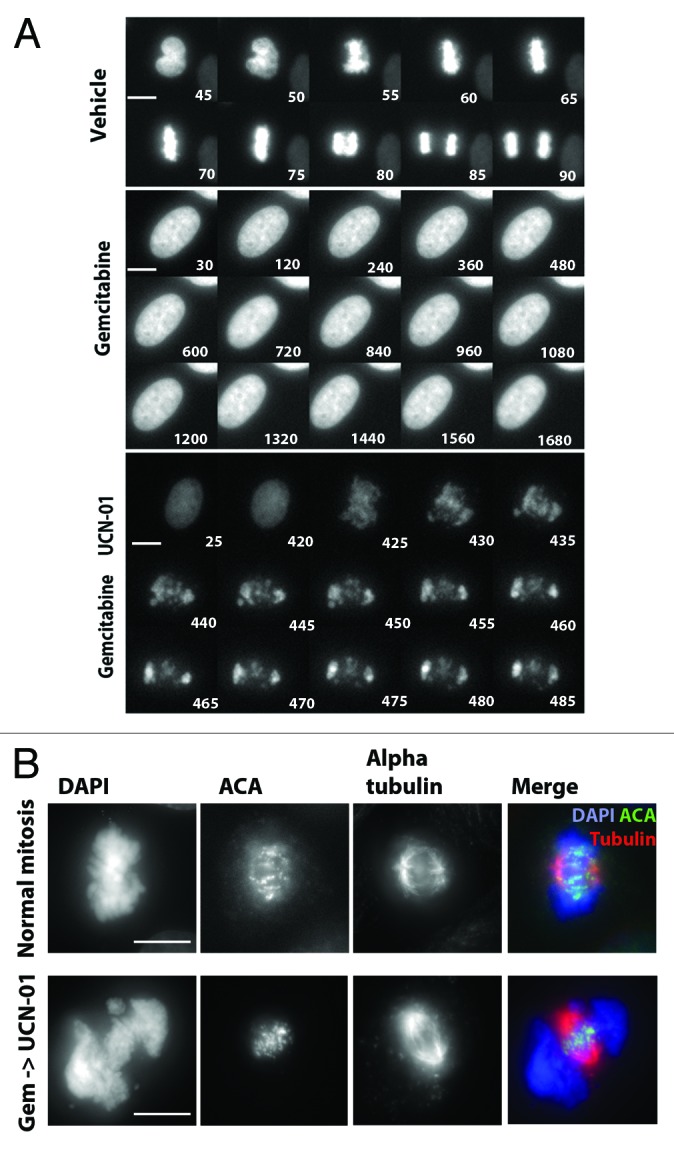
Figure 1. Inhibition of Chk1 causes premature entry into mitosis. (A) A montage from timelapse video microscopy studies performed on PANC1 cells expressing gfp:H2B. Images were obtained from control, gemcitabine alone or gemcitabine followed by UCN-01 drug treatments. Representative cells are shown. Time stamps are relative to the start of filming (minutes). Scale bar represents 10 µm. (B) PANC1 cells were used to generate normal mitotic and MUGs (gemcitabine + UCN-01). Figures were immunostained for DNA (DAPI, blue), mitotic spindles (α tubulin, red) and centromeres (ACA, green). Scale bar represents 10 µm.
Forced entry into mitosis results in mitosis with unreplicated genomes (MUGs)
The distinctive mitotic figures produced by forcing gemcitabine treated cells with under-replicated genomes into mitosis were reminiscent of mitosis with unreplicated genomes (MUGs) that were originally described in Chinese hamster ovary (CHO) cells.15 Addition of caffeine to CHO cells that were arrested in S phase with hydroxyurea caused them to enter mitosis with highly fragmented chromosomes. A defining feature was that the unreplicated centromere with its associated kinetochore were physically separated from the rest of the chromatin mass. Efforts to generate MUGs in human (Hela) cells were not very successful and required overexpression of cyclin A to induce mitotic entry.16 More recently, Hela cells that stably expressed gfp:centrin spontaneously underwent MUG after prolonged (> 40 h) HU treatment.17 To ascertain whether our treatment regimen was generating MUGs, we used electron microscopy (EM) to examine kinetochores at the ultrastructural level. In normal mitotic cells, the kinetochore appears as a tri-laminar structure that is associated on opposite sides of the centromeric heterochromatin, with microtubules connected to the surface of the kinetochore (Fig. 2A, top left shows one of a pair of kinetochores). However, in gemcitabine and UCN-01-treated cells, only small C-shaped structures with distinct laminar plates of the kinetochore were seen. These structures are not visibly connected to the more electron-dense chromatin and were seen in clusters that were concentrated in the center of the cell, as shown above. These structures were very similar to the fragmented kinetochores in CHO cells that underwent MUGs.15 We used immunofluorescence staining to determine the localization of centromere (ACA) and associated kinetochore proteins, including Plk1, BubR1, Aurora A, Sgo1, Sgo2 as well as chromosome-associated proteins (CAP G, G2, H, H2) (Fig. S1B and C and data not shown). Taken together, we show that the centromere/kinetochore complex was dissociated from the bulk of the chromatin mass. Indeed, all detectable centromere/kinetochores were found to lie in the center of the spindle, consistent with the possibility that they were attached to microtubules (Fig. 1B), consistent with previous observations.17 Thus, the combination of gemcitabine and a Chk1 inhibitor can efficiently generate MUGs in human PANC1 cells. The detachment of the centromere/kinetochore complex from chromosomes in CHO cells was reported to depend on forces generated by the attached microtubules.15 However, we were able to generate MUGs in PANC1 cells both in the presence and absence of microtubules (Fig. S1C and 2).

Figure 2. UCN-01 forces gemcitabine-arrested cells to undergo mitosis with unreplicated genomes (MUGs). (A) Electron microscopy micrographs of kinetochores from control (inset, top left) and gemcitabine+ UCN-01 (inset, left, panels 2–4). Main panel shows kinetochores are detached from the bulk chromatin, which is also highly fragmented. Kinetochores are indicated by arrowheads. (B) Chromosome spreads from untreated mitoses and from mitoses generated by gemcitabine followed by UCN-01 treatment. Images shown are representative of metaphase figures observed, with areas highlighted magnified. Scale bars shown are 10 µm and 5 µm, respectively. (C) The alkaline comet assay was performed on control, gemcitabine treated, untreated mitotic shake-offs and mitoses generated by gemcitabine + UCN-01. Quantification of comet assay is represented by the average Olive moment (arbitrary units) in parentheses of approximately 70 individual nuclei. Representative comets generated from treatments are shown.
We performed chromosome spreads to assess chromosome integrity. Unlike normal mitotic cells, which show paired chromatids attached at the primary constriction, cells treated with gemcitabine and UCN-01 failed to produce discrete chromosomes but appeared highly pulverized (Fig. 2B), consistent with early images obtained from fusing S phase cells to mitotic cells.18 In a second approach, we performed alkaline comet assays, which detect both single- and double-stranded breaks, on cells treated with various agents. As shown in Figure 2C, gemcitabine treatment alone generated an increase in the Olive moment, ~4 times higher that than observed in control interphase cells. Vehicle-treated mitotic cells had a comparable Olive moment to vehicle-treated interphase cells. However, the Olive moment of cells treated with gemcitabine and UCN-01 was ~11 times greater than control interphase cells, 6 times higher than normal mitotic cells and 2.5 times higher than interphase cells that were treated with gemcitabine alone. Taken together, these findings suggest that forcing cells with incompletely replicated DNA results in severe fragmentation of the mitotic chromatin. The most prominent structure detected by EM and IF were centromere/kinetochore complexes that had separated from the rest of the chromatin.
Biochemical purification of MUGs
We next sought physical evidence that the kinetochores were detached from bulk chromatin. Lysates from cells treated with gemcitabine and UCN-01 were fractionated through a sucrose gradient. Mitotic cells were lysed by douncing, and the chromatin was pelleted by a low speed spin. The supernatant, which might contain the kinetochore fragments, was fractionated through a 20–50% sucrose gradient by ultracentrifugation. The fractions were probed with antibodies to CENP-A, Mis12 and Bub3, which represented discrete domains within the kinetochore complex. Two major peaks, at the top and bottom of the gradient were found to contain these proteins. As there was significantly more protein in the top of the gradient, the equivalent signal intensities reflect enrichment of kinetochore proteins in the bottom fractions. Notably, using the low-speed chromosome-free sup from normal mitotic lysates only yielded kinetochore protein subcomplexes that migrated near the top of the gradient. The lack of detectable signal at the bottom of the gradient suggested there were no MUGs (Fig. S3). Fractions from the MUGs were also fixed and pelleted onto coverslips and stained for kinetochore proteins Aurora B and Mis12. Only the bottom fractions contained material that produced discrete foci of staining (Fig. 3B). These complexes are likely MUGs, as they also contain DNA that was also detectable in the MUGs observed in cells. These findings provide further evidence that the centromere/kinetochore complex can be separated from bulk chromatin.

Figure 3. Biochemical purification of MUGs. (A) Lysates from MUGs were separated by sucrose gradient fractionation and fractions were subjected to western blot analysis with the indicated antibodies. Protein in each fraction was first concentrated by TCA and resuspended in the same volume, so that the fractions near the top of the gradient contained more total protein than the bottom fractions. Fraction #1 represents the bottom of the gradient, while fraction #23 is the top. (B) Fraction 4 was fixed, pelleted onto coverslips and used for the detection of Aurora B, Mis12 and DNA. Scale bar is 2 μm.
Variable response of cell lines to S phase checkpoint override
We next tested a panel of pancreatic cancer cell lines for their ability to prematurely enter mitosis after treatment with gemcitabine and UCN-01. PANC1, MiaPaCa-2 as well as Hela cells that were treated with gemcitabine entered mitosis within 6–9 h after addition of UCN-01 (100 nM). In contrast, BxPC3 and CFPAC cells could not be forced into mitosis after inhibition of Chk1 (UCN-01 at 100, 500 or 1,000 nM) or ATM/ATR (caffeine) (Fig. S4A and data not shown). We tested other drugs (thymidine, aphidicolin, cisplatin) that arrested cells in S phase in combination with UCN-01 and confirmed the inability of BxPC3 and CFPAC cells to override an S phase checkpoint arrest (Fig. S4B and data not shown). Thus, the failure to override the S phase arrest in certain cells is not idiosyncratic of gemcitabine but more likely a cellular defect.
We next tested whether the limiting factors that prevented the BxPC3 and CFPAC cell lines to override the S phase checkpoint might also prevent them from overriding a G2 checkpoint arrest. We used the alkylating agent methyl methanesulfonate (MMS) as well as the topo II inhibitors etoposide and doxorubicin to induce a G2 arrest (Fig. S5A and data not shown). Interestingly, we found that BxPC3 cells arrested in G2 by MMS were able to enter mitosis upon addition of UCN-01. The same response was observed for PANC1 cells treated with MMS and UCN-01. Examination of the mitotic figures at the LM and EM levels in PANC1 and BxPC3 cells showed that they appeared to contain properly condensed chromosomes (Fig. 4A and B, top panel). We next treated PANC1 and BxPC3 with either etoposide or doxorubicin followed by UCN-01, and found that the G2-arrested cells were also forced into premature mitosis (Fig. 4B and data not shown). Examination of the mitotic figures showed extensive fragmentation and, unexpectedly, evidence of centromere fragmentation as seen in cells that underwent MUGs (Fig. 4A and B). The presence of dissociated centromere/kinetochore complexes were confirmed by EM, which showed C-shaped kinetochore fragments that were indistinguishable from those generated from an S phase checkpoint override.
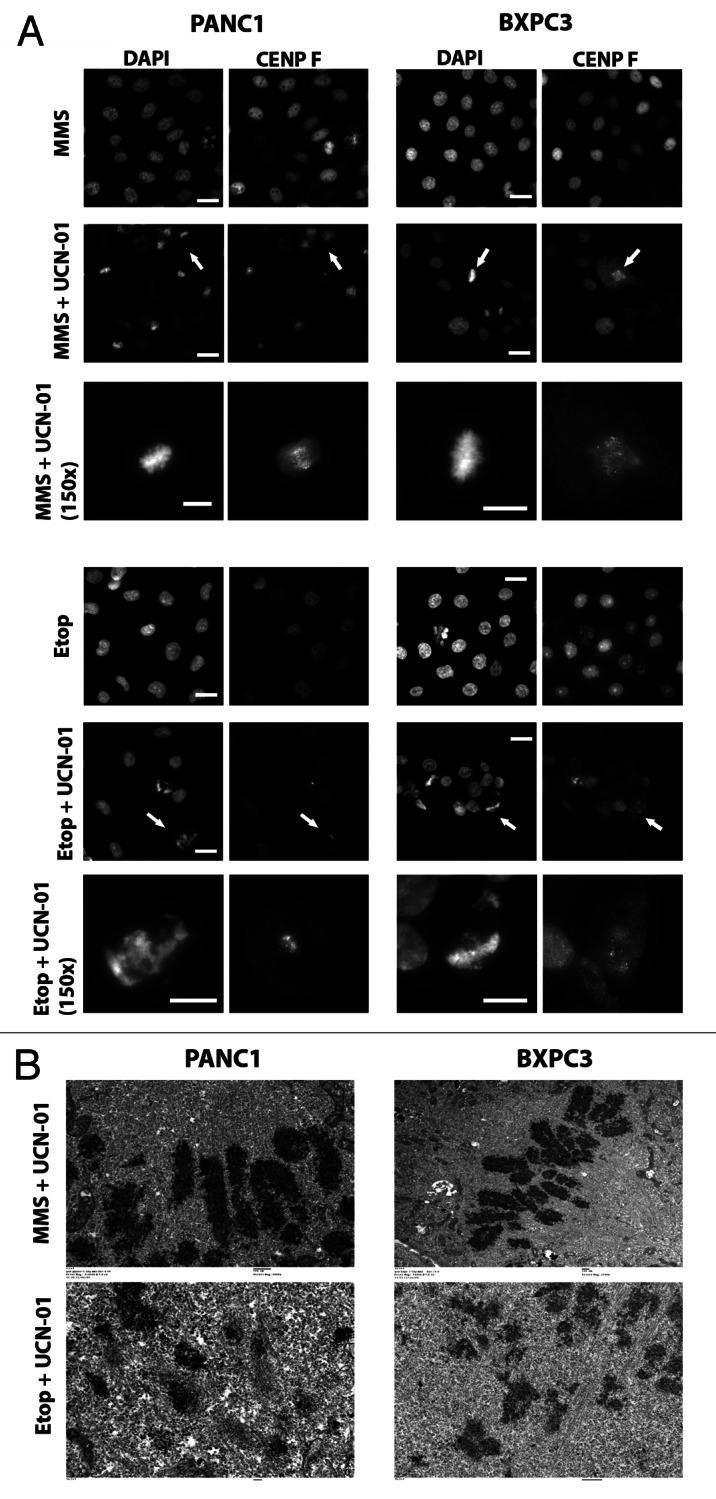
Figure 4. Variable response of cell lines of S phase checkpoint override. (A) PANC1 and BXPC3 cells were synchronized and treated in G1 with MMS or etoposide. Sixteen hours later, cells were treated ± UCN-01 for a further 9 h. Cells were fixed and immunostained with CENP F and counterstained with DAPI. Scale bars are 20 μm. White arrows denote which mitotic figures are shown at higher magnification (150×, scale bar is 10 μm). (B) Electron microscopy micrographs of PANC1 and BXPC3 cells treated as outlined in (A and B)
Inhibitors of replication or topoisomerase II impair centromere replication
MUGs, according to the original observations,15 were a consequence of forcing cells with unreplicated centromeres to enter mitosis. Thus, we were surprised as to how topoII inhibitors, which do not appear to block DNA replication (Fig. S4), would generate MUGs when forced into mitosis. The EM images clearly show single C-shaped kinetochores, rather than a pair of kinetochores that would be expected to assemble onto replicated centromeres. The discrepancy maybe resolved if one posits that centromere replication is uniquely sensitive to topo II inhibition. Indeed, studies have shown that DNA replication can continue into G2 and that replication of centromeric DNA has been shown to occur even in metaphase of primary murine X and Y chromosomes.19 We therefore used a centromere-specific probe that hybridizes to the α-satellite DNA of chromosome 7 (CEP7) to perform fluorescence in situ hybridization (FISH) on HCT116 cells. This cell line was used, because they are a diploid cell line and thus should facilitate interpretation of the experiment. In contrast, PANC1 are highly aneuploid, which could complicate interpretation of the FISH signals if there are unknown numbers of chromosome 7. Thus, two distinct spots, each corresponding to the centromere of the chromosome 7 homologs, should be observed in cells before DNA replication. After replication, the duplicated centromeres are visible as a pair of FISH signals. As a control, we treated cells with MMS alone, which is not expected to inhibit centromere replication. Consistent with our hypothesis, we found that 41.5% of the MMS-treated cells showed duplicated centromeres. These cells were likely those in the population that were arrested in G2 after MMS treatment. After gemcitabine treatment, 88% of the cells exhibited a single FISH signal, which was consistent with unreplicated centromeres. Significantly, 67.9% of doxorubicin-treated cells exhibited a single FISH signal that indicated unreplicated centromeres, despite the fact that FACs analysis showed cells had 4N DNA content (Fig. S4A). As an aside, we also observed an increase in the percentage of abnormal looking FISH signals in MMS (~3.3-fold) and gemcitabine (~9.6-fold) treated cells when compared with doxorubicin-treated cells, which may reflect aberrant replication intermediates.
To further verify that MUGs obtained after topoII inhibition were products of impaired centromere replication (S phase event) rather than DNA strand breaks induced after replication (G2 event), cells were first arrested in G2 (post-replication) with MMS. Gemcitabine or doxorubicin was added for 1 h, followed by the addition of UCN-01 for a further 8 h. Addition of UCN-01 caused these cells to enter mitosis but, importantly, did not generate MUGs. Instead, we observed normal metaphases that were indistinguishable from metaphases seen after overriding the MMS alone G2 arrest (Fig. 5B). Thus, strand breaks caused by doxorubicin do not directly cause MUGs. Instead, doxorubicin likely impairs centromere replication, and these become MUGs upon forced entry into mitosis.
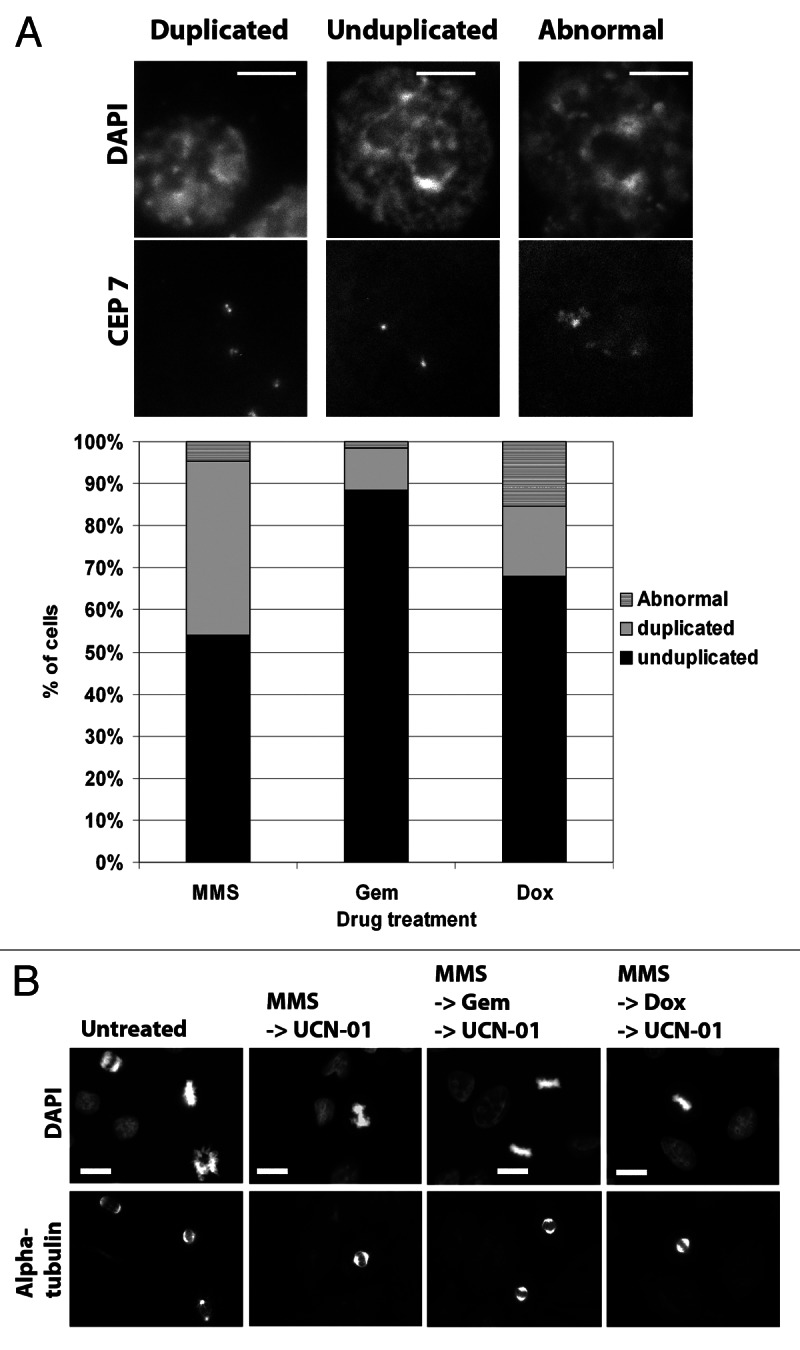
Figure 5. Topoisomerase II inhibitors impair centromere replication. (A) HCT116 cells were synchronized in G1 and treated with either MMS (200 μM), gemcitabine (100 nM) or doxorubicin (250 nM) for 16 h. Cells were then fixed and FISH performed for the centromeric probe CEP7. A minimum of 50 cells were scored for centromere number and integrity. Examples of duplicated, unduplicated or abnormal CEP7 signals are shown. Scale bar is 5 μm. Quantification of the replication status of centromeres is shown (below). (B) PANC1 cells were treated with MMS followed by either gemcitabine (100 nM) or doxorubicin (250 nM) for 1 h before the addition of UCN-01 (100 nM). Cells were fixed 9 h later and stained with α-tubulin and counter-stained with DAPI. Scale bar is 10 μm.
Checkpoint override occurs in a patient-derived pancreatic tumor
To explore whether our observations in pancreatic cancer cell lines were applicable in a clinical setting, we tested the response of EGF1 cells that were isolated from a primary human pancreatic adenocarcinoma. EGF-1 cells were treated with gemcitabine (100 nM) or doxorubicin (250 nM) for 24 h, and FACs analysis confirmed the cells were arrested in S phase or G2, respectively (Fig. 6A). UCN-01 (100 nM) was added to drug-treated cells for 9 h before determining the mitotic index. Figure 6B shows that there were no mitotic figures were observed in EGF-1 cells treated with either gemcitabine or doxorubicin alone, consistent with the cell cycle data. Addition of UCN-01 to drug-treated cells increased the mitotic index of both gemcitabine- and doxorubicin-arrested cells. Using IF, we confirmed that the mitotic EGF-1 cells observed after checkpoint override were indeed MUGs, as they showed the characteristic kinetochores that were positioned within the spindle but dissociated from the bulk of the chromatin as observed in pancreatic cancer cell lines (see Fig. 1B). These data provide clear evidence that the checkpoint override response observed with established cell lines is not only a cell line phenomenon, but can occur in primary human pancreatic tumor cells.

Figure 6. Checkpoint override occurs in a primary pancreatic tumor. (A) Cell cycle profiles of EGF1 cells treated with gemcitabine (100 nM) or doxorubicin (250 nM) as determined by FACs analysis. (B) Immunofluorescence of EGF1 cells treated with gemcitabine or doxorubicin for 24 h, followed by UCN-01 for a further 9 h. Scale bar is 20 μm. The percentage of mitotic cells observed is quantified. Error bars are SD.
Discussion
Inhibitors of Chk1 and Wee1 kinases have been validated in preclinical studies as effective chemosensitizers for gemcitabine and other DNA damaging drugs.7-12 The mechanism of sensitization is due to mitotic catastrophe that arises from forcing cells with damaged DNA into mitosis.12 In this study, we detail the mitotic defects that result from checkpoint override and assess the response of multiple cell lines. As expected, all cell lines treated with gemcitabine failed to complete DNA replication and were arrested in S phase. Interestingly, we found that not all cell lines tested were competent to prematurely enter mitosis after Chk1 was inhibited, consistent with previous studies.10,20 Of the gemcitabine-arrested cells that responded to Chk1 inhibition, their mitotic figures were highly reminiscent of those reported 30 y ago describing MUGs in CHO cells.15 Using a combination of immunofluorescence and electron microscopy, we confirmed that forcing gemcitabine-arrested cells into mitosis with Chk1 inhibitors generated the MUGs. The characterization of MUGs in human cell lines has been limited due to the apparent inefficiency of human cells to undergo MUGs. Indeed, using hydroxyurea followed by caffeine, it was shown that cells from rat, hamster and deer could effectively undergo MUGging, while murine and human (Hela) cell lines could not.16 However, a Hela cell line that stably expressed gfp:centrin was reported to spontaneously generate MUGs at low frequency after exposure to hydroxyurea for ~40 h. Whether addition of Chk1 inhibitors enhanced MUGs frequency was not tested.17 Here we describe a simple drug combination that reliably generates a high percentage of MUGs in a variety of human cell lines. Drugs such as gemcitabine, thymidine, cisplatin or aphidicolin all activate the S phase checkpoint and block DNA synthesis in a Chk1-dependent manner, and pharmacologically inhibiting Chk1 forces arrested cells to prematurely enter mitosis and to generate MUGs. Given the number of previously published studies using Chk1 inhibitors such as UCN-01,11-13,20 EXEL-9844,21 PF-004777367 and SCH90077622 in combination with DNA damaging agents, the mitotic catastrophe observed in those studies likely resulted in fragmentation of the kinetochore/centromere complex.
One of the defining features of the MUGs in CHO cells was detached kinetochores that had assembled onto unreplicated centromeric chromatin and had somehow physically separated from the bulk of the chromatin.15 These kinetochore fragments were found to lie within the bipolar spindle, while the rest of the chromatin was excluded from the spindle. Our studies using the comet assay and metaphase spreads confirmed that the chromatin in MUGs was severely fragmented. Furthermore, ultrastructral analysis using IF and EM demonstrated dissociation of the unreplicated centromere, with its assembled kinetochore complex, from the bulk of the chromatin. A study using CHO cells demonstrated that microtubules were required for MUGs formation, suggesting that microtubules physically ripped the centromere/kinetochore complex away from the chromatin.15 However, we were able to generate MUGs both in the presence and absence of microtubules. We speculate that chromatin condensation that occurs during mitotic entry may generate torsional forces that contribute to fragmentation. Another plausible mechanism is that inhibition of Chk1 by UCN-01 leads to DNA breakage,23 which could putatively force cells into mitosis with broken DNA, manifesting as MUGs. Our studies show that not all cells were able to override the S phase checkpoint when Chk1 was inhibited. BxPC3 and CFPAC cells that were unable to override an S phase checkpoint arrest were able to override a G2 checkpoint arrest, despite the fact that Chk1 is activated in both arrest points.20 Using FACs and IF, we confirmed that the extent of DNA damage and cell cycle perturbations induced by gemcitabine, doxorubicin and MMS was similar in between PANC1 and BxPC3 cells (Fig. S5). Furthermore, BXPC3 cells failed to override a gemcitabine-induced arrest when UCN-01 was increased by 10-fold (1,000 nM, data not shown). Given that 100 nM UCN-01 was sufficient to force BxPC3 cells into mitosis from doxorubicin-induced arrest, their inability to override a gemcitabine-induced arrest maybe due to the presence of a cdk inhibitor that is not directly regulated by Chk1.
Although all cell lines tested were able to override a G2 checkpoint (5/5 cell lines tested) the type of damage induced led to different outcomes. Cells treated with the alkylator MMS overcame their G2 arrest after addition of UCN-01. Time-lapse studies showed that these cells progressed normally through mitosis with no significant delays or evidence of lagging chromosomes. In contrast, all cells lines forced into mitosis after treatment with the topoII inhibitors exhibited fragmented chromosomes that included dissociated centromere/kinetochore complex as seen in MUGs. These observations argue the type of damage induced by MMS has a different effect on DNA replication than either of the topoII inhibitors, leading to normal mitosis in cells treated with MMS, as opposed to the formation of MUGs in cells treated with doxorubicin or etoposide. Indeed, MMS is not known to generate strand breaks that are detected by yH2AX staining.24 Furthermore, in agreement with this notion are our studies using bendamustine (BDM), a bi-functional molecule that can act as an alkylator or anti-metabolite. When cells were treated with 50 μM BDM they arrested in G2 upon UCN-01 addition cells prematurely entered and exited mitosis displaying normal chromosome integrity. However, when cells were treated with 200 μM BDM, a concentration that induces S phase arrest, and thus unreplicated DNA/centromeres, forced entry into mitosis generated mitotic figures consistent with the generation of MUGs.25 The simplest explanation for the overt difference in chromosome integrity after cells are treated with MMS vs. treatment with topoII inhibitors is that topoII is critical for proper centromere replication. Indeed, using centromeric FISH probes, we found that cells arrested in G2 by topoII inhibitors, but not MMS, exhibited a single FISH spot that showed unreplicated centromeres. In accordance, topoII is implicated in the establishment of the centromere/ kinetochore structure.27 How inhibition of topoII blocks centromere replication remains unclear, although studies have shown centromere replication in various organisms can occur from late S through mitosis. Our findings strongly suggest that a defining feature of mitotic catastrophe is fragmentation of the centromere. This results in acentric genomes that likely account for the mechanism of chemosensitization by Chk1 inhibitors.
From a translational standpoint, our studies of different cell lines show that the choice of DNA damaging agent used for therapy is a critical determinant in considering the use of checkpoint inhibitors. We propose that therapies using DNA damaging agents that impair centromere replication and arrest cells in G2, such as etoposide or doxorubicin, in conjunction with Chk1 inhibitors would be the most effective strategy as compared with those based on gemcitabine, where variable responses are seen. Our finding that the checkpoint override ability of a pancreatic cancer cell line, EGF-1, obtained from a patient undergoing therapy, was greater with doxorubicin as compared with gemcitabine supports this notion. In the future, we plan to expand the number of cell lines tested as well as test cancer from other organs to see whether this observation holds true.
Materials and Methods
Reagents
UCN-01 was generously provided by Kyowa Hakko Kirin Co., Ltd. and the National Cancer Institute, NIH. The following reagents were obtained from Sigma: methyl methanesulfonate, doxorubicin, etoposide and cisplatin. Gemcitabine was obtained from the FCCC pharmacy.
Cell culture
Cell lines were obtained from ATCC and banked at Fox Chase Cancer Center (FCCC) until use. Mycoplasma testing was conducted at FCCC prior to studies. HeLa, PANC1, BxPC3, Mia PaCa II, CFPAC, RPE1-hTERT cells were grown in DMEM supplemented with 10% FBS, 2 mM glutamine and 1% penicillin, streptomycin and kanamycin (PSK). HCT116 cells were grown in RPMI supplemented with 10% FBS, 2 mM glutamine and 1% PSK. All cells were maintained at 37°C, 5% CO2.
EGF-1 cells were derived by Igor Astsaturov MD, PhD at FCCC and were provided as an F2 passage pancreatic cancer tumorgraft-derived cell line. Cells were grown in DMEM supplemented with 15% FBS, 2 mM glutamine and 1% PSK, non-essential amino acids and sodium pyruvate. All experiments were conducted on p8-15 cells.
Time-lapse video microscopy
Cells stably expressing H2B:gfp were seeded into 6 well-plates and thymidine (2 mM) added for 18 h to block cells at the G1/S boundary. Thymidine was washed out, and 18 h later, cells were treated with the indicated drugs. The concentration of gemcitabine (100 nM) and UCN-01 (100 nM) was the same for all studies. The concentration of gemcitabine was biologically relevant, as it was sufficient to induce DNA damage and cell cycle arrest.25 The concentration of UCN-01 was determined previously to induce checkpoint override13,25 and not to have adverse effects on cell viability.25 Cells were placed into a heated chamber and using a Nikon TE2000S microscope (Nikon) controlled by Metamorph software (Molecular Devices); brightfield and fluorescent images were taken every 5 min for up to 48 h. Images were compiled into movies, which were used to track fates of individual cells. A minimum of 100 cells were counted for each movie, and movies were conducted at least three times. For montages, selected frames representing different cell morphologies were chosen.
Metaphase spreads
Mitotic PANC1 cells were collected from untreated and gemcitabine (100 nM) + UCN-01 (100 nM) treatments. Cells for metaphase spreads were prepared as outlined in reference 27 and assessed as described previously.25 Images shown are representative of those observed.
Immunofluorescence
Cells were seeded onto coverslips 24 h prior to drug treatment. Cells were synchronized with thymidine prior to drug treatment. Following drug treatments, cells were fixed and stained as previously described.25 Commercial antibodies to α-tubulin (Sigma-Aldrich), Plk (Santa Cruz Biotechnology, Inc.), phospho-H3 and γ-H2AX (Upstate, now EMD Millipore) were used. Antibodies to human CENP-F and BubR1 were generated and used as previously described.28,29 CAP-G, -G2, -H and -H2 were kindly provided by K. Yokomori (University of California). Alexa Fluor-conjugated secondary antibodies (488, 555, 647) (Invitrogen) were used at a final concentration of 1 μg/ml. Slides were counterstained with DAPI. Images were captured using a 40× or 100× objective mounted on an inverted microscope (Eclipse TE2000S; Nikon) with a charge-coupled device camera (Photometrics Cascade 512F; Roper Scientific). Using Metamorph software (Molecular Devices) stacks were taken at 0.25–1 μm and images presented as maximum projections.
Comet assay
Drug-treated cells were collected via trypsinization or mechanical shake off to isolate mitotic cells. Cells were embedded in low-melting point agarose and spotted onto Trevigen® Comet slides. Slides were placed into lysis solution (NaCl 146.1 g/L, EDTA 37.2 g/L pH10, Tris 1.21 g/L, Sarkosyl 10 g/L, Triton X-100 1%) for 1 h at 4°C, placed into alkali solution (NaOH 12 g/L, EDTA 2 ml of 0.25 M pH8) for 1 h at 4°C before electrophoresis at 25 V for 15 min. Slides were fixed with 70% ethanol before being air-dried. For visualization of comets, slides were stained with Sybr Green (Molecular Probes) and observed and captured on an inverted microscope (Eclipse TE2000S; Nikon) with a charge-coupled device camera (Photometrics Cascade 512F) using a 20× objective (Nikon). A minimum of 70 comets were analyzed from each treatment using TriTek CometScore™ software. The Olive moment, defined as the product of the tail length and the fraction of total DNA in the tail, was chosen to determine the extent of fragmented DNA.
Fluorescent in situ hybridization for centromere replication assay
HCT116 cells were synchronized with thymidine (2 mM) for 20 h before being washed out. Twelve hours after washout, when cells were in G1, cells were treated with MMS (200 μM), doxorubicin (250 nM) or gemcitabine (100 nM) for an additional 18 h. Slides were then placed into denaturant solution (70% formamide/2% SSC) followed by rehydration in ethanol. CEP 7 SpectrumGreen probe (Abbott; Vysis CEP 7) to detect α-satellite DNA of chromosome 730 was denatured and used to hybridize to cells on the slides. Following hybridization, slides were washed and counterstained with DAPI. Cells were examined on an inverted TE2000 epifluorescence microscope (Nikon). For analysis, DAPI was used to detect interphase cells and the FITC channel used to indentify unduplicated, duplicated or abnormal centromeres. A minimum of 100 cells per drug treatment were counted and scored as shown.
Electron microscopy
Cells were seeded into 6 cm dishes, synchronized with a double thymidine block (2 mM), washed and treated with gemcitabine (100 nM), MMS (200 μM) or doxorubicin (250 nM) for 18 h. Cells were treated with UCN-01 (100 nM) for an additional 9 h to obtain aberrant mitotic cells. Cells were fixed with glutaraldehyde solution (3%, pH 7.4). Mitotic cells were identified and processed for electron microscopy as previously described.31
Biochemical purification of MUGs
PANC1 cells were infected with pLenti GFP-CenpA and were treated with gemcitabine (100 nM) followed by UCN-01 (100 nM) to generate MUGs, which were collected by mechanical disruption. Cells were lysed using 10–12 strokes of ice-cold dounce homogenizer in 3.75 mM Tris-7.4, 20 mM KCl, 0.5 mM EDTA with 0.1% digitonin and 0.5 mM DTT and spun at 300 g for 5 min at 4°C. Low-speed supernatant was collected and subjected to high-speed centrifugation through 20–50% sucrose gradients prepared in 10 mM Tris7.4, 150 mM KCl and 2 mM MgCl2. The gradients were centrifuged at 100,000 g in a Beckman SW40 rotor for 18 h at 4°C. Gradient fractions were collected, TCA precipitated and subjected to western blotting with indicated antibodies. Sucrose gradient fractions were also fixed in 3% paraformaldehyde, and centrifuged through 10 ml of filtered buffer onto poly-lysine coated coverslips placed on glass chucks in the bottom of 30 ml Corex tubes. Tubes were centrifuged in a Sorvall HB4 swinging bucket rotor for 30 min at 25,000 g at 25°C. Coverslips containing pelleted fractions were removed from tubes, post-fixed with 3% paraformaldehyde and then processed for immunofluorescence microscopy.
Western blots of gradient fractions were probed with antibodies against Bub3, Mis12 and CenpA. Immunofluorescence of fraction 4 and a negative control coverslip (with no fraction added) was performed with antibodies against Aurora B and Mis12. Secondary antibodies were Alexa 555 anti-mouse and Alexa 647 anti-rabbit. DNA was stained with DAPI.
Supplementary Material
Acknowledgments
We wish to thank the NCI/Kyowa Hakko Kirin Co., Ltd for the use of UCN-01 for these studies. Hela GFP-CENP A cells, GFP and CENP-A antibodies were kindly provided by Dr Ben Black, University of Pennsylvania. We also thank the following facilities at Fox Chase Cancer Center for providing assistance with equipment and technical advice: Imaging Facility, Flow Cytometry Facility and Tissue Culture Facility. This work was supported in part by NIH GM086877, core grant CA06927, DoD OC100172 and Appropriation from Common Wealth of PA, the PA CURE and the Greenburg Foundation (T.J.Y.), and the Plain and Fancy Fellowship, FCCC (N.B.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/24740
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24740
References
- 1.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 2.Kawabe T. G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther. 2004;3:513–9. [PubMed] [Google Scholar]
- 3.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–39. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furnari B, Blasina A, Boddy MN, McGowan CH, Russell P. Cdc25 inhibited in vivo and in vitro by checkpoint kinases Cds1 and Chk1. Mol Biol Cell. 1999;10:833–45. doi: 10.1091/mbc.10.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bucher N, Britten CD. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br J Cancer. 2008;98:523–8. doi: 10.1038/sj.bjc.6604208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lau CC, Pardee AB. Mechanism by which caffeine potentiates lethality of nitrogen mustard. Proc Natl Acad Sci USA. 1982;79:2942–6. doi: 10.1073/pnas.79.9.2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blasina A, Hallin J, Chen E, Arango ME, Kraynov E, Register J, et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol Cancer Ther. 2008;7:2394–404. doi: 10.1158/1535-7163.MCT-07-2391. [DOI] [PubMed] [Google Scholar]
- 8.Prevo R, Fokas E, Reaper PM, Charlton PA, Pollard JR, McKenna WG, et al. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol Ther. 2012;13:1072–81. doi: 10.4161/cbt.21093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012;2:524–39. doi: 10.1158/2159-8290.CD-11-0320. [DOI] [PubMed] [Google Scholar]
- 10.Morgan MA, Parsels LA, Parsels JD, Mesiwala AK, Maybaum J, Lawrence TS. Role of checkpoint kinase 1 in preventing premature mitosis in response to gemcitabine. Cancer Res. 2005;65:6835–42. doi: 10.1158/0008-5472.CAN-04-2246. [DOI] [PubMed] [Google Scholar]
- 11.Monks A, Harris ED, Vaigro-Wolff A, Hose CD, Connelly JW, Sausville EA. UCN-01 enhances the in vitro toxicity of clinical agents in human tumor cell lines. Invest New Drugs. 2000;18:95–107. doi: 10.1023/A:1006313611677. [DOI] [PubMed] [Google Scholar]
- 12.Tse AN, Schwartz GK. Potentiation of cytotoxicity of topoisomerase i poison by concurrent and sequential treatment with the checkpoint inhibitor UCN-01 involves disparate mechanisms resulting in either p53-independent clonogenic suppression or p53-dependent mitotic catastrophe. Cancer Res. 2004;64:6635–44. doi: 10.1158/0008-5472.CAN-04-0841. [DOI] [PubMed] [Google Scholar]
- 13.Vogel C, Hager C, Bastians H. Mechanisms of mitotic cell death induced by chemotherapy-mediated G2 checkpoint abrogation. Cancer Res. 2007;67:339–45. doi: 10.1158/0008-5472.CAN-06-2548. [DOI] [PubMed] [Google Scholar]
- 14.Parsels LA, Morgan MA, Tanska DM, Parsels JD, Palmer BD, Booth RJ, et al. Gemcitabine sensitization by checkpoint kinase 1 inhibition correlates with inhibition of a Rad51 DNA damage response in pancreatic cancer cells. Mol Cancer Ther. 2009;8:45–54. doi: 10.1158/1535-7163.MCT-08-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brinkley BR, Zinkowski RP, Mollon WL, Davis FM, Pisegna MA, Pershouse M, et al. Movement and segregation of kinetochores experimentally detached from mammalian chromosomes. Nature. 1988;336:251–4. doi: 10.1038/336251a0. [DOI] [PubMed] [Google Scholar]
- 16.Balczon RC. Overexpression of cyclin A in human HeLa cells induces detachment of kinetochores and spindle pole/centrosome overproduction. Chromosoma. 2001;110:381–92. doi: 10.1007/s004120100157. [DOI] [PubMed] [Google Scholar]
- 17.O’Connell CB, Loncarek J, Kaláb P, Khodjakov A. Relative contributions of chromatin and kinetochores to mitotic spindle assembly. J Cell Biol. 2009;187:43–51. doi: 10.1083/jcb.200903076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rao PN, Johnson RT. Mammalian cell fusion: studies on the regulation of DNA synthesis and mitosis. Nature. 1970;225:159–64. doi: 10.1038/225159a0. [DOI] [PubMed] [Google Scholar]
- 19.Madan K, Allen JW, Gerald PS, Latt SA. Fluorescence analysis of late DNA replication in mouse metaphase chromosomes using BUdR and 33258 Hoechst. Exp Cell Res. 1976;99:438–44. doi: 10.1016/0014-4827(76)90604-2. [DOI] [PubMed] [Google Scholar]
- 20.Rodríguez-Bravo V, Guaita-Esteruelas S, Salvador N, Bachs O, Agell N. Different S/M checkpoint responses of tumor and non tumor cell lines to DNA replication inhibition. Cancer Res. 2007;67:11648–56. doi: 10.1158/0008-5472.CAN-07-3100. [DOI] [PubMed] [Google Scholar]
- 21.Matthews DJ, Yakes FM, Chen J, Tadano M, Bornheim L, Clary DO, et al. Pharmacological abrogation of S-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo. Cell Cycle. 2007;6:104–10. doi: 10.4161/cc.6.1.3699. [DOI] [PubMed] [Google Scholar]
- 22.Montano R, Chung I, Garner KM, Parry D, Eastman A. Preclinical development of the novel Chk1 inhibitor SCH900776 in combination with DNA-damaging agents and antimetabolites. Mol Cancer Ther. 2012;11:427–38. doi: 10.1158/1535-7163.MCT-11-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Syljuåsen RG, Sørensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, et al. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol. 2005;25:3553–62. doi: 10.1128/MCB.25.9.3553-3562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lundin C, North M, Erixon K, Walters K, Jenssen D, Goldman ASH, et al. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 2005;33:3799–811. doi: 10.1093/nar/gki681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beeharry N, Rattner JB, Bellacosa A, Smith MR, Yen TJ. Dose dependent effects on cell cycle checkpoints and DNA repair by bendamustine. PLoS ONE. 2012;7:e40342. doi: 10.1371/journal.pone.0040342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rattner JB, Hendzel MJ, Furbee CS, Muller MT, Bazett-Jones DP. Topoisomerase II alpha is associated with the mammalian centromere in a cell cycle- and species-specific manner and is required for proper centromere/kinetochore structure. J Cell Biol. 1996;134:1097–107. doi: 10.1083/jcb.134.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henegariu O, Heerema NA, Lowe Wright L, Bray-Ward P, Ward DC, Vance GH. Improvements in cytogenetic slide preparation: controlled chromosome spreading, chemical aging and gradual denaturing. Cytometry. 2001;43:101–9. doi: 10.1002/1097-0320(20010201)43:2<101::AID-CYTO1024>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 28.Jablonski SA, Chan GK, Cooke CA, Earnshaw WC, Yen TJ. The hBUB1 and hBUBR1 kinases sequentially assemble onto kinetochores during prophase with hBUBR1 concentrating at the kinetochore plates in mitosis. Chromosoma. 1998;107:386–96. doi: 10.1007/s004120050322. [DOI] [PubMed] [Google Scholar]
- 29.Liu ST, Hittle JC, Jablonski SA, Campbell MS, Yoda K, Yen TJ. Human CENP-I specifies localization of CENP-F, MAD1 and MAD2 to kinetochores and is essential for mitosis. Nat Cell Biol. 2003;5:341–5. doi: 10.1038/ncb953. [DOI] [PubMed] [Google Scholar]
- 30.Casorzo L, Corigliano M, Ferreo P, Venesio T, Risio M. Evaluation of 7q31 region improves the accuracy of EGFR FISH assay in non small cell lung cancer. Diagn Pathol. 2009;4:4. doi: 10.1186/1746-1596-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rattner JB, Wang T. Kinetochore formation and behaviour following premature chromosome condensation. J Cell Sci. 1992;103:1039–45. doi: 10.1242/jcs.103.4.1039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.