

Abstract
Fanconi anemia (FA) is a human syndrome characterized by genomic instability and increased incidence of cancer. FA is a genetically heterogeneous disease caused by mutations in at least 15 different genes; several of these genes are conserved in the yeast Saccharomyces cerevisiae. Elg1 is also a conserved protein that forms an RFC-like complex, which interacts with SUMOylated PCNA. The mammalian Elg1 protein has been recently found to interact with the FA complex. Here we analyze the genetic interactions between elg1Δand mutants of the yeast FA-like pathway. We show that Elg1 physically contacts the Mhf1/Mhf2 histone-like complex and genetically interacts with MPH1 (ortholog of the FANCM helicase) and CHL1 (ortholog of the FANCJ helicase) genes. We analyze the sensitivity of double, triple, quadruple and quintuple mutants to methylmethane sulfonate (MMS) and to hydroxyurea (HU). Our results show that genetic interactions depend on the type of DNA damaging agent used and show a hierarchy: Chl1 and Elg1 play major roles in the survival to these genotoxins and exhibit synthetic fitness reduction. Mph1 plays a lesser role, and the effect of the Mhf1/2 complex is seen only in the absence of Elg1 on HU-containing medium. Finally, we dissect the relationship between yeast FA-like mutants and the replication clamp, PCNA. Our results point to an intricate network of interactions rather than a single, linear repair pathway.
Keywords: Fanconi anemia, Elg1, PCNA, genome stability, DNA damage
Fanconi anemia (FA) is a genomic instability syndrome characterized by bone marrow failure, developmental abnormalities and increased incidence of cancers.1 Clinically, FA is very heterogeneous; patients exhibit congenital abnormalities including skeletal defects and hypopigmentation, bone marrow failure and early onset of cancer.2 This wide range of clinical findings can be explained by the fact that FA is a chromosomal instability disorder, and cells from FA patients accumulate DNA damage at an increased rate. Unrepaired and misrepaired DNA damage can randomly give rise to mutations and translocations that result in blood cancer and solid tumors, or may sometimes activate pro-apoptotic pathways leading to depletion of hematopoietic stem cells. Thus, the same population of cells may sometimes be hyper-represented (as cancerous cells) or lacking (causing anemia).3
Cells of FA patients are hypersensitive to a class of DNA damaging agents that create DNA interstrand crosslinks (ICLs), such as diepoxybutane (DEB), cisplatin or mitomycin C (MMC).4 ICLs bind to both strands of DNA, preventing DNA unwinding and thus blocking both DNA replication and transcription. The toxicity of crosslinking agents to dividing cells is currently being exploited as an anticancer therapeutic methodology. However, FA patients are extremely sensitive to these agents, and this fact makes the treatment of their cancers extremely difficult.
FA is a genetically heterogeneous disease, caused by mutations in at least 15 different genes (although the total number of genes involved is likely to increase). The gene products of all these genes are believed to function in a common DNA repair signaling pathway, which closely cooperates with other DNA repair proteins for resolving DNA ICLs during replication. Several of the FA proteins assemble into a large nuclear E3 ubiquitin ligase complex termed the “FA core complex.” Upon DNA damage, the core complex causes the monoubiquitination of FANCD2 and FANCI.5 The monoubiquitinated FANCD2/FANCI heterodimer was shown to play multiple roles in the pathway6 and to functionally interact with downstream FA proteins such as FANCD1 (or BRCA2), FANCN, FANCJ and their associated protein, BRCA1. In addition to these core FA proteins, there are several FA pathway-associated proteins whose functions are critical to the pathway; however, mutations have not yet been found in the corresponding genes in FA patients. These include Fanconi-associated proteins 24 and 100 (FAAP24 and FAAP100), FANCM-associated histone fold protein 1 (MHF1) and 2 (MHF2),7-9 FAN1,10 USP1 and UAF1.11 All of these proteins are required for efficient activation of FANCD2 monoubiquitination. The activated FA pathway must be inactivated for completion and recycling of the functional pathway, and this event is regulated by the USP1/UAF1 deubiquitinating enzyme complex, which deubiquitinates FANCD2 and FANCI. Disruption of the USP1/UAF1 complex leads to DNA repair defects similar to those of mutants in the FA pathway.12,13 ELG1 (enhanced levels of genomic instability) is a new addition to this list: it was recently found associated with USP1/UAF1 and may play a role in the completion of the FA activity.14,15
The current models propose that the activity of the FANC pathway allows a fork regression or lesion bypass mechanism, followed by a homologous recombination (HR) event, to restore the integrity of the replication fork. Indeed, FANCM and FANCJ have helicase activity, whereas two FANC proteins (FANCD1/BRCA2, FANCN/PALB2) participate in HR. Monoubiquitylated FANCD2-FANCI is also required for unhooking and translesion bypass of ICLs in a cell-free system.6 Recent work suggests that this complex is also required for histone management during DNA repair.16
During DNA replication the activity of the DNA polymerases may be impaired by the presence of secondary structures, bound proteins or DNA lesions; this may lead to stalling or even collapse of replication forks. In response, cellular mechanisms are activated that arrest cell cycle progression, induce DNA repair and restore replication.17,18 These repair mechanisms act on lesions to promote their repair and to prevent them from being converted into fatal genomic rearrangements. In some cases, cells may overcome the damage without actually repairing it; the post-replication repair (PRR) pathway19 is such a mechanism. Genetic analysis has uncovered two main mechanisms of PPR: an error-prone pathway employs damage-tolerant DNA polymerases capable of synthesizing DNA past the damaged template. These are usually called trans-lesion synthesis (TLS) polymerases.20 In addition, an error-free mechanism bypasses the lesion by utilizing the information encoded by the undamaged sister chromatid (possibly by some sort of template switch). This mechanism is strikingly similar to the FA pathway: upon fork stalling, a complex series of signals leads to the modification of the replication clamp, PCNA, by ubiquitin. As in FA, most genes in the PRR pathway encode E2 and E3 enzymes required for the pathway regulation.19 As in FA, helicases and a yet-uncharacterized HR event are needed to bypass the lesion.
In addition to its modification by ubiquitin, PCNA can also be modified by SUMO, mainly at the K164 residue. This modification takes place at each S-phase; in addition, SUMOylation is observed following DNA damage; this modification requires the SUMO-specific E2 Ubc9 and the SUMO ligase Siz1.21 In addition, PCNA is SUMOylated at K127. SUMOylation of PCNA strongly affects the choice of pathway used for processing the lesions. SUMOylation seems to prevent homologous recombination, favoring ubiquitin-dependent lesion bypass.22,23 Thus, mutants that prevent SUMOylation of PCNA suppress the sensitivity of PRR mutants to DNA damaging agents.
The ELG1 gene was identified as a yeast mutant that causes enhanced levels of genomic instability.24-26 Deletion of ELG1 leads to increased recombination levels25,27 as well as elevated levels of chromosome loss25,28 and gross chromosomal rearrangements.28 elg1 mutants also exhibit elongated telomeres29 and increased levels of Ty transposition.30 Elg1 function is thus clearly required for maintaining genome stability during normal growth, and its absence has severe genetic consequences. The human Elg1 ortholog, which physically interacts with USP1/UAF1 (see above) has been recently shown to play an important role in maintaining genome stability in S phase.31 Targeted gene knockdown of ELG1 resulted in spontaneous foci formation of γ-H2AX, 53BP1 and phosphorylated-ATM that usually mark chromosomal breaks, as well as increased levels of recombination and chromosomal aberrations, such as chromosomal fusions and inversions.31 Mice with mutations in ELG1 are embryonic lethal, and heterozygotes show an increased level of tumor formation. Moreover, ELG1 has been shown to be mutated in 5% of human sporadic endometrial tumors tested,32 underscoring its important role in tumor suppression.
The yeast Elg1 interacts physically and genetically with PCNA in a manner that depends on PCNA modification.33 Deletion of the Elg1 gene suppresses the sensitivity to DNA damaging agents of mutants of the PRR (error-free branch). The sensitizing activity seems to be the unloading of SUMOylated PCNA molecules from the fork; indeed, Elg1 exhibits preferential affinity for SUMOylated PCNA, as demonstrated in vitro with purified proteins.33 This interaction is mediated by three SUMO-interacting motifs (SIM) and a PCNA-interacting protein (PIP) box close to the N terminus of Elg1. The interaction with PCNA is evolutionarily conserved.31 Thus, in both yeast and in humans, Elg1 plays a central role in lesion bypass.
The FA pathway is conserved in all mammals. Several orthologs of FA proteins can be found in yeast. These genes include MPH1 (FANCM), CHL1 (FANCJ), MHF1 (MHF1) and MHF2 (MHF2). Mph1 and Chl1 encode DNA helicases with roles in genome maintenance.34-36 The Mhf1 and Mhf2 are recent additions to this family.9 Their biochemical function is still unknown.
Here we investigate the physical and genetic interactions between the yeast Elg1 protein and the other members of the FA pathway in yeast. Our results show complex genetic relations, which are dependent on the type of DNA damage analyzed.
Results
We performed a screen for proteins that interact with Elg1 in a yeast-two hybrid assay. For this purpose we divided the Elg1 protein into an N-terminal, a C-terminal and a central AAA domain.24-26 This last domain carries most of the RFC-like motifs of the protein. Among the clones that exhibited an interaction with the AAA region of Elg1 (aas 235–514), we identified two independent clones containing the then unknown ORF YOL086W-A, now re-named Mhf18,9 (Fig. 1A). Mhf1 and Mhf2 encode two small conserved proteins that were recently found to interact in humans with FANCM, and in yeast with its ortholog, Mph1.37 The Mph1 protein also showed a positive result in the yeast two hybrid assay when tested against Elg1, although the signal was weaker (Fig. 1A).
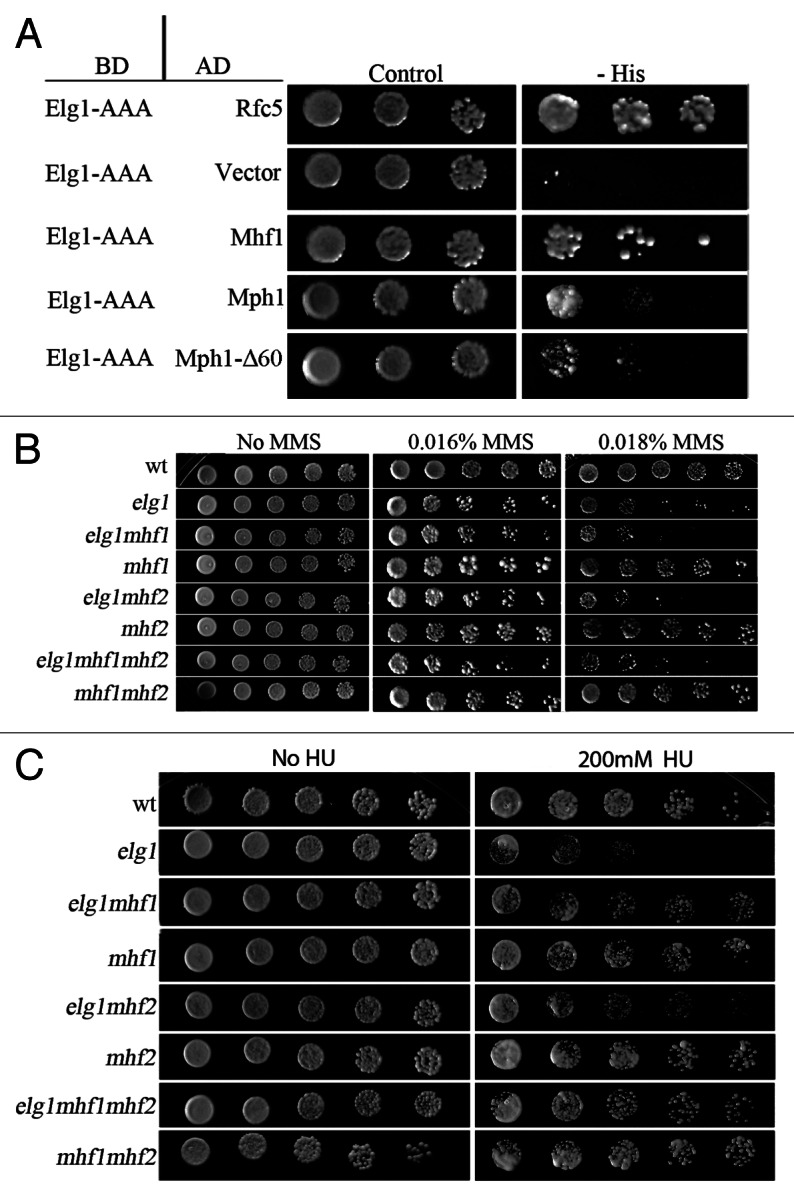
Figure 1. Interactions between Elg1 and Mhf1/2.(A) Yeast-two hybrid experiment. A plasmid carrying the AAA domain of Elg1 (aas 235 to 514) fused to GAL4 DNA binding domain (BD) shows interactions (growth on plates lacking histidine) with the following proteins fused to GAL4 activating domain (AD): Rfc5, Mhf1, Mph1 and Mph1-Δ60. (B) Genetic interactions between elg1Δ, mhf1Δ and mhf2Δ mutants on MMS (drop test). Serial 10-fold dilutions are shown on plates with increasing concentrations of MMS. (C) Genetic interactions between elg1Δ, mhf1Δ and mhf2Δ mutants on HU (drop test).
Mutations in ELG1 result in mild sensitivity to the DNA methylating agent methyl methane sulfonate (MMS) and to hydroxyurea (HU), an inhibitor of the enzyme ribonucleotide reductase. We tested whether mhf1 and mhf2 also showed sensitivity or resistance to DNA damaging agents, and whether they interact with elg1Δ. Figure 1B shows that mutation of MHF1, MHF2 or their combination did not have an effect on the sensitivity of the cells to MMS. In addition, no additive sensitivity could be observed when each gene, or both together, were deleted in a elg1Δ background (Fig. 1B).
The response to HU was very different: whereas mhf mutants (individually or combined) did not show sensitivity, mutation in either MHF1, MHF2 or both, suppressed the sensitivity of elg1Δ to HU (Fig. 1C). This result indicates that the sensitivity to HU of the elg1Δ mutant depends on the activity of these proteins.
Interactions between mph1Δ and elg1Δ
Mutations in ELG1 and MPH1 cause a mild sensitivity to MMS. The biochemical function controlled by these genes are very different: whereas Elg1 may function as an unloader of SUMOylated PCNA,38 Mph1 has been suggested to act as an helicase with a role in D-loop stabilization.35 The double mutant elg1Δ mph1Δ shows higher sensitivity to this alkylating agent, indicating that Elg1 and Mph1 participate in alternative repair pathways (Fig. 2A). Inactivation of MHF1, MHF2 or both did not affect the sensitivity of each of the single mutants or of the double mutant, consistent with the idea that the Mhf proteins do not play an important role in the repair of DNA damage caused by MMS. In contrast, mutation in each of the MHF genes or in both completely suppressed the sensitivity to HU of the elg1Δ and the elg1Δ mph1Δ mutants, again stressing the importance of the Mhf1 and Mhf2 proteins in the survival to HU (Fig. 2B).

Figure 2. Genetic interactions between elg1Δ, mph1Δ, mhf1Δ and mhf2Δ mutants. (A) Drop test on MMS. (B) Drop test on HU. (C) Effect of mutants that inactivate Mph1’s helicase activity or its interactions with the Smc5/6 complex.
To try to understand better the nature of the interaction between Elg1 and Mph1, we analyzed the effect of specific mutations in MPH1 on the sensitivity to MMS of an elg1Δ mutant. The mph1-Δ60 allele deletes amino acids 751–810 of the protein, which are required for the interactions between Mph1 and the Smc5/6 complex. This complex plays a still-undefined role in a repair mechanism that involves sister chromatids.39,40Figure 2C shows that the mph1-Δ60 allele is able to complement the sensitivity of a double elg1Δ mph1Δ mutant. This implies that the interaction with Smc5/6 is not required for the complementation by the Mph1protein and suggests that the interactions between Elg1 and Mph1 are independent of the Smc5/6 complex. This result is in agreement with the fact that the mph1-Δ60-encoded protein, which does not bind Smc5/6, can still bind to Elg1 (albeit at a lower strength) (Fig. 1A). In contrast, the helicase-defective mph1-DE (D,E209–210N,Q) allele and the mph1-KQ (K113Q) allele, which affects a DEXDc conserved motif, completely abolished the ability of the protein to complement the elg1Δ mph1Δ synthetic phenotype (Fig. 2C). We thus conclude that the helicase activity of Mph1 can compensate for lack of activity of Elg1.
In contrast to the increased sensitivity to MMS, the double mutant elg1Δ mph1Δ was as sensitive as the elg1Δ mutant to HU (Fig. 2B), implying that Mph1 plays no role in the sensitivity to this drug. This result is in striking contrast to the results obtained by combining elg1Δ with mhf1Δ and mhf2Δ (Fig. 1C) and implies that Mph1 and the Mhf proteins act independently to provide resistance to HU (see “Discussion”).
Interactions between chl1Δ and elg1Δ:
Since Mph1 is the yeast ortholog of FancM, and recent results showed a connection between human Elg1 and the FA pathway,14,41 we decided to check whether there are genetic interactions between Elg1 and another member of the FA family, Chl1, the yeast ortholog of the FancJ helicase. A cross between an elg1Δ mutant and a chl1Δ strain was performed, and tetrads were dissected. Figure 3A shows that the double mutant elg1Δ chl1Δ spores formed small colonies. Indeed, growth curves confirmed that whereas the single mutant strains had doubling times similar to that of the wild type, the double mutant exhibited very slow growth, with a doubling time of 270 min, compared with 170 for elg1Δ and 148 for the chl1Δ single mutant (Fig. 3A). In addition to this synthetic loss of fitness, the two mutations displayed synergistic interactions with respect to their sensitivity to MMS (Fig. 3C) and hydroxyurea (Fig. 3D).

Figure 3. Genetic interactions between elg1Δ, chl1Δ and mhf1Δ and mhf2Δ mutants. (A) Tetrad analysis of a cross between a elg1Δ and a chl1Δ strain showing a synthetic fitness phenotype for the elg1Δ chl1Δ double mutant. Generation times were measured for six independent spores of each phenotype. (B) Tetrad analysis show no synthetic fitness interactions between chl1Δ and mutations that abolish the PCNA interacting motif (PIP), the SUMO interacting motif of Elg1 or both. (C) Drop test on MMS. (D) Drop test on HU.
As explained, the Elg1 protein contains a central AAA domain, which shows similarity to Rfc1, and unique N- and C-terminal domains. To determine which of these domains contributes to the slow-growth and the MMS hypersensitivity of the elg1Δ chl1Δ cells, we used a series of constructs containing the full-length and the truncated versions of Elg142 and examined their ability to complement the above-mentioned phenotypes (Fig. 4). All proteins were expressed at similar levels (data not shown).

Figure 4. Determining the region of Elg1 required to complement the synthetic sickness between elg1Δ and chl1Δ.(A) The 791 aa long Elg1 protein was divided into fragments and used to complement either the growth rate or the MMS sensitivity of a elg1Δ chl1Δ double mutant. (B) Generation time of a elg1Δ chl1Δ strain carrying various regions of Elg1 on a centromeric plasmid. Generation times were measured for 6 independent transformants of each plasmid. (C) Drop test on MMS of the same strains.
As expected, the full-length Elg1 protein was able to restore both rapid growth and MMS resistance to the elg1Δ chl1Δ strain. The construct lacking the first 216 amino acid residues (a.a. 216–791) was able to fully complement the growth defect and rescued almost completely the MMS sensitivity. C-terminal truncations of the last 40–60 amino acids (a.a. 216–751, a.a. 216–741 and a.a. 216–731) of the above constructs partially complemented the slow growth of the double mutants; however, these alleles allowed only limited growth on MMS. A construct with a larger C-terminal deletion (a.a. 1–519) was completely defective in rescuing both the growth defect and the MMS hypersensitivity (Fig. 4). We thus conclude that both the N- and the C-termini of Elg1 are dispensable for growth in the absence of Chl1. Consistent with these results, a cross between an elg1Δ mutant lacking either the PIP (PCNA-interacting) motif, the SIM (SUMO-interacting motif) and both, located at the N terminus,33 did not result in a synthetic phenotype when crossed to a chl1Δ mutant (Fig. 3B). We conclude that the region between amino acids 519 and 731 of Elg1 is important for viability in the absence of Chl1. Repair of MMS damage, on the other hand, requires either a functional N terminus (probably through interactions with PCNA) or a functional C terminus (which probably allows binding to a still unknown protein). Whereas the Elg1 1–731 or 216–791 constructs were able to fully complement the MMS sensitivity of a elg1Δ chl1Δ strain; the plasmid carrying only the 216–731 region was MMS-sensitive.
Interactions between elg1Δ, mph1Δ and chl1Δ
Having established that ELG1 genetically interacts with two yeast FA orthologs, MPH1 and CHL1, we were interested in determining the relationship between the three genes. We therefore analyzed the interactions between elg1Δ, mph1Δ and chl1Δ. Figure 5 shows that deleting MPH1 in the absence of Chl1 slightly sensitizes the cells to MMS, but it has no further effect on a elg1Δ chl1Δ background: the triple elg1Δ chl1Δ mph1Δ mutant is not more sensitive to MMS than the elg1Δ chl1Δ mutant, although it is more sensitive than the chl1Δ mph1Δ mutant. Thus, in the absence of Chl1, Elg1 plays a more important role than Mph1. Similar results were observed in HU: a deletion of MPH1, which by itself does not confer sensitivity to HU, sensitizes chl1Δ, but not elg1Δ cells. However, in HU the triple elg1Δ mph1Δ chl1Δ mutant strain has the same sensitivity as the mph1Δ chl1Δ or elg1Δ chl1Δ strain, which are much more sensitive than the elg1Δ mph1Δ strain (Fig. 5). Thus, Chl1 plays a pivotal role, and in its absence either Elg1 or Mph1 can take over, as two secondary pathways. Interestingly, deleting MHF1, MHF2 or both have no further effect on the resistance of these strains to either MMS or HU (Fig. 5).

Figure 5. Genetic interactions between elg1Δ, mph1Δ, chl1Δ and mhf1Δ and mhf2Δ mutants. (A) Drop test on MMS. (B) Drop test on HU.
Interactions with PCNA
Given the physical and genetic interactions of ELG1 and CHL1 with the clamp, PCNA and the emerging evidence that these three molecules are present at the replication fork, we sought to examine the functional relationships among them. PCNA is a key regulator of the response to DNA damage. Depending on the cell cycle location, type of lesion and other, still-not-understood factors, PCNA undergoes various modifications (mainly ubiquitination and SUMOylation), which lead to repair by different mechanisms. In order to map the possible roles played by PCNA in choosing between the Elg1-, Chl1- and Mph1-dependent repair pathways, we chose two widely studied alleles of PCNA (Pol30), pol30–104 and pol30-RR. The first allele43 changes an alanine in the interdomain region of PCNA, directly beneath a loop connecting two monomers (A251V) known to be required for interactions with various DNA repair and replication partners. In pol30-RR lysine 164, which can get ubiquitinated or SUMOylated, and lysine 127, which can undergo SUMOylation, are changed to arginines. Thus, in pol30-RR mutants there is no post-translational modification of PCNA. Some of the synthetic genetic interactions of elg1, such as those with mutations in the Srs2 helicase, were shown to be alleviated by this mutation.33
Tetrad analysis of a diploid strain heterozygous for elg1Δ, chl1Δ and pol30–104 showed that the elg1Δ chl1Δ pol30–104 triple mutant cells exhibited a slight increase in colony size compared with elg1Δ chl1Δ double mutant colonies (Fig. 6A), indicating that the pol30–104 allele partially rescues the growth defect of elg1Δ chl1Δ mutant cells. Growth rate measurements confirmed this observation (Fig. 6B). Interestingly, the suppression effect was specific for the double mutant: elg1Δ pol30–104 strains showed the same growth rate as elg1Δ single mutants, whereas chl1Δ pol30–104 strains grew more slowly than the single chl1Δ cells (Fig. 6B).

Figure 6. Genetic interactions between elg1Δ, mph1Δ, chl1Δ and mutants in PCNA. (A) Example of tetrads of a elg1Δ chl1Δ strain crossed to a pol30–104 and pol30-RR haploid, showing suppression/aggravation of the fitness defect. (B) Generation times were measured for six independent spores of each phenotype. (C) Drop test on MMS. (D) Drop test on HU. (E) Schematic model of the activities controlled by Elg1, Chl1 and Mph1. If, during DNA replication, a lesion halts progression of the DNA polymerase, Elg1 may allow repair by unloading the SUMOylated PCNA molecule; Mph1 can promote D-loop formation with the sister chromatid; Chl1’s helicase activity may promote fork reversion.
In contrast to these results, combining the double mutant elg1Δ chl1Δ with pol30-RR led to even slower growth, suggesting that the common function inactivated in the elg1Δ chl1Δ mutants acts in parallel to PCNA modification to take care of spontaneous DNA damage.
We next examined the sensitivities of these strains to DNA damaging agents. Consistent with previous reports,44 pol30–104 exhibited sensitivity to MMS and HU. Deletion of ELG1 had no additional effect on HU and only slightly sensitized the cells to MMS. In contrast, addition of the chl1Δ mutation caused high MMS and HU sensitivity. Interestingly, this phenotype was not affected by further mutation of ELG1 (Fig. 6C and D). These results place elg1Δ and pol30–104 in the same epistasis group, which has negative genetic interactions with chl1Δ.
In contrast to these results, pol30-RR exhibited increased sensitivity to MMS when combined with elg1Δ or chl1Δ, and even more sensitivity when the three mutants were combined (Fig. 6C). The results in HU were slightly different: elg1Δ, pol30-RR and the pol30-RR elg1Δ double mutant showed the same sensitivity to the drug, locating elg1Δ and pol30-RR in the same epistasis group. In contrast, pol30-RR and chl1Δ showed additive results. However, the triple mutant showed a dramatic decrease in resistance to HU. Thus, modification of PCNA becomes extremely important for surviving to HU exposure in the absence of both Elg1 and Chl1.
Discussion
The integrity of the genome is under continuous attack from external insults as well as a result of the normal cellular metabolism or errors that take place during DNA replication or repair. Cells have therefore evolved a large arsenal of mechanisms that help them cope with various forms of DNA damage. The FA pathway has been shown to play an important role in repairing inter-strand cross links (ICLs).4 This pathway, composed of at least 15 proteins in mammalian cells, appears to be at least partially conserved in yeast. Two recent publications indeed have shown that the yeast orthologs of human FA proteins participate in ICL repair.45,46 Interestingly, a connection was found between the yeast FA pathway and portions of the post-replication repair (PRR) pathway as well as with PCNA modifications.45 The yeast Elg1 protein has been suggested to act as an unloader of SUMOylated PCNA, and it also genetically interacts with the PRR pathway: for example, mutations in ELG1 suppress the sensitivity of rad5 mutants to DNA damaging agents.33 Here, we have analyzed the physical and genetic interactions between Elg1 and members of the FA pathway in yeast.
We have uncovered a physical interaction between Elg1 and the Mhf1 protein (Fig. 1A). Mhf1 and Mhf2 are small conserved proteins.9 Their crystal structure has recently been solved, and it shows that the two proteins form a tetramer, that resembles the histone (H3-H4)2 heterotetramer.41 Deletion of any of the two proteins, or of both, does not result in sensitivity to any DNA damaging agent tested in S. pombe9,41 or in budding yeast (this work). Indeed, in previous publications, the effect of mhf1Δ and mhf2Δ could be discerned only on the background of an srs2Δ mutant.9,41 Here, we show an additional phenotype for these mutants: they suppress the sensitivity to hydroxyurea of elg1Δ mutants (Fig. 1C).
The Mhf proteins have been shown to form a complex with the FANCM/Mph1 helicase.9,45 Consistent with this proposal, Elg1 showed physical interactions with both Mhf1 and Mph1 (Fig. 1A). However, our genetic results suggest that the Mhf proteins do not always act in a complex with Mph1. The phenotypes of mph1Δ mutants were very different from those observed in strains mutated for the MHF genes. For example, although mutations in MPH1 do not affect the sensitivity of elg1Δ mutants to HU, this sensitivity was rescued by mutations in the MHF genes. Furthermore, the rescue was independent of Mph1. These findings suggest that the Mhf proteins can function independently of Mph1 to modulate HU resistance. Moreover, mutations in MPH1 increase the sensitivity of elg1 mutants to MMS, whereas mutations in MHF1, MHF2 or both have no additional effect (Fig. 2A).
The results of our analysis show complex genetic interactions between the components of the FA pathway (Fig. 6E), which depend on the DNA damaging agent tested. Below, we discuss each of the damaging agents separately.
Methyl methanesulfonate (MMS) methylates DNA on N7-deoxyguanine and N3-deoxyadenine, and is believed to stall the replication fork. Mutations in ELG1, MPH1 or CHL1 cause a mild sensitivity to this agent, similar for all single mutants. However, when combined, a synergistic effect can be observed: elg1Δ mph1Δ and chl1Δ mph1Δ double mutants are sensitive to 0.010% MMS (Fig. 5). In contrast, elg1Δ chl1Δ mutants shows an even stronger sensitivity, being unable to form colonies on plates containing as little as 0.003% MMS. Deletion of MPH1 in this strain does not confer further sensitivity. Our results thus point to a hierarchical order between these DNA repair proteins. It is clear from our results that Chl1 is a major player and constitutes a strong alternative to Elg1: in the absence of both Chl1 and Elg1, cells are very MMS-sensitive. Moreover, under these circumstances mutations in Mph1 have no further effect (Fig. 5). These results can be explained (Fig. 6E) by assuming that there are two alternative pathways to bypass the replication stalling, one Chl1-dependent and another Elg1- (and Mph1-) dependent. The helicase activity of Chl1 could be involved in fork reversal, whereas Elg1 is necessary to remove SUMOylated PCNA from the stalled fork38. Mph1 has been implicated in D-loop formation and may contribute to a pathway of homologous recombination involving the recently synthesized sister chromatid35 (Fig. 6E). It should also be noted that both Elg1 and Chl1 have known roles in sister chromatid cohesion.47,48 Cohesion between the sisters, or some kind of interaction with the cohesin complexes, could constitute a pre-requisite for the activity of Mph1; without the activity provided either by Chl1 or by Elg1, mutations in MPH1 have no effect. Interestingly, as noted above, the Mhf proteins do not seem to play any role in the repair of MMS-caused lesions.
The picture is slightly different for hydroxyurea (HU). This drug causes inactivation of the ribonucleotide reductase complex, effectively depleting the pool of dNTPs and possibly causing fork stalling. Despite the fact that elg1 mutants have increased levels of dNTPs,49 these cells are sensitive to HU at high concentrations. Interestingly, this sensitivity depends on the presence of the Mhf proteins, as deletion of any of them or both, suppresses the HU sensitivity of elg1 strains (Fig. 1). As explained above, it has been proposed that the Mhf proteins and Mph1 form a complex. However, in contrast to the expectation from a single protein complex, a double mutant elg1 mph1 is as sensitive to HU as the single elg1 mutant (Fig. 2), whereas mutations in the MHF genes suppress the sensitivity of elg1, suggesting that only the Mhf proteins, and not Mph1, play roles in HU resistance. We thus suggest that Mhf1 and Mhf2 can form a complex with Elg1, which may control their loading or activity. In the absence of Elg1, the Mhf1/2 activity becomes toxic, and deletion of any of these two proteins alleviates the sensitivity of elg1 mutants to HU (Fig. 2B). The toxicity of the Mhf proteins could be related to their resemblance to the histone (H3-H4)2 heterotetramer,41 which may be required as a molecular decoy during DNA repair but could impede normal genomic activity if left unchecked. Interestingly, recent work has suggested that the FA pathway in mammals may play also a role in controlling histone deposition and its regulation during DNA repair.16
Mutations in CHL1 confer sensitivity to HU similar to that of the elg1Δ mutant. The two mutations showed an additive phenotype, which was not further affected by mutations in MPH1. This again suggests the existence of two parallel pathways, one ruled by Chl1 and the other by Elg1. The role of Mph1 in resistance to HU could be seen only in the absence of Chl1: the double mutant chl1Δ mph1Δ was more sensitive than the single chl1Δ strain (Fig. 5). The triple elg1Δ chl1Δ mph1Δ was not more sensitive than the elg1Δ chl1Δ double mutant, which supports the idea that Mph1 plays a role in the Chl1-independent Elg1 pathway (Fig. 6E). Fork reversal by the Chl1 helicase seems to be the preferred mechanism of replication fork re-initiation in the presence of HU, with Elg1 serving as a backup by controlling the activity of Mhf1/2. The Mph1-dependent homologous recombination sub-pathway, however, is not used much in the presence of HU, if the Chl1 pathway is active (Fig. 6E).
Our results thus show that Chl1 and Elg1 play alternative roles with respect to survival of both MMS and HU. The need for either Elg1 or Chl1 is seen not only in the sensitivity to DNA damaging agents: the elg1Δ and chl1Δ mutations exhibit a synthetic fitness defect (Figs. 3A and 4). We have investigated what region in Elg1 is responsible for the essential function in the absence of Chl1. Our results (Fig. 4A) show that neither the N terminus, which has been implicated in the interactions between Elg1 and SUMOylated proteins,38,48 nor the C terminus, which is important for its repair function (Fig. 4 and ref. 42) are necessary. The region of Elg1 defined by our studies (between aas 517 and 731) has been shown to be important for the incorporation of Elg1 into an RFC-like complex,42 suggesting that its interactions with the small Rfc subunits (Rfc2–5) are important here.
We have explored the interactions between the yeast FA pathway members and PCNA. Interestingly, the two PCNA mutations analyzed behaved in very different fashion: pol30–104 mutants, carrying the A251V substitution, showed epistatic interactions with elg1Δ with respect to the sensitivity to MMS and HU, and was able to slightly suppress the synthetic sick phenotype of elg1Δ chl1Δ double mutants. This PCNA mutant indeed shares with elg1Δ a number of synthetic genetic interactions, suggesting that either the region of PCNA affected (the inter-domain loop) is responsible for the attachment of Elg1, or, alternatively, that binding of a still unknown factor to this region is essential to carry out Elg1’s function. The suppression of the synthetic sickness of an elg1Δ chl1Δ mutant supports the second model: binding of the unknown factor may be toxic in a strain devoid of both Elg1 and Chl1; a mutation that prevents its binding alleviates the synthetic sickness.
In striking contrast, mutations in the lysines 127 and 164 of PCNA, which abrogate post-translational modifications of the clamp, exhibited increased toxicity and sensitivity to MMS. Lysine 164 can undergo both mono- and poly-ubiquitination, which direct lesion bypass by trans-lesion synthesis (mono-ubiquitination) or template-switch synthesis (poly-ubiquitination). In addition, lysine 164 (and lysine 127 as well) can also undergo SUMOylation. This modification is believed to prevent homologous recombination events at the fork (reviewed in ref. 50). Thus, in a pol30-RR mutant, the bypass pathways are abolished and recombination is unchecked. The additional synthetic sickness and sensitivity to MMS conferred to the elg1Δ chl1Δ mutant by the pol30-RR mutation suggests that the bypass pathways abolished play a central role when both Chl1 and Elg1 are inactivated; alternatively, the unchecked recombination may be toxic. Deletion of RAD52 in a elg1Δ chl1Δ background enhanced the synthetic sickness (data not shown), indicating that the low fitness is not caused by increased recombination. Surprisingly, the epistasis observed in HU suggests that Elg1 might cooperate with one of the bypass mechanisms to deal with the effects of HU.
Two papers have very recently characterized the role of the yeast FA orthologs in the repair of ICLs. Daee and coworkers found that the pathway is required for the repair of nitrogen mustard-induced ICLs by a mechanism that is independent of the Pso2 protein, previously identified as essential for ICL repair,51 but relies on the Rad5 component of the post-replication repair pathway. Moreover, they also found evidence for a role of Mph1 in preventing ICL-stalled replication intermediates from collapsing into double-strand breaks.45 Ward and coworkers also identified the yeast FA pathway as an alternative to Pso2 in the repair of ICLs, and suggested that this pathway includes the yeast mismatch repair system (Mutsα).46 In both papers, the yeast FA genes appear genetically to work as a single pathway. We have shown here that this is not true when dealing with other forms of DNA damage.
Materials and Methods
Yeast strains, plasmids, primers and genetic manipulations
Yeast strains used in this study are shown in Table 1. All strains are derived from the BY4741/BY4742 background, unless otherwise noted.
Table 1. List of strains used in this study.
| Source | Genotype | Strain |
|---|---|---|
| ATCC 4040002 |
MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 |
BY4741 |
| This study |
MAT@ elg1:: URA3 |
yTS20 |
| This study |
MATa mhf1:: KanMX |
ySF681 |
| This study |
MATa mhf2:: HYG |
ySF680 |
| This study |
MATa elg1:: URA3 mhf1:: KanMX |
ySF863 |
| This study |
MATa elg1:: URA3 mhf2:: HYG |
ySF678 |
| This study |
MATa elg1:: URA3 mhf1:: KanMX mhf2:: HYG |
ySF684 |
| This study |
MAT@ mhf1:: KanMX mhf2:: HYG |
ySF132 |
| This study |
MATa mph1:: KanMX |
ySF827 |
| This study |
MATa elg1:: URA3 mph1:: KanMX |
ySF829 |
| This study |
MAT@ elg1:: URA3 mhf1:: KanMX mph1:: KanMX |
ySF713 |
| This study |
MAT@ elg1:: URA3 mhf2:: HYG mph1:: KanMX |
ySF842 |
| This study |
MAT@ elg1:: URA3 mhf1:: KanMX mhf2:: HYG mph1:: KanMX |
ySF129 |
| This study |
MATa mhf1:: KanMX mph1:: KanMX |
ySF715 |
| This study |
MATa mhf2:: HYG mph1:: KanMX |
ySF719 |
| This study |
MATa mhf1:: KanMX mhf2:: HYG mph1:: KanMX |
ySF128 |
| This study |
MATa chl1:: KanMX |
ySF836 |
| This study |
MATa chl1:: KanMX elg1:: URA3 |
ySF317 |
| This study |
MAT@ chl1:: KanMX elg1:: URA3 mph1:: KanMX |
ySF849 |
| This study |
MATa chl1:: KanMX mph1:: KanMX |
ySF717 |
| This study |
MATa elg1:: URA3 chl1:: KanMX mhf1:: KanMX mhf2:: HYG |
ySF687 |
| This study |
MATa chl1:: KanMX mhf1:: KanMX mhf2:: HYG |
ySF709 |
| This study |
MATa elg1:: URA3 chl1:: KanMX mph1:: KanMX mhf1:: KanMX mhf2:: HYG |
ySF869 |
| This study |
MATa pol30-RR(K127R,K164R)::LEU2 |
ySF405 |
| This study |
MAT@ elg1:: URA3 pol30-RR::LEU2 |
ySF410 |
| This study |
MATa chl1:: KanMX pol30-RR::LEU2 |
ySF415 |
| This study |
MAT@ elg1:: URA3 chl1:: KanMX pol30-RR::LEU2 |
ySF408 |
|
43 |
MATa pol30–104::LEU2 |
CH2166 |
| This study |
MATa elg1:: URA3 pol30–104::LEU2 |
ySF381 |
| This study |
MAT@ chl1:: KanMX pol30–104::LEU2 |
ySF386 |
| This study |
MATa elg1:: URA3 chl1:: KanMX pol30–104::LEU2 |
ySF389 |
| This study |
MAT@ elg1-SIM::13 myc::KanMX |
yTS112 |
| This study |
MAT@ elg1-SIMPIP:: 13 myc::KanMX |
yTS115 |
|
52 |
W1588–4C, MATa MPH1-YFP::HIS3 |
T497–1 |
|
52 |
W1588–4C, MAT@ mph1-Q603D-YFP::HIS5 |
T597–1 |
|
52 |
W1588–4C, MAT@ mph1-E210Q-YFP::HIS5 |
T617 |
| This study |
T597–1 elg1::HYG mph1-Q603D-YFP::HIS5 |
ySF791 |
| This study |
T617 elg1::HYG mph1-E210Q-YFP::HIS5. |
ySF798 |
| This study |
T497–1 elg1::HYG |
ySF801 |
| This study |
T497–1 mph1::KanMX |
ySF804 |
| This study |
T497–1 elg1::HYG mph1::KanMX |
ySF793 |
| 53 | trp1–901 leu2–3,112 ura3–52 his3–200 gal4del gal80del GAL2-ADE2 LYS2:: GAL1-HIS3 met2::GAL7-lacZ. | PJ69–4α |
Standard yeast protocols were used for strain construction, growth and medium preparation. Spot assay plates were incubated at 30°C and photographed after 2–3 d, unless otherwise indicated.
Drop assays: logarithmically growing yeast cells were serially diluted 10-fold and plated on SD-complete plates carrying different concentrations of methyl methanesulfonate (MMS) (Sigma Aldrich) or hydroxyurea (Sigma-Aldrich).
Doubling time measurement: Six independent cultures of each genotype were grown to mid-logarithmic phase, diluted to ~1 × 106 cells/ml and incubated in 96-well plates at 30°C. OD600 was measured automatically every 30 min. Generation time was calculated from the growth curve in the logarithmic growth period.
We performed 2H screens and pair-wise testing as described in Parnas et al., 2010.33 Primer sequences are available upon request.
Plasmids
To construct the plasmids used in the yeast two hybrid assay, ELG1's AAA domain (AA 235–514) was cloned into pGBU9.33
The pRS315-ELG1–13-myc::KanMX plasmid (WT), pRS315-ELG1(1–519)-13-myc::KanMX, pRS315-ELG1(216–791)-13-myc::KanMX and pRS315-ELG1(216–731)-13-myc::KanMX are a generous gift from Grant W. Brown.42 The other deletion constructs pRS315-ELG1(1–731)-13-myc::KanMX, pRS315-ELG1(1–741)-13-myc::KanMX, pRS315-ELG1(1–751)-13-myc::KanMX, pRS315-ELG1(216–741)-13-myc::KanMX and pRS315-ELG1(216–751)-13-myc::KanMX were constructed by using Phusion® High-Fidelity PCR Master Mix with appropriate primer oligonucleotides. All constructs were verified by nucleotide sequencing.
The plasmids AC616 (pRS415-MPH1), AC617 (pRS415-Mph1-K113Q), AC618 (pRS415-Mph1-DE), AC431 (control vector containing the MPH1 promoter but no insert), AC605 (AD of MPH1), AC1069 (AD of MPH1-Δ60), AC306 (BD alone control) and AC305 (AD alone control) are a kind gift from F. Brad Johnson (University of Pennsylvania School of Medicine).
Acknowledgments
We thank Brad Johnson, Grant Brown, Robert Skibbens and Xiaolan Zhiao for plasmids and yeast strains, and all members of the Kupiec group for ideas and support. This work was supported by grants from the Fritz Thyssen Foundation and the Israel Science Fund to M.K.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24756
References
- 1.Kee Y, D’Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010;24:1680–94. doi: 10.1101/gad.1955310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bagby GC, Alter BP. Fanconi anemia. Semin Hematol. 2006;43:147–56. doi: 10.1053/j.seminhematol.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Niedernhofer LJ, Lalai AS, Hoeijmakers JH. Fanconi anemia (cross)linked to DNA repair. Cell. 2005;123:1191–8. doi: 10.1016/j.cell.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 4.Moldovan GL, D’Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–49. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kennedy RD, Chen CC, Stuckert P, Archila EM, De la Vega MA, Moreau LA, et al. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest. 2007;117:1440–9. doi: 10.1172/JCI31245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knipscheer P, Räschle M, Smogorzewska A, Enoiu M, Ho TV, Schärer OD, et al. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol Cell. 2007;25:331–43. doi: 10.1016/j.molcel.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Singh TR, Saro D, Ali AM, Zheng XF, Du CH, Killen MW, et al. MHF1-MHF2, a histone-fold-containing protein complex, participates in the Fanconi anemia pathway via FANCM. Mol Cell. 2010;37:879–86. doi: 10.1016/j.molcel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan Z, Delannoy M, Ling C, Daee D, Osman F, Muniandy PA, et al. A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol Cell. 2010;37:865–78. doi: 10.1016/j.molcel.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou W, Otto EA, Cluckey A, Airik R, Hurd TW, Chaki M, et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet. 2012;44:910–5. doi: 10.1038/ng.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Villamil MA, Chen J, Liang Q, Zhuang Z. A noncanonical cysteine protease USP1 is activated through active site modulation by USP1-associated factor 1. Biochemistry. 2012;51:2829–39. doi: 10.1021/bi3000512. [DOI] [PubMed] [Google Scholar]
- 12.Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D’Andrea AD, et al. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 2005;17:331–9. doi: 10.1016/j.molcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 14.Lee KY, Yang K, Cohn MA, Sikdar N, D’Andrea AD, Myung K. Human ELG1 regulates the level of ubiquitinated proliferating cell nuclear antigen (PCNA) through Its interactions with PCNA and USP1. J Biol Chem. 2010;285:10362–9. doi: 10.1074/jbc.M109.092544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang K, Moldovan GL, Vinciguerra P, Murai J, Takeda S, D’Andrea AD. Regulation of the Fanconi anemia pathway by a SUMO-like delivery network. Genes Dev. 2011;25:1847–58. doi: 10.1101/gad.17020911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sato K, Ishiai M, Toda K, Furukoshi S, Osakabe A, Tachiwana H, et al. Histone chaperone activity of Fanconi anemia proteins, FANCD2 and FANCI, is required for DNA crosslink repair. EMBO J. 2012;31:3524–36. doi: 10.1038/emboj.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolodner RD, Putnam CD, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 2002;297:552–7. doi: 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- 18.Aroya SB, Kupiec M. The Elg1 replication factor C-like complex: a novel guardian of genome stability. DNA Repair (Amst) 2005;4:409–17. doi: 10.1016/j.dnarep.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Ulrich HD. Regulating post-translational modifications of the eukaryotic replication clamp PCNA. DNA Repair (Amst) 2009;8:461–9. doi: 10.1016/j.dnarep.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 20.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–53. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 21.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–41. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 22.Papouli E, Chen S, Davies AA, Huttner D, Krejci L, Sung P, et al. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell. 2005;19:123–33. doi: 10.1016/j.molcel.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 23.Pfander B, Moldovan GL, Sacher M, Hoege C, Jentsch S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature. 2005;436:428–33. doi: 10.1038/nature03665. [DOI] [PubMed] [Google Scholar]
- 24.Bellaoui M, Chang M, Ou J, Xu H, Boone C, Brown GW. Elg1 forms an alternative RFC complex important for DNA replication and genome integrity. EMBO J. 2003;22:4304–13. doi: 10.1093/emboj/cdg406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ben-Aroya S, Koren A, Liefshitz B, Steinlauf R, Kupiec M. ELG1, a yeast gene required for genome stability, forms a complex related to replication factor C. Proc Natl Acad Sci USA. 2003;100:9906–11. doi: 10.1073/pnas.1633757100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanellis P, Agyei R, Durocher D. Elg1 forms an alternative PCNA-interacting RFC complex required to maintain genome stability. Curr Biol. 2003;13:1583–95. doi: 10.1016/S0960-9822(03)00578-5. [DOI] [PubMed] [Google Scholar]
- 27.Ogiwara H, Ui A, Enomoto T, Seki M. Role of Elg1 protein in double strand break repair. Nucleic Acids Res. 2007;35:353–62. doi: 10.1093/nar/gkl1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith S, Hwang JY, Banerjee S, Majeed A, Gupta A, Myung K. Mutator genes for suppression of gross chromosomal rearrangements identified by a genome-wide screening in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2004;101:9039–44. doi: 10.1073/pnas.0403093101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smolikov S, Mazor Y, Krauskopf A. ELG1, a regulator of genome stability, has a role in telomere length regulation and in silencing. Proc Natl Acad Sci USA. 2004;101:1656–61. doi: 10.1073/pnas.0307796100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scholes DT, Banerjee M, Bowen B, Curcio MJ. Multiple regulators of Ty1 transposition in Saccharomyces cerevisiae have conserved roles in genome maintenance. Genetics. 2001;159:1449–65. doi: 10.1093/genetics/159.4.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sikdar N, Banerjee S, Lee KY, Wincovitch S, Pak E, Nakanishi K, et al. DNA damage responses by human ELG1 in S phase are important to maintain genomic integrity. Cell Cycle. 2009;8:3199–207. doi: 10.4161/cc.8.19.9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bell DW, Sikdar N, Lee KY, Price JC, Chatterjee R, Park HD, et al. NISC Comparative Sequencing Program Predisposition to cancer caused by genetic and functional defects of mammalian Atad5. PLoS Genet. 2011;7:e1002245. doi: 10.1371/journal.pgen.1002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parnas O, Zipin-Roitman A, Pfander B, Liefshitz B, Mazor Y, Ben-Aroya S, et al. Elg1, an alternative subunit of the RFC clamp loader, preferentially interacts with SUMOylated PCNA. EMBO J. 2010;29:2611–22. doi: 10.1038/emboj.2010.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scheller J, Schürer A, Rudolph C, Hettwer S, Kramer W. MPH1, a yeast gene encoding a DEAH protein, plays a role in protection of the genome from spontaneous and chemically induced damage. Genetics. 2000;155:1069–81. doi: 10.1093/genetics/155.3.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schürer KA, Rudolph C, Ulrich HD, Kramer W. Yeast MPH1 gene functions in an error-free DNA damage bypass pathway that requires genes from Homologous recombination, but not from postreplicative repair. Genetics. 2004;166:1673–86. doi: 10.1534/genetics.166.4.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laha S, Das SP, Hajra S, Sau S, Sinha P. The budding yeast protein Chl1p is required to preserve genome integrity upon DNA damage in S-phase. Nucleic Acids Res. 2006;34:5880–91. doi: 10.1093/nar/gkl749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, et al. International Consortium for Blood Pressure Genome-Wide Association Studies. CARDIoGRAM consortium. CKDGen Consortium. KidneyGen Consortium. EchoGen consortium. CHARGE-HF consortium Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–9. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parnas O, Amishay R, Liefshitz B, Zipin-Roitman A, Kupiec M. Elg1, the major subunit of an alternative RFC complex, interacts with SUMO-processing proteins. Cell Cycle. 2011;10:2894–903. doi: 10.4161/cc.10.17.16778. [DOI] [PubMed] [Google Scholar]
- 39.Chavez A, Agrawal V, Johnson FB. Homologous recombination-dependent rescue of deficiency in the structural maintenance of chromosomes (Smc) 5/6 complex. J Biol Chem. 2011;286:5119–25. doi: 10.1074/jbc.M110.201608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang RY, Eddy M, Vujcic M, Kowalski D. Genome-wide screen identifies genes whose inactivation confer resistance to cisplatin in Saccharomyces cerevisiae. Cancer Res. 2005;65:5890–7. doi: 10.1158/0008-5472.CAN-04-4093. [DOI] [PubMed] [Google Scholar]
- 41.Yang H, Zhang T, Tao Y, Wu L, Li HT, Zhou JQ, et al. Saccharomyces cerevisiae MHF complex structurally resembles the histones (H3-H4)₂ heterotetramer and functions as a heterotetramer. Structure. 2012;20:364–70. doi: 10.1016/j.str.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 42.Davidson MB, Brown GW. The N- and C-termini of Elg1 contribute to the maintenance of genome stability. DNA Repair (Amst) 2008;7:1221–32. doi: 10.1016/j.dnarep.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 43.Amin NS, Holm C. In vivo analysis reveals that the interdomain region of the yeast proliferating cell nuclear antigen is important for DNA replication and DNA repair. Genetics. 1996;144:479–93. doi: 10.1093/genetics/144.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Merrill BJ, Holm C. The RAD52 recombinational repair pathway is essential in pol30 (PCNA) mutants that accumulate small single-stranded DNA fragments during DNA synthesis. Genetics. 1998;148:611–24. doi: 10.1093/genetics/148.2.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daee DL, Ferrari E, Longerich S, Zheng XF, Xue X, Branzei D, et al. Rad5-dependent DNA repair functions of the Saccharomyces cerevisiae FANCM protein homolog Mph1. J Biol Chem. 2012;287:26563–75. doi: 10.1074/jbc.M112.369918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ward TA, Dudášová Z, Sarkar S, Bhide MR, Vlasáková D, Chovanec M, et al. Components of a Fanconi-like pathway control Pso2-independent DNA interstrand crosslink repair in yeast. PLoS Genet. 2012;8:e1002884. doi: 10.1371/journal.pgen.1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skibbens RV. Chl1p, a DNA helicase-like protein in budding yeast, functions in sister-chromatid cohesion. Genetics. 2004;166:33–42. doi: 10.1534/genetics.166.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parnas O, Zipin-Roitman A, Mazor Y, Liefshitz B, Ben-Aroya S, Kupiec M. The ELG1 clamp loader plays a role in sister chromatid cohesion. PLoS ONE. 2009;4:e5497. doi: 10.1371/journal.pone.0005497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davidson MB, Katou Y, Keszthelyi A, Sing TL, Xia T, Ou J, et al. Endogenous DNA replication stress results in expansion of dNTP pools and a mutator phenotype. EMBO J. 2012;31:895–907. doi: 10.1038/emboj.2011.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karras GI, Fumasoni M, Sienski G, Vanoli F, Branzei D, Jentsch S. Noncanonical Role of the 9-1-1 Clamp in the Error-Free DNA Damage Tolerance Pathway. Mol Cell. 2013;49:536–46. doi: 10.1016/j.molcel.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 51.Ruhland A, Haase E, Siede W, Brendel M. Isolation of yeast mutants sensitive to the bifunctional alkylating agent nitrogen mustard. Mol Gen Genet. 1981;181:346–51. doi: 10.1007/BF00425609. [DOI] [PubMed] [Google Scholar]
- 52.Chen YH, Choi K, Szakal B, Arenz J, Duan X, Ye H, et al. Interplay between the Smc5/6 complex and the Mph1 helicase in recombinational repair. Proc Natl Acad Sci USA. 2009;106:21252–7. doi: 10.1073/pnas.0908258106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.James P, Halladay J, Craig EA. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144:1425–36. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]