

Abstract
The proton-coupled folate transporter (PCFT, SLC46A1) mediates folate transport across the apical brush-border membrane of the proximal small intestine and the basolateral membrane of choroid plexus ependymal cells. Two loss-of-function mutations in PCFT, which are the basis for hereditary folate malabsorption, have been identified within the fourth transmembrane domain (TMD4) in subjects with this disorder. We have employed the substituted Cys accessibility method (SCAM) to study the accessibilities of all residues in TMD4 and their roles in folate substrate binding to the carrier. When residues 146–167 were replaced by Cys, all except R148C were expressed at the cell surface. Modification of five of these substituted Cys residues (positions 147, 152, 157, 158, and 161) by methanethiosulfonate (MTS) reagents led to reduction of PCFT function. All five residues could be labeled with N-biotinylaminoethyl-MTS, and this could be blocked by the high-affinity PCFT substrate pemetrexed. Pemetrexed also protected PCFT mutant function from inhibitory modification of the substituted Cys at positions 157, 158, and 161 by a MTS. The findings indicate that these five residues in TMD4 are accessible to the aqueous translocation pathway, play a role in folate substrate binding, and are likely located within or near the folate binding pocket. A homology model of PCFT places three of these residues, Phe157, Gly158, and Leu161, within a breakpoint in the midportion of TMD4, a region that likely participates in alterations in the PCFT conformational state during carrier cycling.
Keywords: folates, homology model, folate transporters, hereditary folate malabsorption, hereditary folate malabsorption, proton-coupled folate transporter, heme carrier protein 1, intestinal folate absorption
the proton-coupled folate transporter (PCFT, SLC46A1) is the mechanism by which folates are transported across the apical brush-border membrane of the proximal small intestine and the basolateral membrane of ependymal cells at the blood-choroid plexus-cerebrospinal fluid barrier (36). Both processes are markedly impaired when there are loss-of-function mutations in this gene, as occurs in the autosomal recessive disorder hereditary folate malabsorption (HFM) (6, 36). Because of the key role of this transporter in the maintenance of folate homeostasis in humans, the impact of disease-causing mutations, and PCFT's pharmacological potential in antifolate cancer treatment (4, 5), understanding the structural basis for PCFT function is of considerable interest. It has been established that PCFT has 12 transmembrane domains (TMDs), with its COOH and NH2 termini facing the cytoplasm (43), and exists in a homo-oligomeric configuration (12, 41). The major focus of studies on PCFT has been on the impact of single mutations on the expression and function of this transporter. The residues chosen for study have been 1) identified in subjects with HFM (14, 19, 26–28, 39), 2) charged residues in TMDs (31, 32), and 3) identified by random mutagenesis (40). Although a variety of functionally important PCFT residues have been identified in this way, there is very limited information on residues directly involved in folate binding and translocation.
Most recently, Ile188 in TMD5 of PCFT was identified as a residue that may be directly involved in folate binding (28). The functional role of this residue was discovered because of its close proximity to the irreplaceable G(189)XXG(192) motif. The I88C PCFT mutant was labeled with membrane-impermeant N-biotinylaminoethylmethanethiosulfonate (MTSEA-biotin), indicating that this residue is in the aqueous translocation pathway and is accessible from the extracellular compartment. The activity of the I188C mutant was inhibited by modification with a membrane-impermeant sulfhydryl-specific 2-(trimethylammonium)ethyl methanethiosulfonate, bromide (MTSET), and this inhibition was prevented by pemetrexed, an antifolate substrate with high affinity for PCFT (34, 38). These observations were consistent with the localization of I188C in or near an element of the folate binding pocket.
TMD4 of PCFT has been a region of considerable interest. The Gly147 and Asp156 residues were mutated in subjects with HFM (26, 39), and the L161R mutant, with a marked decrease in affinity for methotrexate (MTX), was identified by random mutagenesis (40). In the current study the substituted Cys accessibility method (SCAM) was employed to explore the accessibility of all 21 residues of TMD4, from positions 146 to 167, and their potential role in binding of folates to this transporter (13). All Cys mutations could be generated in wild-type (WT) PCFT, since none of the seven native Cys residues react with membrane-impermeant sulfhydryl-specific reagents (41). In five mutants, G147C, A152C, F157C, G158C, and L161C, the substituted Cys residue was found to be accessible to the aqueous translocation pathway. Pemetrexed blocked the labeling of all five mutants by MTSEA-biotin and protected the inhibitory modification of the latter three mutants by MTSET. These findings are consistent with the localization of the Gly147, Ala152, Phe157, Gly158, and Leu161 residues in or near the folate binding pocket.
EXPERIMENTAL PROCEDURES
Chemicals.
[3H]MTX {3′,5′,7-[3H](N)} was obtained from Moravek Biochemicals (Brea, CA), purified by liquid chromatography, and stored at -20°C. EZ-Link Sulfo-NHS-LC-Biotin [sulfosuccinimidyl 6-(biotinamido)hexanoate], streptavidin-agarose beads, and methylmethanethiosulfonate (MMTS) were purchased from Thermo Scientific (Rockford, IL), MTSEA-biotin from Biotium (Hayward, CA), MTSET and 2-sulfonatoethyl methanesulfonate sodium salt (MTSES) from Affymetrix (Santa Clara, CA), 2-aminoethyl methanethiosulfonate hydrobromide (MTSEA) from Toronto Research Chemicals (Toronto, ON, Canada), and protease inhibitor cocktail from Roche Applied Science (Mannheim, Germany).
Construction of Cys PCFT mutant plasmids by site-directed mutagenesis.
Amino acid residues at positions 146–167 of PCFT were individually replaced with Cys using the QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). An expression vector, pcDNA3.1(+), that encodes hemagglutinin (HA)-tagged PCFT at the COOH terminus was used as a template. The coding sequence of PCFT was verified by DNA sequencing in the Albert Einstein Cancer Center Genomics Shared Resource.
Cell lines, cell culture conditions, and transient transfection.
HeLa-R1-11 cells that lack reduced folate carrier expression due to a genomic deletion (37) and do not express PCFT due to silencing of its promoter by methylation (7) were used as transfection recipients. R1-11 cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified atmosphere of 5% CO2. R1-11 cells (3.5 × 105 cells/vial) were seeded into 17-mm liquid scintillation vials in preparation for transport studies or six-well plates (6 × 105 cells/well) in preparation for labeling experiments. After 2 days, the cells were transiently transfected with PCFT constructs (0.8 μg/vial or 2 μg/well of a 6-well-plate) with Lipofectamine 2000 (Invitrogen, Carlsbad, CA). After 2 additional days, the cells were processed for transport studies or biotin labeling.
Transport measurements.
Cells were washed twice with 2 ml of HBS (20 mM HEPES, 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, and 5 mM dextrose, pH 7.4) buffer and incubated with the same buffer (2 ml) in a water bath (37°C) for 20 min. After incubation, the buffer was aspirated, and 500 μl of MBS (20 mM MES, 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, and 5 mM dextrose, pH 5.5) containing 0.5 μM [3H]MTX were added. Transport was terminated after 1 min by the addition of 5 ml of ice-cold HBS buffer (pH 7.4); over this interval, uptake was unidirectional. Cells were washed three times with 5 ml of ice-cold HBS, 500 μl of 0.2 M NaOH were added, and cells were digested for 1 h at 65°C. A 400-μl portion of the hydrolysate was analyzed on a liquid scintillation spectrometer; another portion of the hydrolysate (10 μl) was analyzed for protein with the Pierce kit (Thermo Scientific). Influx is expressed as picomoles of [3H]MTX per milligram of protein per minute or percentage of WT activity.
Modification of substituted Cys residues with methanethiosulfonate reagents.
Cells were washed with HBS buffer twice and treated with freshly made methanethiosulfonate (MTS) reagent solutions at room temperature for 30 min. MTSES and MTSET are water-soluble and were dissolved in HBS buffer. MMTS, MTSEA, and MTSEA-biotin are more hydrophobic and were dissolved in DMSO first and then diluted into HBS buffer at a ratio of 1:100. After 30 min, cells were washed twice with 2 ml of HBS, and [3H]MTX influx was determined as described above.
Biotinylation of PCFT.
For analysis of PCFT accessibility at the cell surface, cells in HBS were treated for 30 min with 1 mg/ml EZ-Link Sulfo-NHS-LC-Biotin, which reacts with Lys residues on the cell surface. For SCAM, cells were treated with MTSEA-biotin for 30 min at room temperature or at 0°C. The extent to which pemetrexed blocks Cys biotinylation with MTSEA-biotin was also assessed by Western blotting. Cells were treated with pemetrexed before MTSEA-biotin was added at room temperature or 0°C. Pemetrexed was present throughout the biotinylation reaction. MTSEA-biotin concentrations were adjusted to optimize pemetrexed protection on the basis of the prior observation that pemetrexed protection was greater at the lowest MTS concentration that still achieved near-maximal inhibition of PCFT function (41): 100 μM MTSEA-biotin for F157C and L161C, 170 μM for G158C, and 500 μM for G147C and A152C PCFT mutants. After these reactions, cells were washed twice with 2 ml of HBS buffer and treated with hypotonic buffer (0.5 mM Na2HPO4 and 0.1 mM EDTA, pH 7.0) containing protease inhibitors and kept on ice for 30 min. Cells were then scraped from the plates with a cell lifter and centrifuged at 14,000 rpm for 15 min at 4°C. The supernatant was aspirated, and the membrane fraction was pelleted by centrifugation at 14,000 rpm for 15 min. The pellet was resuspended with 400 μl of lysis buffer (50 mM Tris-base, 150 mM NaCl, 1% NP-40, and 0.5% sodium deoxycholate, pH 7.4) containing protease inhibitors and rotated in a cold room at 4°C for 30–120 min. A 25-μl portion was taken for Western blot analysis and designated the crude membrane protein fraction. The remaining sample was centrifuged at 14,000 rpm at 4°C for 15 min. The supernatant was then mixed with streptavidin-agarose beads (50 μl) that had been washed three times with lysis buffer overnight at 4°C on the rotator. The next day, the beads were washed twice with lysis buffer (500 μl) and twice with lysis buffer (500 μl) containing 2% SDS; each wash cycle consisted of a 20-min rotation at room temperature. Protein bound to the beads was stripped by heating for 5 min at 95° in 2× SDS-PAGE loading buffer containing DTT.
Western blot analyses.
The protein samples obtained as described above were loaded directly onto 12% Tris·HCl polyacrylamide gels. The crude membrane fraction was mixed with 2× SDS-PAGE loading buffer (1:1) containing DTT at room temperature before Western blot analysis. After SDS-PAGE, proteins were transferred to Amersham Hybond membranes (GE Healthcare, Piscataway, NJ) and blocked with 10% dry milk in TBST (20 mM Tris, 135 mM NaCl, and 1% Tween 20, pH 7.6) overnight at 4°C. The blots of crude membrane fractions from cells subjected to surface biotinylation were probed with a rabbit β-actin antibody (Cell Signaling Technology, Danvers, MA) and then stripped with buffer (100 mM 2-β-mercaptoethanol, 2% SDS, and 62.5 mM Tris·HCl, pH 6.7) and reprobed with anti-HA antibody (Sigma, St. Louis, MO; 1:4,000 in TBST-0.1% milk). The blots of crude membrane fractions of Cys-substituted residues exposed to MTSEA-biotin were probed directly with anti-HA antibody. For samples obtained from beads, blots were probed directly with anti-HA antibody. After application of the first antibody, the blot was probed with an anti-rabbit IgG-horseradish peroxidase conjugate (Cell Signaling Technology; 1:5,000 in TBST). Blots were developed with the Amersham ECL Plus reagent (GE Healthcare).
Statistical analysis.
Differences were considered significant at P ≤ 0.05 by one-tailed Student's paired t-test or one-way ANOVA using GraphPad Prism software.
Molecular modeling.
A homology model has been built for PCFT (15, 28, 31, 41). A hidden Markov model-based fold-recognition and alignment method, HHpred, was used to establish the sequence alignment between PCFT and its template (31). The best scoring template remained the crystal structure of the Escherichia coli glycerol-3-phosphate transporter (PDB code 1pw54), consistent with earlier studies (15, 26, 31); 1pw54 was utilized as template in comparative protein structure modeling with the Modeller program using the HHpred alignment as input (9, 24). The resulting model was verified through energetic analyses, using statistical pair potentials implemented in Prosa function (29), by correlating the location of transmembrane segments as obtained from the HMMTOP prediction (30) with the observed location of transmembrane helices in the three-dimensional model and by checking the distribution of electrostatic charges along the transporter using the GRASP program (http://www.ncbi.nlm.nih.gov/pubmed/14696386). The previous workflow described the most successful model built, but other alignment techniques [such as Muscle (8), ClustalW (3), MMM (21, 22), and Align2D (17, 18)] and their corresponding comparative models were also explored.
RESULTS
Function and expression of Cys-substituted PCFT residues of TMD4.
Figure 1A illustrates the established topological structure of PCFT, magnifying TMD4 (43). A Cys residue was substituted for each of the amino acids at positions 146–167 in the WT PCFT. Although the Cys-less PCFT might be considered a more desirable template to generate these mutants than the WT PCFT, prior studies indicated that while function is preserved in the Cys-less PCFT, it is vulnerable to loss-of-function with additional mutations that do not affect WT PCFT activity (42). Also, none of the seven native Cys residues in the WT PCFT could be modified by membrane-impermeable sulfhydryl reagents (41). There is one endogenous Cys residue at position 151 that is not accessible. D156C was excluded, since it was previously shown to be an unstable protein (26). Figure 1B indicates the partial sequence alignment among species of this TMD; Gly147, Arg148, Gly155, Asp156, Leu161, Phe165, and Ala166 are fully conserved, and Leu146, Leu153, Phe157, and Leu160 are semiconserved.
Fig. 1.
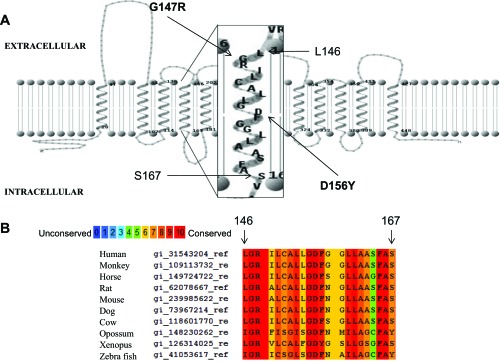
Fourth transmembrane domain (TMD4) within the context of the proton-coupled folate transporter (PCFT) topological structure: partial sequence alignment of PCFT among species. A: PCFT topology has been established by the substituted Cys accessibility method (SCAM) (43). TMD4 is magnified. Leu146 and Ser167 are at the extracellular and intracellular interfaces, respectively. These residues and the residues between them were evaluated by the SCAM. Two residues mutated in subjects with hereditary folate malabsorption, G147R and D156Y, are indicated. B: human PCFT TMD4 protein sequence was aligned with 9 different species. PRALINE sequence alignment program (http://www.ibi.vu.nl/programs/) was utilized. Numbers above the sequence are derived from human PCFT.
MTX influx was assessed for all mutants to determine if they were appropriate for subsequent analysis by the SCAM. As Fig. 2A indicates, more than half of these Cys PCFT mutants (total 12) preserved >50% of WT PCFT activity. Western blot analyses of the crude membrane preparation and cell surface labeling by biotinylation were assessed for the mutants that preserved <50% of WT activity (Fig. 2B; total 8 Cys mutants). Consistent with the lack of function, the R148C PCFT mutant protein was barely detectable in either fraction and, therefore, was not amenable to SCAM; however, all the other Cys mutant proteins were expressed, detected at the cell surface, and, hence, suitable for further study.
Fig. 2.

Function and expression of Cys PCFT mutants encompassing the entire TMD4. A: functional assessment. [3H]methotrexate (MTX) influx (0.5 μM) was measured at pH 5.5 at 37°C over 1 min. The horizontal line indicates 50% of wild-type (WT) activity. Values are means ± SE from 3 independent experiments. *Statistically significant reduction in activity compared with WT PCFT (P < 0.05). B: Western blot analysis of PCFT expression in the crude membrane preparation and biotinylated at the cell surface; the actin loading control is also shown. The blot is representative of 2 independent experiments. Vertical lines indicate repositioned gel lanes.
Impact of MTS reagents on Cys PCFT mutants.
Membrane-impermeable hydrophilic sulfhydryl-reactive reagents were used to modify Cys residues accessible to the extracellular aqueous milieu (13). Several MTS reagents, MMTS, MTSEA, MTSET, MTSES, and MTSEA-biotin (23), were first assessed for their effects on the WT and Cys-less PCFT to exclude reagents that nonspecifically suppress PCFT activity. MMTS is the smallest and most hydrophobic, MTSEA and MTSET carry a positive charge, MTSES carries a negative charge, and MTSEA-biotin is the largest molecule and does not penetrate the cell membrane (2).
To assess the effects of MTS reagents on PCFT activity, cells expressing WT or Cys-less PCFT were incubated with MTS reagents for 30 min at 20°C in HBS buffer at pH 7.4. Then influx of [3H]MTX (0.5 μM) was measured at 37°C, pH 5.5, over 1 min. On the basis of two separate experiments, each performed in duplicate, treatment of WT PCFT with MMTS (2 mM), MTSEA (2 mM), MTSET (3 mM), MTSES (3 mM), or MTSEA-biotin (500 μM) resulted in a decrease in MTX influx to 60, 12, 85, 95, or 89%, respectively, of the untreated cells that express WT PCFT. Treatment of the cells that express the Cys-less PCFT with MMTS (2 mM), MTSEA (2 mM), MTSET (3 mM), MTSES (3 mM), or MTSEA-biotin (500 μM) resulted in a decrease in MTX influx to 84, 44, 96, 114, or 108%, respectively, of the untreated cells that express Cys-less PCFT. Hence, MTSEA treatment markedly inhibited [3H]MTX influx in WT cells and, to a lesser extent, in cells that express Cys-less PCFT. MMTS was a weaker inhibitor of transport mediated by WT PCFT, from which all the Cys mutants in the current study were generated. Therefore, MMTS and MTSEA were excluded in the subsequent studies.
The effect of MTSET on PCFT function was then assessed. As illustrated in Fig. 3A, treatment of the 19 mutants with 3 mM MTSET resulted in modest (>25%) inhibition of MTX influx mediated by G147C, A152C, G158C, L161C, and F157C PCFT. Figure 3B indicates the effects of modification of these five mutants by MTSES and MTSEA-biotin. There was negligible inhibition of the G147C and A152C PCFT mutants by these two reagents. MTSES and MTSEA-biotin produced the most potent inhibition of F157C, while the two reagents produced comparable and significant inhibition of G158C. MTSET and MTSEA-biotin produced >70% inhibition of L161C-PCFT function, whereas MTSES had no effect on L161C PCFT function. Hence, while it might be expected that the largest MTS reagent, MTSEA-biotin, would have less accessibility than smaller reagents, particularly at residues located near the inner cell membrane-cytosolic interface, this was not to be the case. Neither MTSES nor MTSEA-biotin inhibited transport mediated by the other Cys mutants that were not inhibited by MTSET, as indicated in Fig. 3A (data not shown).
Fig. 3.

Impact of methanethiosulfonate modification on transport function of PCFT mutants. A: all mutants were treated with 3 mM 2-(trimethylammonium)ethyl methanethiosulfonate, bromide (MTSET). B: G147C, A152C, F157C, G158C, and L161 mutants were treated with 3 mM 2-sulfonatoethyl methanesulfonate sodium salt (MTSES) or 500 μM N-biotinylaminoethylmethanethiosulfonate (MTSEA-biotin). C: the L161C PCFT mutant was treated with MTSET, MTSES, or MTSES followed by MTSET before influx assays. PCFT transient transfectants were treated with MTS reagents for 30 min at room temperature prior to assessment of [3H]MTX influx, as described Fig. 2 legend. Values are means ± SE from 3 independent experiments. *P < 0.05 vs. respective control group. #P < 0.05 vs. MTSET alone.
Of all the inhibitors tested, MTSES was the only one that did not inhibit L161C PCFT. To examine the possibility that this reagent reacts with this residue but this modification does not result in an alteration of function, the L161C PCFT mutant was treated with MTSES prior to treatment with MTSET. As indicated in Fig. 3C, this sequence of treatment eliminated the effect of MTSET, indicating that the Cys residue was already blocked by MTSES and was not available for reaction with MTSET. Hence, MTSES modifies L161C PCFT, but without a functional consequence.
Analysis of the protective effect of pemetrexed on biotinylation of the Cys-substituted PCFT mutants by MTSEA-biotin.
To assess the proximity of the Gly147, Ala152, Phe157, Gly158, and Leu161 residues to the folate binding pocket, experiments were designed to determine the inhibitory effect of folate substrate on biotinylation of the Cys-substituted PCFT mutants. If the biotinylation is abrogated or diminished in the presence of a folate, then the residue is likely located in or near the folate binding pocket. Among folate/antifolate substrates, pemetrexed has the highest affinity for PCFT at pH 5.5 and, in particular, at pH 7.4; hence, pemetrexed was used as the substrate in these experiments (34, 38). These experiments were performed at room temperature, which allows conformational changes of the carrier, and at 0°C, which prevents the carrier from cycling between its conformational states (1, 11, 16).
Cells were treated with MTSEA-biotin in the presence or absence of pemetrexed at room temperature. As shown in Fig. 4A, pemetrexed completely blocked MTSEA-biotin labeling of the G147C and A152C mutants, with substantial, although incomplete, protection of labeling of the F157C and L161C PCFT mutants. No protection was observed for the G158C mutant. When these experiments were performed at 0°C (Fig. 4B), all five Cys PCFT mutants were biotinylated, and this was completely abrogated by the presence of pemetrexed. These observations are consistent with a role for these five residues in folate binding to PCFT.
Fig. 4.

Western blot analysis of the impact of pemetrexed on biotinylation of Cys-substituted residues at room temperature (20°C, A) and 0°C (B and C). In A and B, cells were exposed to HBS (20 mM HEPES, 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, and 5 mM dextrose, pH 7.4) or 1 mM pemetrexed in HBS for 10 min, MTSEA-biotin was added, and cells were incubated for 30 min. In C, cells were exposed to different concentrations of pemetrexed, or with 1 mM PT523, for 10 min and then labeled with MTSES-biotin for 30 min. Cells were exposed to HBS in control groups. The MTSEA-biotin concentration was adjusted to optimize pemetrexed protection while sustaining a high level of reaction of MTSEA-biotin in the immunoprecipitate: 100 μM MTSEA-biotin for F157C, L161C, and G207C Cys-less, 170 μM for G158C, and 500 μM for G147C and A152C PCFT mutants. Blots are representative of ≥2 independent experiments. White vertical lines in C indicate positions at which different X-ray films are aligned.
Several control experiments were conducted to verify that the block of MTSEA-biotin labeling by pemetrexed was specific (Fig. 4C). Labeling of the A152C mutant with 500 μM MTSEA-biotin could be blocked not only by 1 mM pemetrexed, but by 0.3 and 0.1 mM pemetrexed as well. On the other hand, the labeling was not blocked by 1 mM PT523, an antifolate that has the lowest, if any, affinity for PCFT (20, 34, 38). Similar observations were also made with the L161C mutant, in which labeling with 100 μM MTSEA-biotin was markedly reduced by 1 and 0.3 mM pemetrexed but was not affected by 1 mM PT523. The G207C mutant on a Cys-less background (Gly207 Cys-less), generated previously for the analysis of PCFT membrane topology (43), was also labeled with 100 μM MTSEA-biotin in the absence or presence of 1 mM pemetrexed or PT523. Neither pemetrexed nor PT523 had an effect on the labeling of this mutant, which is located in the third extracellular loop of PCFT (43).
To exclude the possibility that residues other than Gly147, Ala152, Phe157, Gly158, and Leu161 in TMD4 react with MTSET, MTSES, or/and MTSEA-biotin, but these modifications do not alter function, biotinylation with MTSEA-biotin was also assessed with these mutants. None of the substituted Cys residues were biotinylated except for the minimal biotinylation of the L160C PCFT (data not shown), which is not conserved.
Analysis of the protective effect of pemetrexed on inhibition of G147C, A152C, F157C, G158C, and L161C mutant function by MTSET.
Further studies were undertaken to assess whether pemetrexed protection of the PCFT mutants from modification by MTSET preserved transport function. These experiments were performed at 0°C, a temperature at which pemetrexed blockage of labeling by MTSEA-biotin was optimal, as shown in Fig. 4. As indicated in Fig. 5, treatment of the G147C, A152C, F157C, G158C, and L161C variants with MTSET reduced function for all the mutants. However, the presence of pemetrexed in the modification step by MTSET preserved function of only the F157C, G158C, and L161C mutants. Therefore, from a functional perspective, at least for F157C, G158C, and L161C mutant PCFTs, pemetrexed could protect the substituted Cys residues from modification by MTSEA-biotin and MTSET.
Fig. 5.

Pemetrexed preservation of transport function after modification of PCFT mutants by MTSET. Transient transfectants of G147C, A152C, F157C, G158C, and L161C mutants were divided into 3 groups for MTSET (3 mM) treatment at 0°C for 30 min: incubation in HBS (no treatment), MTSET, and MTSET + 1 mM pemetrexed. After MTSET treatment, cells from all groups were washed 3 times with HBS to remove MTSET and pemetrexed before [3H]MTX (0.5 μM) influx was measured at pH 5.5 and 37°C over 1 min. Values are means ± SE from 4 independent experiments. *P < 0.05 vs. MTSET. Differences in influx between cells treated with MTSET + pemetrexed and cells not treated with MTSET (no treatment) are not statistically significant (P > 0.05).
DISCUSSION
Since the cloning of PCFT, a variety of experimental approaches have been utilized to explore the structure and function of this carrier. These approaches encompass characterization of the PCFT residues mutated in subjects with HFM, along with random and site-directed mutagenesis. The current study was focused on application of SCAM to identify residues in TMD4 that are accessible to the aqueous translocation pathway and located within or near the folate binding pocket. All the Cys mutants, except R148C, which was unstable, were expressed at the plasma membrane and retained some degree of function. D156C was previously shown to be unstable (26). These observations are consistent with the notion that Cys substitutions in TMDs are almost always tolerated. A previous study showed that the lack of expression of R148C was related to the specific residue substituted. Substitutions with like-charged Lys or His were fully functional; some function was preserved with the R148A, but not the R148L, PCFT mutants (32), while 50% of function was preserved when Arg148 was replaced with the oppositely charged Asp (unpublished observation). Hence, the positive charge at this residue is not absolutely required to sustain the tertiary structure and intrinsic function, so this residue is not involved in a charge-pair interaction or a substrate binding.
Five of the 21 residues in TMD4 could be modified by MTS reagents, including MTSET, MTSES, and MTSEA-biotin, and, on this basis, are considered to be accessible to the aqueous translocation pathway and, through this channel, to the extracellular compartment. There was no strict correlation between inhibition of transport function and the size or charge of the sulfhydryl inhibitor. In general, the most potent inhibitor was MTSET, a molecule in the midrange of size with a positive charge. Interestingly, MTSEA-biotin, the largest of the MTS reagents, was a potent inhibitor of residues deep within this helix. Hence, the electrostatic potential at each residue along the pathway and local steric constraints may be more important determinants of the effects of these reagents than size (13).
To further define the extent to which residues accessible to the sulfhydryl reagents might be located within the folate binding pocket, the protective effect of pemetrexed, a folate analog with high affinity for PCFT at low and physiological pH (38), was assessed by its impact on biotinylation of the Cys-substituted residues. The pemetrexed protection of MTSEA-biotin labeling appeared to be specific on the basis of the following observations. 1) The protection was achieved at a low concentration of pemetrexed (0.1 mM), even when the concentration of MTSEA-biotin was high (500 μM). 2) The protection was not affected by PT523, an antifolate with very high affinity for the reduced folate carrier and a very low affinity for PCFT at low or neutral pH (20, 34, 38). 3) Labeling of a PCFT mutant located in the extracellular loop (G207C) was not altered by 1 mM pemetrexed or by 1 mM PT523.
These experiments were performed at two temperatures on the basis of the current supposition that 1) if substrate binding physically prevents access of the MTS reagent to a specific Cys residue, this should occur irrespective of temperature, while 2) if a conformational change occurs after substrate binding, this should render the substituted Cys residue inaccessible to the reagent; this would not occur at 0°C, where conformational changes occur very slowly, if at all (1, 11, 16). These data suggest that the Gly147, Ala152, Phe157, and Leu161 residues physically interact with folate substrate. The complete protection of G158C biotinylation at 0°C by pemetrexed is also consistent with direct involvement of this residue in folate binding. It is unclear why protection by pemetrexed of biotinylation of this residue was not observed at room temperature. It is possible that a conformational change triggered by pemetrexed binding produces a conformation shift of the G158C residue, such that it remains accessible to MTSEA-biotin. However, at 0°C, this conformational change does not occur, and, thus, pemetrexed blocks the reaction of MTSEA-biotin with the residue (G158C).
Further evidence to support the direct involvement of Phe157, Gly158, and Leu161 in folate binding came from protection of PCFT function from inhibition by MTSET. Although some protection was also observed for the G147C and A152 mutants, the difference did not reach statistical significance probably because of the low level of reduction in function achieved in the modification by MTSET. The relatively small inhibition by MTSET of the G147C and A152C mutants could be attributed to the incomplete modification of the Cys residues with this reagent and/or by a small disturbance in folate binding, even when the Cys residues were completely modified. The latter case was best illustrated for the L161C mutant, in which modification by MTSES did not have an impact on function, as shown in Fig. 3C. Pemetrexed protection of MTSEA-biotin labeling could be readily measured, even when the modification by this reagent was incomplete and had no functional consequence. Hence, Western blot analysis of the protective effect of pemetrexed on MTSEA-biotin labeling is more sensitive than a functional analysis of pemetrexed protection.
To further relate the residues associated with folate binding to PCFT structure, we used a homology model of this carrier based on a best fit to the glycerol-3-phosphate bacterial transporter, for which a crystal structure is available, as previously reported (15, 28, 31). This TMD is predicted to be broken (discontinuous or unwound helix) in the midregion (Fig. 6). The G147C and A152C residues are located within the external half of this TMD. The accessibility of the G147C mutant to biotinylation may be related to its relative proximity to the extracellular compartment; the accessibility of the Ala152 mutant appears to be due to its proximity to the aqueous translocation pathway. Three residues, Phe157, Gly158, and Gly159, are located within a loop that separates the internal and external TMD segments. Leu161 is close by and, along with Gly158, appears to be accessible to the aqueous translocation pathway, consistent with the biotinylation results. Phe157 appears to point into the lipid bilayer and should not be accessible. However, the accessibility of the F157C mutant to biotinylation could be due to the impact of the Cys substitution that alters the usual conformation of this residue, or this residue could be transiently accessible to the aqueous channel because of the flexibility of this region. Modeling of this midregion of the carrier within the lipid bilayer is particularly reliable, since all alternative models, using different programs to align the structural template of the glycerol-3-phosphate bacterial transporter with the target PCFT sequence, returned an identical solution.
Fig. 6.

A homology model representation of PCFT. Helices are shown in cylinders and colored from the NH2 to the COOH terminal using a red-to-blue color transition. Cys mutated residues are shown in ball-and-stick representation: Gly147 in yellow, Ala152 in blue, Phe157 in tan, Gly158 in orange, Gly159 in red, Leu161 in green. A and B: top views of PCFT looking in from the extracellular space. B: enlarged view of a segment of A. C: side view of PCFT, as it is embedded in the membrane bilayer. The second transmembrane domain is translucent to enhance visibility of this region. D: enlarged view of C.
The break in TMD4 is distinct from the other 11 helices, except TMD8, which is also broken in the midregion in the PCFT homology model. Two Gly residues (Gly158 and Gly159) located in the breakpoint in TMD4 provide flexibility, suggesting that this region plays an important role in conformational changes of the carrier that occur during the transport cycle. While these Gly residues are not well conserved, on the basis of the partial sequence alignment of Fig. 1B, one or the other would provide flexibility and motion in this region. Broken or discontinuous (unwound) helices in other solute transporter structures have roles in substrate binding (25). There are breaks in the midportion of the helical structure of TMD1 and TMD6 of the Na+-Cl--dependent neurotransmitter transporters, based on the structure of the bacterial homolog. In TMD1, two adjacent residues (Val and Gly) adopt an extended conformation, linking the two segments. The interruption was greater in TMD6 with Ser and Gly residues. These breaks expose main-chain carbonyl oxygen and nitrogen atoms for hydrogen bonding, ion coordination, or substrate binding (35). Cys mutagenesis and molecular modeling of the mammalian neurotransmitter transporter [serotonin transporter (SERT)], based on the LeuT bacterial transporter structure, suggested helical breaks in TMD1 and TMD6 that contain residues within the translocation pathways involved in substrate binding (10). This was also observed for the APC (amino acid-polyamine-organification) amino acid transporter (33). Taken together, these findings are consistent with an important role for TMD4 of PCFT in providing residues accessible to the aqueous translocation pathway and within or close to the folate binding pocket. The break in this helix further suggests a role for this region in the conformational changes that occur during oscillation of the carrier between its conformational states.
GRANTS
This work was supported by National Cancer Institute Grant CA-82621 (I. D. Goldman).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.S.S., R.Z., and I.D.G. are responsible for conception and design of the research; D.S.S. and R.Z. performed the experiments; D.S.S., R.Z., and I.D.G. analyzed the data; D.S.S., R.Z., A.F., and I.D.G. interpreted the results of the experiments; D.S.S. and A.F. prepared the figures; D.S.S., R.Z., and A.F. drafted the manuscript; D.S.S., R.Z., A.F., and I.D.G. edited and revised the manuscript; R.Z. and I.D.G. approved the final version of the manuscript.
REFERENCES
- 1. Androutsellis-Theotokis A, Ghassemi F, Rudnick G. A conformationally sensitive residue on the cytoplasmic surface of serotonin transporter. J Biol Chem 276: 45933–45938, 2001 [DOI] [PubMed] [Google Scholar]
- 2. Chen R, Wei H, Hill ER, Chen L, Jiang L, Han DD, Gu HH. Direct evidence that two cysteines in the dopamine transporter form a disulfide bond. Mol Cell Biochem 298: 41–48, 2007 [DOI] [PubMed] [Google Scholar]
- 3. Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31: 3497–3500, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Desmoulin SK, Wang L, Hales E, Polin L, White K, Kushner J, Stout M, Hou Z, Cherian C, Gangjee A, Matherly LH. Therapeutic targeting of a novel 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolate to human solid tumors based on selective uptake by the proton-coupled folate transporter. Mol Pharmacol 80: 1096–1107, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Desmoulin SK, Wang Y, Wu J, Stout M, Hou Z, Fulterer A, Chang MH, Romero MF, Cherian C, Gangjee A, Matherly LH. Targeting the proton-coupled folate transporter for selective delivery of 6-substituted pyrrolo[2,3-d]pyrimidine antifolate inhibitors of de novo purine biosynthesis in the chemotherapy of solid tumors. Mol Pharmacol 78: 577–587, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Diop-Bove N, Kronn D, Goldman ID. Hereditary folate malabsorption. In: GeneReviews, edited by Pagon RA, Bird TD, Dolan CR, Stephens K. Seattle, WA: University of Washington, 2011 [Google Scholar]
- 7. Diop-Bove NK, Wu J, Zhao R, Locker J, Goldman ID. Hypermethylation of the human proton-coupled folate transporter (SLC46A1) minimal transcriptional regulatory region in an antifolate-resistant HeLa cell line. Mol Cancer Ther 8: 2424–2431, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5: 113, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol 374: 461–491, 2003 [DOI] [PubMed] [Google Scholar]
- 10. Forrest LR, Zhang YW, Jacobs MT, Gesmonde J, Xie L, Honig BH, Rudnick G. Mechanism for alternating access in neurotransmitter transporters. Proc Natl Acad Sci USA 105: 10338–10343, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Henry LK, Adkins EM, Han Q, Blakely RD. Serotonin and cocaine-sensitive inactivation of human serotonin transporters by methanethiosulfonates targeted to transmembrane domain I. J Biol Chem 278: 37052–37063, 2003 [DOI] [PubMed] [Google Scholar]
- 12. Hou Z, Desmoulin SK, Etnyre E, Olive M, Hsiung B, Cherian C, Wloszczynski PA, Moin K, Matherly LH. Identification and functional impact of homo-oligomers of the human proton-coupled folate transporter. J Biol Chem 287: 4982–4995, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karlin A, Akabas MH. Substituted-cysteine accessibility method. Methods Enzymol 293: 123–145, 1998 [DOI] [PubMed] [Google Scholar]
- 14. Lasry I, Berman B, Glaser F, Jansen G, Assaraf YG. Hereditary folate malabsorption: a positively charged amino acid at position 113 of the proton-coupled folate transporter (PCFT/SLC46A1) is required for folic acid binding. Biochem Biophys Res Commun 386: 426–431, 2009 [DOI] [PubMed] [Google Scholar]
- 15. Lasry I, Berman B, Straussberg R, Sofer Y, Bessler H, Sharkia M, Glaser F, Jansen G, Drori S, Assaraf YG. A novel loss of function mutation in the proton-coupled folate transporter from a patient with hereditary folate malabsorption reveals that Arg113 is crucial for function. Blood 112: 2055–2061, 2008 [DOI] [PubMed] [Google Scholar]
- 16. Lopez-Corcuera B, Nunez E, Martinez-Maza R, Geerlings A, Aragon C. Substrate-induced conformational changes of extracellular loop 1 in the glycine transporter GLYT2. J Biol Chem 276: 43463–43470, 2001 [DOI] [PubMed] [Google Scholar]
- 17. Madhusudhan MS, Marti-Renom MA, Sanchez R, Sali A. Variable gap penalty for protein sequence-structure alignment. Protein Eng Des Sel 19: 129–133, 2006 [DOI] [PubMed] [Google Scholar]
- 18. Madhusudhan MS, Webb BM, Marti-Renom MA, Eswar N, Sali A. Alignment of multiple protein structures based on sequence and structure features. Protein Eng Des Sel 22: 569–574, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mahadeo K, Diop-Bove N, Shin D, Unal E, Teo J, Zhao R, Chang MH, Fulterer A, Romero MF, Goldman ID. Properties of the Arg376 residue of the proton-coupled folate transporter (PCFT-SLC46A1) and a glutamine mutant causing hereditary folate malabsorption. Am J Physiol Cell Physiol 299: C1153–C1161, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay S, Tsai E, Sandoval C, Zhao R, Akabas MH, Goldman ID. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 127: 917–928, 2006 [DOI] [PubMed] [Google Scholar]
- 21. Rai BK, Fiser A. Multiple mapping method: a novel approach to the sequence-to-structure alignment problem in comparative protein structure modeling. Proteins 63: 644–661, 2006 [DOI] [PubMed] [Google Scholar]
- 22. Rai BK, Madrid-Aliste CJ, Fajardo JE, Fiser A. MMM: a sequence-to-structure alignment protocol. Bioinformatics 22: 2691–2692, 2006 [DOI] [PubMed] [Google Scholar]
- 23. Riegelhaupt PM, Frame IJ, Akabas MH. Transmembrane segment 11 appears to line the purine permeation pathway of the Plasmodium falciparum equilibrative nucleoside transporter 1 (PfENT1). J Biol Chem 285: 17001–17010, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779–815, 1993 [DOI] [PubMed] [Google Scholar]
- 25. Screpanti E, Hunte C. Discontinuous membrane helices in transport proteins and their correlation with function. J Struct Biol 159: 261–267, 2007 [DOI] [PubMed] [Google Scholar]
- 26. Shin DS, Min SH, Russell L, Zhao R, Fiser A, Goldman ID. Functional roles of aspartate residues of the proton-coupled folate transporter (PCFT; SLC46A1); a D156Y mutation causing hereditary folate malabsorption. Blood 116: 5162–5169, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shin DS, Zhao R, Fiser A, Goldman DI. Functional roles of the A335 and G338 residues of the proton-coupled folate transporter (PCFT-SLC46A1) mutated in hereditary folate malabsorption. Am J Physiol Cell Physiol 303: C834–C842, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shin DS, Zhao R, Yap EH, Fiser A, Goldman ID. A P425R mutation of the proton-coupled folate transporter causing hereditary folate malabsorption produces a highly selective alteration in folate binding. Am J Physiol Cell Physiol 302: C1405–C1412, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sippl MJ. Recognition of errors in three-dimensional structures of proteins. Proteins 17: 355–362, 1993 [DOI] [PubMed] [Google Scholar]
- 30. Tusnady GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics 17: 849–850, 2001 [DOI] [PubMed] [Google Scholar]
- 31. Unal ES, Zhao R, Chang MH, Fiser A, Romero MF, Goldman ID. The functional roles of the His247 and His281 residues in folate and proton translocation mediated by the human proton-coupled folate transporter SLC46A1. J Biol Chem 284: 17846–17857, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Unal ES, Zhao R, Goldman ID. Role of the glutamate 185 residue in proton translocation mediated by the proton-coupled folate transporter SLC46A1. Am J Physiol Cell Physiol 297: C66–C74, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vangelatos I, Vlachakis D, Sophianopoulou V, Diallinas G. Modelling and mutational evidence identify the substrate binding site and functional elements in APC amino acid transporters. Mol Membr Biol 26: 356–370, 2009 [DOI] [PubMed] [Google Scholar]
- 34. Wang Y, Zhao R, Goldman ID. Characterization of a folate transporter in HeLa cells with a low pH optimum and high affinity for pemetrexed distinct from the reduced folate carrier. Clin Cancer Res 10: 6256–6264, 2004 [DOI] [PubMed] [Google Scholar]
- 35. Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature 437: 215–223, 2005 [DOI] [PubMed] [Google Scholar]
- 36. Zhao R, Diop-Bove N, Visentin M, Goldman ID. Mechanisms of membrane transport of folates into cells and across epithelia. Annu Rev Nutr 31: 177–201, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao R, Gao F, Hanscom M, Goldman ID. A prominent low-pH methotrexate transport activity in human solid tumor cells: contribution to the preservation of methotrexate pharmacological activity in HeLa cells lacking the reduced folate carrier. Clin Cancer Res 10: 718–727, 2004 [DOI] [PubMed] [Google Scholar]
- 38. Zhao R, Hanscom M, Chattopadhyay S, Goldman ID. Selective preservation of pemetrexed pharmacological activity in HeLa cells lacking the reduced folate carrier: association with the presence of a secondary transport pathway. Cancer Res 64: 3313–3319, 2004 [DOI] [PubMed] [Google Scholar]
- 39. Zhao R, Min SH, Qiu A, Sakaris A, Goldberg GL, Sandoval C, Malatack JJ, Rosenblatt DS, Goldman ID. The spectrum of mutations in the PCFT gene, coding for an intestinal folate transporter, that are the basis for hereditary folate malabsorption. Blood 110: 1147–1152, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao R, Shin DS, Diop-Bove N, Ovits CG, Goldman ID. Random mutagenesis of the proton-coupled folate transporter (PCFT, SLC46A1), clustering of mutations and the bases for associated losses of function. J Biol Chem 286: 24150–24158, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao R, Shin DS, Fiser A, Goldman ID. Identification of a functionally critical GXXG motif and its relationship to the folate binding site of the proton-coupled folate transporter (PCFT-SLC46A1). Am J Physiol Cell Physiol 303: C673–C681, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao R, Shin DS, Goldman ID. Vulnerability of the cysteine-less proton-coupled folate transporter (PCFT-SLC46A1) to mutational stress associated with the substituted cysteine accessibility method. Biochim Biophys Acta 1808: 1140–1145, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhao R, Unal ES, Shin DS, Goldman ID. Membrane topological analysis of the proton-coupled folate transporter (PCFT-SLC46A1) by the substituted cysteine accessibility method. Biochemistry 49: 2925–2931, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]