

Abstract
Chronic pancreatitis (CP) is a devastating disease characterized by persistent and uncontrolled abdominal pain. Our lack of understanding is partially due to the lack of experimental models that mimic the human disease and also to the lack of validated behavioral measures of visceral pain. The ligand-gated cation channel transient receptor potential ankyrin 1 (TRPA1) mediates inflammation and pain in early experimental pancreatitis. It is unknown if TRPA1 causes fibrosis and sustained pancreatic pain. We induced CP by injecting the chemical agent trinitrobenzene sulfonic acid (TNBS), which causes severe acute pancreatitis, into the pancreatic duct of C57BL/6 trpa1+/+ and trpa1-/- mice. Chronic inflammatory changes and pain behaviors were assessed after 2–3 wk. TNBS injection caused marked pancreatic fibrosis with increased collagen-staining intensity, atrophy, fatty replacement, monocyte infiltration, and pancreatic stellate cell activation, and these changes were reflected by increased histological damage scores. TNBS-injected animals showed mechanical hypersensitivity during von Frey filament probing of the abdomen, decreased daily voluntary wheel-running activity, and increased immobility scores during open-field testing. Pancreatic TNBS also reduced the threshold to hindpaw withdrawal to von Frey filament probing, suggesting central sensitization. Inflammatory changes and pain indexes were significantly reduced in trpa1-/- mice. In conclusion, we have characterized in mice a model of CP that resembles the human condition, with marked histological changes and behavioral measures of pain. We have demonstrated, using novel and objective pain measurements, that TRPA1 mediates inflammation and visceral hypersensitivity in CP and could be a therapeutic target for the treatment of sustained inflammatory abdominal pain.
Keywords: chronic pancreatitis, pain, inflammation, transient receptor potential ankyrin 1, trinitrobenzene sulfonic acid
chronic pancreatitis (CP) is a devastating disease characterized by continuous or recurrent inflammation of the pancreas, progressive and irreversible fibrosis and atrophy, and, in severe cases, loss of exocrine and endocrine function. Severe and uncontrolled abdominal pain is the cardinal feature of CP, but the mechanisms of this pain are poorly understood (7). Our lack of understanding is partly due to the lack of available animal models that mimic the human disease and the lack of validated measures of visceral pain. Increasing evidence (3, 15, 36) supports the importance of the “pancreatic neuropathy theory,” based on the notion that patients with CP have damaged intrapancreatic nerves that are more susceptible to inflammatory mediators, the expression of which directly correlates with pain frequency and intensity (41). This peripheral neural sensitization thereby contributes to sustained pain.
Transient receptor potential ankyrin 1 (TRPA1) is an ion channel expressed by primary afferent nociceptive neurons that is activated by endogenous inflammatory compounds (e.g., products of oxidative stress and cyclopentenone prostaglandins) (4). TRPA1 can also be sensitized by agents found in the inflamed pancreas. Trypsins that are prematurely activated within the inflamed pancreas and play an important role in the pathogenesis of experimental and human CP (44, 54) can activate protease-activated receptor 2 (PAR2) on pancreatic sensory nerves. PAR2 may sensitize TRPA1 to increase neuroexcitability and, thereby, exacerbate neurogenic inflammation and hyperalgesia. We (13) and others (45) previously demonstrated that TRPA1 activation produces early pancreatic inflammation and pain, but nothing is known about the contribution of this channel following the resolution of acute inflammation in the fibrotic, atrophic pancreas.
To study the mechanisms of sustained pain in CP, including the contribution of TRPA1, we adapted a model of painful CP previously established in rats (42, 53) by injecting trinitrobenzene sulfonic acid (TNBS) into the pancreatic duct of healthy mice. After 3 wk, we confirmed the development of pancreatic fibrosis, fatty replacement, chronic inflammatory cell infiltrate, and atrophy. We used graduated von Frey filaments (VFFs) to measure abdominal referred mechanical hypersensitivity as an index of visceral pain. We also evaluated the usefulness of several other, observer-independent, behavioral pain measures. By comparing results in trpa1+/+ and trpa1-/- mice, we were able to determine the contribution of TRPA1 to sustained pancreatic inflammation and pain.
MATERIALS AND METHODS
Animals.
C57BL/6 mice (8–12 wk old) were originally obtained from Charles River Laboratories (Hollister, CA) and bred in our facility; trpa1+/+ and trpa1-/- mice were maintained as heterozygotes, and age-matched littermates were studied (8). All mice were maintained in a temperature-controlled environment with free access to food and water. The University of California San Francisco Institutional Animal Care and Use Committee approved all procedures.
Induction of CP.
To establish a mouse model of painful CP, we induced TNBS pancreatitis, as previously described in rats (42, 52), with several modifications, the most significant of which was a dramatic reduction in the dose and volume of TNBS. Preliminary attempts proved that the protocols used in rats (400 or 500 μl of 2% TNBS in 10% ethanol) had a high mortality rate, even when scaled down by weight. Considerable trial-and-error was needed to adapt the model to the less hearty mice to avoid multiorgan failure and death within the first 48 h after surgery. Ultimately, 35 μl of a 0.75% solution of TNBS, 10% ethanol, and saline produced consistent results and a mortality rate of 22.5% (see below), which is representative of the human situation (38).
After the abdomen was shaved, the mice were anesthetized with isoflurane and antiseptically cleaned, and a laparotomy was performed. It was crucial to the success of the surgery that the tissues, especially duodenum and pancreas, were handled very gently to avoid trauma and nonspecific inflammatory responses. The proximal common bile duct was temporarily occluded with a Hemoclip to prevent flow of infused agents into the liver. The duodenum was identified, very gently exposed, and punctured on the antimesenteric side near the papilla of Vater using a 30-gauge needle. A polyethylene (PE-10) catheter was inserted through the papilla into the pancreatic duct (∼2–3 mm) and held in place with blunted tweezers. Next, a Hamilton syringe was used to infuse 35 μl of a 0.75% solution of TNBS (Sigma, St. Louis, MO) in 10% ethanol in saline, vehicle (10% ethanol in saline), or saline solution slowly and constantly (over a 1-min period) into the pancreatic duct. Success of the procedure was monitored by ensuring the presence of a yellowish color in the pancreas (if TNBS was injected) or enlargement of the gland (if vehicle or saline was injected). The catheter and the Hemoclip were removed before the abdomen was closed. The sham-operated group underwent laparotomy and suture closure of the abdomen, without intraductal injection. All mice were allowed to recover, and buprenorphine (0.1 mg/kg sc) was administered twice a day for 3 days after surgery for postoperative analgesia (per veterinary protocol) and saline solution (0.5 ml ip) was administered for postoperative fluid resuscitation (24). Body weight was monitored daily for up to 3 wk. CP was induced for 3 wk in C57BL/6 mice and for 2 wk in trpa1+/+ and trpa1-/- mice. We chose these 3- and 2-wk time points because they coincided with the greatest differences among groups. The mortality rates in the control groups were 0% for the sham-operated group, 10% for the saline-treated group, and 9% for the ethanol-treated group. In contrast, the mortality rate for the TNBS-treated group was 22.5% within the first 3–4 days, mainly due to severe acute necrotizing pancreatitis, characterized by tissue edema, necrosis, and large areas of hemorrhage. After 2 wk (trpa1+/+ and trpa1-/- mice) or 3 wk (C57BL/6 mice), mice were killed with an overdose of pentobarbital sodium (200 mg/kg ip), and tissues were collected.
Tissue collection.
Blood was collected from the left ventricle and centrifuged (10,000 g for 10 min at 4°C), and the serum was obtained. Pancreatic atrophy was estimated by measurement of the area of an image taken from a consistent distance of the explanted pancreas divided by the area of the adjacent spleen in the same image to adjust for differences in body weight between mice. In one set of mice, pancreata were collected, postfixed in 10% formalin, embedded in paraffin, and processed for histological analysis [hematoxylin-eosin and picrosirius red (PSR) staining]. In another set of mice, fresh pancreata were collected and snap-frozen in liquid nitrogen for Western blot analysis.
Assessment of inflammation.
Four methods were used to establish the presence of chronic inflammatory changes. First, morphological changes were evaluated, and a histological severity score (HSS) was determined as follows. Pancreatic samples were embedded in paraffin, sectioned (5 μm), and stained with hematoxylin-eosin. Pathological changes were evaluated by light microscopy (×20 objective) by an investigator unaware of the experimental groups, as we previously described for acute pancreatitis (13), with some modifications to better characterize the chronicity of this model. The following six categories were assessed on a scale of 0–5: fat replacement, zymogen loss, inflammatory cell infiltration, presence of vacuoles, necrosis, and fibrosis. The scores were tabulated, and the mean value for each group was used as the HSS.
Second, fibrosis was assessed using qualitative and semiquantitative analyses. Pancreatic samples were embedded in paraffin, sectioned (5 μm), and stained with PSR, which has been used to estimate collagen deposition (27). An investigator unaware of the treatment captured two random images per slide with a fluorescence microscope (model IX81, Olympus) fitted with an analyzer (U-ANT, Olympus) and a polarizer (U-POT, Olympus) oriented parallel and orthogonal to each other and equipped with a Spot Flex camera (43). We used two different microscope settings: one for capturing images with the polarizer oriented in parallel and the other with the polarizer oriented orthogonally. Images were then analyzed using Java software (ImageJ, National Institutes of Health, http://rsb.info.nih.gov). Parameters used for analysis and thresholding were maintained across all images taken with the polarizer in each orientation. Quantification of collagen was achieved by dividing the area of the minimal thresholded light passing through the orthogonally oriented polarizer (representing only the fibrous structures) by the thresholded light passing through the parallel-oriented polarizer (representing the whole tissue) and averaged for each experimental group.
Third, since decline in serum amylase has been associated with progression of experimental CP due to the loss of acinar cells (49), serum was assayed for amylase activity, as previously described (13), using Infinity Amylase Liquid Stable Reagent (Thermo Electron, Louisville, CO). Results are expressed in units per liter.
Fourth, since pancreatic stellate cell (PSC) activation and proliferation contribute to extracellular matrix deposition and fibrogenesis in the pancreas (56), activated PSCs were estimated by Western blot analysis using antibodies to α-smooth muscle actin (α-SMA) (see below).
Behavioral assessment of pain.
Pain was assessed using five behavioral approaches. First, we used VFF probing of the abdomen preoperatively and 1–3 wk after intraductal injection/sham. We and others have used VFF probing as a measure of referred abdominal mechanical hypersensitivity (14, 48, 52, 53). Starting 2 days before CP was surgically induced, mice were acclimated for ≥1 h/day in plastic cylinders on a mesh floor. At 1 day before testing, the abdomen was shaved. Abdominal mechanical referred pain was measured by application of calibrated VFFs (North Coast Medical, Morgan Hill, CA) of different applied forces (0.07, 0.16, and 1 g) in ascending order to the upper abdominal area 10 times each for 1–2 s. A response was considered positive when mice raised, retracted, or licked the abdomen (withdrawal response). Data are expressed as the average of the number of withdrawal responses per filament per mouse per experimental group.
Second, since patients with severe chronic pancreatic pain commonly describe hypersensitivity to remote noxious stimuli (10, 18), such as a forearm needle stick, we used VFF probing and techniques previously established in our laboratory (16, 19) to measure somatic pain in the hindpaw. Briefly, VFFs of different applied forces (0.008, 0.04, 0.07, 0.16, 0.4, 0.6, 1, and 2 g) were applied to the plantar aspect of the left hindpaw: if a paw withdrawal response was not observed, a stronger stimulus was applied, and if the withdrawal response was detected, a weaker stimulus was used (the widely employed “up-and-down” paradigm). The responses were tabulated, and the 50% response threshold was determined. Data are expressed as percentage of baseline per mouse.
Third, since patients with severe chronic pancreatic pain have reduced performance status and quality of life (10, 21, 25), which contribute to their overall disability, we tested whether a similar effect could be measured in mice (31). Voluntary wheel running was assessed in regular housing cages equipped with a running wheel (5-in. diameter, Petco, San Diego, CA) fitted with a magnetic switch that allowed recording of the covered distance with an odometer (CatEye Vectra Wireless, Osaka, Japan). Mice were adapted to the running wheel for 1 wk prior to induction of CP. Mice were allowed free access to the running wheel, food, and water at all times. Voluntary wheel-running activity (distance/day) was measured starting 2 days before surgery (average = baseline), and measurements were made daily for up to 3 wk. Results are expressed as a percentage of baseline.
Fourth, we measured catalepsy (time spent immobile within a 10-min period) using the SmartFrame open-field system (Kinder Scientific, Poway, CA), which consisted of a square clear Perspex arena (16 ft × 16 ft) surrounded by clear Perspex walls (15 ft high) in which locomotor and exploratory activities are measured by an automated system of 32 infrared photobeams (16x and 16y). At the same time each day, individual mice were placed in the center of the arena, and exploratory movements were recorded for 10 min. Animals were adapted for 3 days prior to surgery, and the average values served as individual baselines. An open-field test was performed weekly, and the results, normalized to baseline, were analyzed using Motor Monitor Software.
As a fifth behavioral end point, we measured audible and ultrasonic vocalizations. A total of seven mice were recorded before the surgery (baseline) and then weekly after the sham operation (controls) or induction of CP. Vocalizations (1–180,000 Hz) were recorded using an Avisoft Ultrasoundgate microphone (CM16/CMPA) that was placed at a fixed distance (6–8 cm) from the mouse, which was gently restrained and probed on the upper abdomen with a painful (4.56) VFF every 10 s over a 90-s period. The recordings were collected using a National Instruments data acquisition board and processed in Matlab (Natick, MA). Individual calls were detected using a previously described spectral subtraction algorithm (57). Once the background noise was subtracted, the automated algorithm detected all signals >0.01 V. Data are expressed as the average of the number of calls from each mouse.
SDS-PAGE and Western blotting.
Snap-frozen samples (spinal cord and pancreas) were homogenized in Camiolo buffer containing 10 mM NaF, 0.1 mM Na3VO4, complete protease inhibitor cocktail, and phosphatase inhibitor cocktail. Samples (30–40 μg of protein) were separated by SDS-PAGE (12% acrylamide). Membranes were blocked and incubated overnight at 4°C with antibodies to α-SMA (1:1,000 dilution; Abcam, Cambridge, MA) and β-actin (1:10,000 dilution; Sigma). Membranes were treated with the appropriate secondary antibodies conjugated to Alexa Fluor 680 (Invitrogen, Carlsbad, CA) or IRDye 800 (Rockland Immunochemicals, Gilbertsville, PA) and analyzed with an Odyssey Infrared Imaging System (Li-COR Biosciences, Lincoln, NE). To account for loading discrepancies, data are expressed as the ratio of the optical density of the band of interest normalized to the optical density of the β-actin signal.
Statistical analysis.
Results are expressed as means ± SE. Statistical differences were computed using Student's t-test (for comparisons between 2 groups) or 1- or 2-way ANOVA (for comparisons among all 4 groups) followed by Bonferroni's multiple comparisons posttest. P < 0.05 was considered statistically significant.
RESULTS
TNBS induced CP in mice.
Mice in the control groups (sham-operated, saline, and vehicle) lost an average of 6.1% of their original body weight within the first 3 days after the surgery but started to regain weight by day 4 and continued to gain weight until day 21 (105%, 105.2%, and 108.8% of body weight in sham, saline, and vehicle groups, respectively). In contrast, mice treated with TNBS lost 13.9% of their original body weight during weeks 1 and 2 and then slowly started to regain weight during week 3 (101.2% at day 21) (Fig. 1A).
Fig. 1.
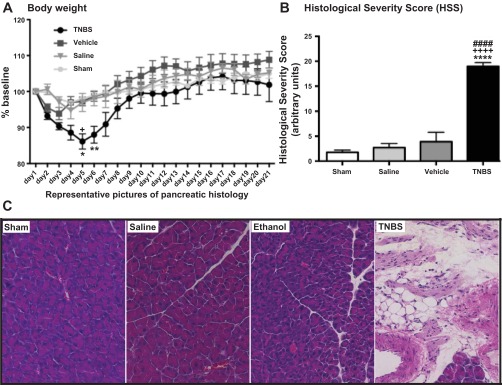
Trinitrobenzene sulfonic acid (TNBS)-induced chronic pancreatitis (CP) decreased body weight (A) and increased histological damage score (B and C) in mice. Mice with TNBS-induced CP lost more body weight (A) and had higher histological severity score (B and C) than did ethanol, saline, and sham-operated control groups. Values are means ± SE (n = 6–10). *P < 0.05, **P < 0.01, ****P < 0.0001 vs. sham. ####P < 0.0001 vs. saline. +P < 0.05, ++++P < 0.0001 vs. vehicle. C: representative hematoxylin-eosin (H & E)-stained histological sections of the pancreas 21 days after instillation of TNBS, vehicle, or saline into the pancreatic duct. Striking features of this CP model were fat replacement, fibrosis, and presence of inflammatory cells.
The pancreas from the sham-operated mice appeared normal, and no morphological changes were detected in the pancreata from saline or vehicle control groups (Fig. 1, B and C). In contrast, pancreata from TNBS-injected animals showed marked inflammatory infiltrates, mainly composed of mononuclear cells (monocytes). There were focal areas of necrosis and other areas with some zymogen loss and vacuolization. The most striking feature of this murine model, however, was the massive loss of acinar cells and their replacement by fat and fibrosis (Fig. 1C). Marked atrophy developed within 3 wk. These findings were present in all animals, and they are consistent with the changes in pancreatic morphology previously reported in rats (42, 52). Quantification of the severity of the histological damage (Fig. 1B) showed that mice treated with TNBS had an 11-fold increase in the HSS compared with the sham group, a 7-fold increase compared with the saline group, and a 5-fold increase compared with the vehicle group.
Serum amylase activity levels ranged from 4,550 to 6,600 U/l for vehicle, saline, and sham-operated animals. Amylase activity was significantly lower in mice treated with TNBS than in the control groups (Fig. 2A). This finding is consistent with loss of exocrine function of the pancreas, which is typical of human patients with CP (1, 2, 20).
Fig. 2.
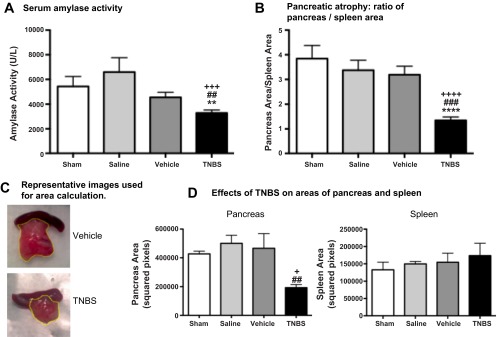
TNBS-induced CP reduced amylase activity (A) and pancreas size (B) in mice. Mice with TNBS-induced CP had significantly less amylase activity than control groups (A), consistent with loss of exocrine functionality of the pancreas. TNBS caused a significant decrease in pancreas size, as quantified (B and D) and shown in representative images (C), consistent with the presence of atrophy in the gland. Spleen size was similar among all experimental groups (D). Values are means ± SE (n = 5–15). **P < 0.05, ****P < 0.0001 vs. sham. ##P < 0.01, ###P < 0.001 vs. saline. +P < 0.05, +++P < 0.001, ++++P < 0.0001 vs. vehicle.
As an index of pancreatic atrophy, using image processing of a photograph taken of the organs removed en bloc, we measured the ratio of the two-dimensional area of the pancreas and spleen. In controls, the pancreas-to-spleen ratio was 3.2–3.8. TNBS injection significantly decreased the ratio to 1.3 (P < 0.0001 vs. sham and vehicle, P < 0.001 vs. saline; Fig. 2, C and D), suggesting marked pancreatic atrophy, which is consistent with findings in humans with severe CP.
Analysis of collagen deposition using the combination of PSR and polarized light showed no fibrosis in sham-operated control mice and minimal change in the saline and vehicle groups. By contrast, mice treated with TNBS had a 40-fold increase in collagen deposition compared with the sham-operated group, a 29-fold increase compared with the saline group, and a 15-fold increase compared with the vehicle group (Fig. 3, A and B). Moreover, TNBS-treated tissues showed a preponderance of larger collagen fibers (deeper red), which have been associated with later stages of scar formation following injury (43). PSCs have been shown to play an important role in promoting pancreatic fibrogenesis, and α-SMA has been used as a marker of activated PSCs (5, 6, 26, 34, 40). Expression of α-SMA by Western blotting was fivefold greater in pancreatic samples from mice treated with TNBS than from vehicle-treated animals (Fig. 3C). Our results confirm that instillation of TNBS into the pancreatic duct of mice induces CP, mainly characterized by acinar atrophy, mononuclear cell infiltrate, and marked fibrosis.
Fig. 3.

TNBS-induced CP increased pancreatic fibrosis in mice. Fibrosis was measured by immunohistochemistry and Western blotting: picrosirius red was used to stain collagen deposition (A and B), and Western blotting to α-smooth muscle actin (α-SMA) was used to measure activation of pancreatic stellate cells (PSCs, C), which have been shown top play a major role in promoting pancreatic fibrosis. Mice with TNBS-induced CP had robust pancreatic collagen deposition, which was absent in all control groups, as shown in representative images in A. EtOH, ethanol. Superficial area of stained collagen was significantly increased in mice with CP (B). Mice with TNBS-induced CP also had increased activation of PSCs compared with vehicle-treated controls (C). Values are means ± SE (n = 7–9). *P < 0.05, ***P < 0.001 vs. sham. #P < 0.05 vs. saline. +++P < 0.001 vs. vehicle.
Pancreatic TNBS increased pain-related behaviors and decreased spontaneous activity. Abdominal VFF probing, a well-established method to measure referred visceral pain in rodents (48, 53), showed a slight increase in abdominal mechanical sensitivity to the thinnest and intermediate filaments at 1 wk in the saline and sham-operated groups, likely due to the effect of the abdominal surgery, since scores returned to baseline by 2 wk. Mice treated with TNBS had markedly higher abdominal pain scores at all time points than sham-operated and saline- and vehicle-treated mice (Fig. 4B). We observed increased sensitivity to a subthreshold stimulus (“allodynia,” thin filament, with applied force of 0.07 g) and increased responses to painful stimuli (“hypersensitivity,” thicker filaments, with applied force of 0.16 and 1 g). We were somewhat surprised to find mildly increased mechanical sensitivity scores at 2 and 3 wk in the vehicle group compared with the other two control groups. While the magnitude was much less than that seen with TNBS, these results suggest that intraductal ethanol causes some degree of nociceptive pathway activation.
Fig. 4.

TNBS-induced CP decreases voluntary wheel-running activity (A) and increases pain behaviors (B–D). In mice with TNBS-induced CP, voluntary wheel-running activity decreased dramatically within the first 2 wk compared with control groups (A). Mechanical sensitivity was measured with calibrated (0.07, 0.16, and 1 g) Von Frey filaments (VFFs). Mice were probed on the abdomen (B) and hindpaw (C). In both cases, mice with TNBS-induced CP had increased mechanical sensitivity compared with control groups. D: mice with CP also showed significantly more catalepsy (immobility measured over a 10-min period using the open-field apparatus) than sham-operated controls at all time points. Thus all the behavioral end points strongly support the presence of abdominal and generalized hypersensitivity in mice with TNBS-induced CP. Values are means ± SE (n = 6–10). *P < 0.5, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. sham-operated.
Voluntary wheel running has been recently established as a nonreflexive test for evaluating somatic inflammatory pain and analgesia (17), but data have not been published for visceral pain. As shown in Fig. 4A, mice treated with TNBS ran the wheel significantly less than mice from all three control groups, indicating that visceral pain was inversely related to spontaneous activity.
Using the open-field apparatus, we found significantly greater periods of immobility at each time point assessed after induction of CP in TNBS-injected mice than sham-operated controls (Fig. 4D). These results support our data obtained using the running wheel and suggest that impaired mobility among animals with CP could be a reflection of visceral pain.
We sought to determine if stimuli remote from the abdomen would also produce enhanced responses. Using the up-down paradigm (16), we found that TNBS-treated mice had a significant decrease in their mechanical threshold to probing of the hindpaw at all time points compared with the three control groups (Fig. 4C), which did not differ from baseline values. These results suggest that mice with CP had “generalized hypersensitivity,” which could reflect the “generalized hyperalgesia” seen in CP patients (K. S. Kirkwood, clinical observation).
Ultrasonic vocalizations have been reported to be a behavioral indicator of pain in adult mice (30, 32). We measured the number of calls and the “type” of call (pain-related call vs. annoyance-related call) in the audible and the ultrasonic range over a 90-s period of upper abdominal probing with a thick VFF during the first 2 wk after TNBS/sham operations, after a pilot study showed no difference in spontaneous calls. There were no significant differences in the overall number of calls or type of call between the two groups (data not shown). Moreover, the calls did not have a consistent pattern over time. Our results suggest that measurement of audible and/or ultrasonic vocalizations may not be a suitable method to assess or quantify chronic pancreatic pain behavior in mice.
TRPA1 mediates TNBS-induced CP.
We recently demonstrated that TRPA1 contributes to inflammation and pain during experimental acute pancreatitis (13) and colitis (12). To evaluate the role of TRPA1 in CP, we injected TNBS into the pancreatic duct of trpa1+/+ and trpa1-/- mice and collected tissue after 2 wk, the time point at which we observed the greatest differences among groups during the first set of experiments. As expected, TNBS caused a robust increase in HSS in trpa1+/+ mice (Fig. 5, A and D), similar to that observed at 3 wk. Striking features were fibrosis, fatty replacement, and cellular infiltration. In trpa1-/- mice, the histological inflammatory damage was markedly reduced, and the HSS was lower (12 vs. 17). Serum amylase activity was reduced after 2 wk in trpa1+/+, but not trpa1-/-, mice (Fig. 5B). No significant differences between trpa1+/+ and trpa1-/- mice were detected when we compared the pancreatic atrophy results (Fig. 5C). Our findings confirm that TRPA1 promotes chronic inflammatory changes in the pancreas.
Fig. 5.
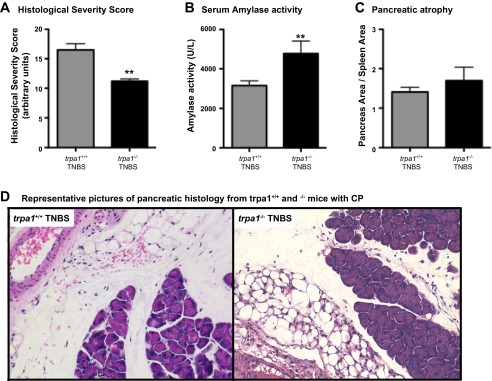
Transient receptor potential ankyrin 1 (TRPA1) mediates TNBS-induced CP. CP was induced by intraductal injection of TNBS in trpa1+/+ and trpa1-/- mice. HSS was decreased (A and D) and amylase activity was increased (B) in trpa1-/- compared with trpa1+/+ mice, suggesting that TRPA1 is involved in mediating inflammation in this CP model. C: no significant differences between the 2 genotypes were detected when pancreatic atrophy was measured. Values are means ± SE (n = 5–10). **P < 0.01 vs. trpa1+/+ TNBS.
TRPA1 mediates TNBS-induced chronic pancreatic pain.
TNBS increased abdominal mechanical hypersensitivity responses in trpa1+/+ mice. By contrast, responses in trpa1-/- mice were markedly reduced and were not significantly different from control animals at all assessed time points (Fig. 6A). Similar results were obtained when mechanical sensitivity was assessed on mouse hindpaw: TNBS significantly decreased the mechanical threshold in trpa1+/+, but not trpa1-/-, mice (Fig. 6B). These findings suggest that TRPA1 is required for TNBS-induced CP inflammatory pain.
Fig. 6.
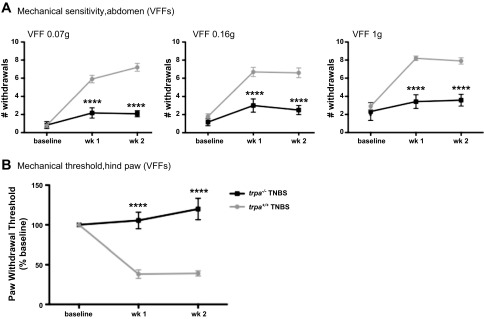
TRPA1 mediates TNBS-induced CP hypersensitivity. CP was induced by intraductal injection of TNBS in trpa1+/+ and trpa1-/- mice: trpa1 -/- mice showed no mechanical sensitivity, as suggested by lack of increase in referred abdominal (A) hypersensitivity and hindpaw sensitivity threshold (B) compared with trpa1+/+ mice. These findings suggest that TRPA1 is involved in activating pain pathways in this model of CP. Values are means ± SE (n = 6–10). ****P < 0.0001 vs. trpa1+/+ TNBS.
DISCUSSION
Sustained visceral pain is the hallmark of human CP. Lack of suitable murine models and reliable behavioral measures of abdominal pain have significantly retarded progress in our understanding of pancreatic pain. In the present study, injection of TNBS into the mouse pancreatic duct produced a model of CP with many similarities to the human condition. The histological picture of severe fibrosis, monocyte infiltration, fatty replacement, and atrophy mimic the findings reported in humans who have had the pancreas removed as a treatment for chronic pancreatic pain (29). TNBS led to physiological changes of weight loss and decline in serum amylase levels. These changes were accompanied by the development of pain-related behaviors, including referred abdominal mechanical hypersensitivity, immobility, and reduced voluntary activity. To evaluate the role of TRPA1 in chronic inflammation and pain, we injected TNBS into the pancreatic duct of trpa1-/- and trpa1+/+ mice. Animals lacking functional TRPA1 channels had significantly less inflammation and fibrosis, as evidenced by a decrease in the HSS, as well as markedly less severe pain-related behaviors. In fact, TNBS-injected trpa1-/- animals had abdominal mechanical sensitivity scores and hindpaw mechanical thresholds similar to those of saline-injected controls (cf. Fig. 6 with Fig. 4). We previously showed a similar effect in a colitis model, and we believe that these findings support the contribution of central mechanisms of sustained visceral pain sensitization (12).
We recently localized functional TRPA1 on pancreatic nociceptive neurons and found that activation causes inflammation and pain in a cerulein model of acute pancreatitis (13). We now show that TRPA1 is important in inflammation and pain in CP. Interestingly, serum amylase was also preserved in trpa1-/- animals with CP, suggesting a possible effect of TRPA1 on pancreatic exocrine function. Given the known contribution of TRPA1 to inflammation, which we and others have previously shown, the simplest explanation seems to be that reduced inflammation contributes to less acinar necrosis in animals without TRPA1, but, indeed, we cannot exclude a direct effect. TRPA1 has been found on lacrimal and submandibular gland acinar cells (9, 23). Nevertheless, we are unaware of any reports in pancreatic acinar cells. We previously identified this channel on nerve bundles within the pancreas running between acinar cells (12).
TRPA1 can be activated by products of lipid membrane peroxidation and oxidative stress (33, 47), and it can be sensitized by activation of PAR2, which responds to a wide variety of noxious inflammatory stimuli. Moreover, the intrinsic properties of TRPA1 are modulated by transient receptor potential vanilloid 1 (TRPV1), most likely through a direct physical interaction between the two channels to form a complex. This complex would account for the functional interactions and cross talk between TRPA1 and TRPV1, leading to a more regulated control of transduction of nociceptive signals (46). In the pancreas, early evidence suggests that TRPA1 and TRPV1 act together to produce acute inflammation and pain, and this synergistic interaction is essential to development of early pancreatitis (45). Further studies are needed to identify which endogenous agonists that are generated in CP directly activate or sensitize TRPA1 to produce pain. To our knowledge, this is the first evidence that TRPA1 contributes to chronic pancreatic inflammation and pain.
Although pancreatic fibrosis and atrophy have been induced in the mouse by repeated serial cerulein injections (37) or genetic modifications (35), these models have not been shown to be painful. In the rat, the TNBS surgical model of CP has been shown to be painful (52, 55), but this model has typically been lethal in mice. Since our pilot necropsy data showed that the mice died from severe acute pancreatitis, we adapted the model by reducing the magnitude of the insult and by providing postoperative fluid resuscitation, which has been shown to reduce early mortality in humans with severe pancreatitis (24). The resultant model is similar to that developed in rats, which has been characterized by a significant decrease in body weight, serum amylase activity (an index of pancreatic exocrine function), and pancreatic area (an index of pancreatic atrophy) with an increase in histological severity score, acinar atrophy, fibrosis, and fat replacement (42, 52). Thus this model has excellent face validity compared with the human condition. As with the other commonly used models of CP, the initiating cause of the inflammatory cascade is unlikely to mimic the human condition. In CP of different etiologies, the subsequent neuroimmune response, acinar injury and loss, stellate cell activation, fibrosis, and clinical sequelae are relatively similar (22). While there are some differences, to a large degree, these various initial insults seem to converge on a common pathway, suggesting that this is a useful model for the study of pain in CP in mice (52).
We found several quantitative tools to be useful techniques for grading the severity of the chronic inflammatory and fibrotic changes in this model. The gross observation of pancreatic atrophy was pronounced after 2–3 wk. We developed a quick and inexpensive method to estimate atrophy by taking a photo at a fixed distance from the explanted organs to compare the ratio of the area of pancreas to spleen in the acquired image, thereby normalizing for differences in animal size. We also found that PSR staining with polarized light and α-SMA Western blotting provided useful measures of collagen deposition and activation of myofibroblasts, which were similar in our model at 2 wk. There is increasing evidence that activated stellate cells have an important role in expression of α-SMA, instead of desmin, and as the main source of collagen in the pancreas of humans and rats with CP (5, 26).
VFF probing of the abdomen confirmed that TNBS-induced CP is painful. VFF probing is an established measure of visceral referred mechanical hypersensitivity (48, 52, 53) that mimics abdominal tenderness reported when pressure is applied to the abdomen of patients with CP. This technique requires extensive experience to produce reliable results, especially in mice. We used VFF probing to confirm that our model is painful and to test the usefulness of other potential measures of chronic visceral pain, including voluntary wheel running, immobility, and acoustic and ultrasonic vocalizations, all of which are less operator-dependent. Our results suggest that recordings of spontaneous activity and immobility are useful for the assessment of chronic pancreatic pain. Voluntary wheel running has been recently demonstrated to be a nonreflexive test to evaluate somatic inflammatory pain and analgesia in mice (17). Our results suggest that it may also be a useful, operator-independent, measure of visceral pain. We noticed that spontaneous activity slightly increased at week 3 compared with week 2, while the pain levels did not change. This apparent discrepancy could be due to physiological adaptation to pain to maintain functions such as eating behaviors. Alternatively, it is possible that, with continued atrophy of the pancreas, pain improves, as it does in some patients with prolonged atrophy, and that mobility is an early predictor of this improvement. Studies conducted over a longer duration would be necessary to determine if other measures of pain also improved.
There is one report in which parameters measured using open-field testing were used as indexes of early inflammatory visceral pain (45). To our knowledge, ours is the first report in which open-field measures, which we validated using VFF probing, were used to measure sustained visceral pain. In contrast, although ultrasonic vocalizations have been used to measure cancer pancreatic pain (32), we did not detect any difference when we compared calls between sham-operated and TNBS mice. Our results are consistent with other studies (50, 51) that concluded that vocalizations are poor metrics for pain in laboratory rodents. Our findings of reduced voluntary wheel running and greater immobility likely reflect the integration of supraspinal centers and, thus, may more accurately reflect the impact of pain on behavior than do the reflexive tests.
Since patients with CP often report a generalized status of hyperalgesia (10, 11, 39), we measured the mechanical threshold not just in the abdominal area, but also in the hindpaw, of mice with and without CP. Our results show a significant decrease in hindpaw mechanical threshold in mice treated with TNBS compared with all control groups. These findings are in disagreement with prior studies in rats (28, 42). This discrepancy could be due to our use of mice, instead of rats, the relative severity of chronic inflammation or pain, or other experimental differences. Hypersensitivity to stimuli remote from the chronically inflamed organ suggests the presence of central mechanisms of sensitization.
A major advantage of a validated painful model of CP in mice is the potential to study genetically modified animals in which a pain pathway mediator of interest has been deleted to determine its contribution. This technique is particularly helpful in the study of chronic inflammatory pain, since repeated administration of antagonists, if available, may be impractical or ineffective due to the pharmacokinetics of the agents.
To summarize, we have characterized a painful murine model of CP that resembles the human features of the disease and established investigator-independent behavioral measures of visceral pain. We have also demonstrated, for the first time, that TRPA1 plays a major role in mediating chronic inflammatory changes in the pancreas and sustained visceral pain. In conclusion, our findings provide new tools to better investigate visceral pain in mice and suggest a potential therapeutic target for the treatment of chronic pancreatic pain.
GRANTS
This work was supported by National Institutes of Health Grants DK-046285 (K. S. Kirkwood), T32 DK-007573 (K. S. Kirkwood), R01 AR-059402-01 (M. Steinhoff), and University of California San Francisco Clinical and Translational Science Institute Grant TL1 RR-024129 (C. Johnson).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
F. Cattaruzza and K.S.K. are responsible for conception and design of the research; F. Cattaruzza, C.J., A.L., F. Cevikbas, W.J.C., S.B., and R.K. performed the experiments; F. Cattaruzza, C.J., A.L., E.F.G., A.K.S., B.J.M., and M.S. analyzed the data; F. Cattaruzza and K.S.K. interpreted the results of the experiments; F. Cattaruzza prepared the figures; F. Cattaruzza drafted the manuscript; F. Cattaruzza, B.J.M., N.W.B., and K.S.K. edited and revised the manuscript; F. Cattaruzza and K.S.K. approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank David Julius for trpa1 knockout mice, Irene Acerbi and Valerie Weaver for assistance with PSR quantification, Anna Brodski for assistance with mice handling, and Pankaj Jay Pasricha for helpful discussions in developing the model.
REFERENCES
- 1. Aghdassi AA, Mayerle J, Christochowitz S, Weiss FU, Sendler M, Lerch MM. Animal models for investigating chronic pancreatitis. Fibrogenesis Tissue Repair 4: 26, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ammann RW, Akovbiantz A, Largiader F, Schueler G. Course and outcome of chronic pancreatitis. Longitudinal study of a mixed medical-surgical series of 245 patients. Gastroenterology 86: 820–828, 1984 [PubMed] [Google Scholar]
- 3. Anaparthy R, Pasricha PJ. Pain and chronic pancreatitis: is it the plumbing or the wiring? Curr Gastroenterol Rep 10: 101–106, 2008 [DOI] [PubMed] [Google Scholar]
- 4. Andersson DA, Gentry C, Moss S, Bevan S. Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci 28: 2485–2494, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 43: 128–133, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Apte MV, Haber PS, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut 44: 534–541, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barreto SG, Saccone GT. Pancreatic nociception—revisiting the physiology and pathophysiology. Pancreatology 12: 104–112, 2012 [DOI] [PubMed] [Google Scholar]
- 8. Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124: 1269–1282, 2006 [DOI] [PubMed] [Google Scholar]
- 9. Bessac BF, Jordt SE. Breathtaking TRP channels: TRPA1 and TRPV1 in airway chemosensation and reflex control. Physiology (Bethesda) 23: 360–370, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buscher HC, van Goor H, Sweep CG, Lenders JW, Wilder-Smith OH. Increased sympathetic activity in chronic pancreatitis patients is associated with hyperalgesia. J Pain Palliat Care Pharmacother 24: 362–366, 2010 [DOI] [PubMed] [Google Scholar]
- 11. Buscher HC, Wilder-Smith OH, van Goor H. Chronic pancreatitis patients show hyperalgesia of central origin: a pilot study. Eur J Pain 10: 363–370, 2006 [DOI] [PubMed] [Google Scholar]
- 12. Cattaruzza F, Spreadbury I, Miranda-Morales M, Grady EF, Vanner S, Bunnett NW. Transient receptor potential ankyrin-1 has a major role in mediating visceral pain in mice. Am J Physiol Gastrointest Liver Physiol 298: G81–G91, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ceppa E, Cattaruzza F, Lyo V, Amadesi S, Pelayo JC, Poole DP, Vaksman N, Liedtke W, Cohen DM, Grady EF, Bunnett NW, Kirkwood KS. Transient receptor potential ion channels V4 and A1 contribute to pancreatitis pain in mice. Am J Physiol Gastrointest Liver Physiol 299: G556–G571, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ceppa EP, Lyo V, Grady EF, Knecht W, Grahn S, Peterson A, Bunnett NW, Kirkwood KS, Cattaruzza F. Serine proteases mediate inflammatory pain in acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 300: G1033–G1042, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ceyhan GO, Demir IE, Rauch U, Bergmann F, Muller MW, Buchler MW, Friess H, Schafer KH. Pancreatic neuropathy results in “neural remodeling” and altered pancreatic innervation in chronic pancreatitis and pancreatic cancer. Am J Gastroenterol 104: 2555–2565, 2009 [DOI] [PubMed] [Google Scholar]
- 16. Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53: 55–63, 1994 [DOI] [PubMed] [Google Scholar]
- 17. Cobos EJ, Ghasemlou N, Araldi D, Segal D, Duong K, Woolf CJ. Inflammation-induced decrease in voluntary wheel running in mice: a nonreflexive test for evaluating inflammatory pain and analgesia. Pain 153: 876–884, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Drewes AM, Gratkowski M, Sami SA, Dimcevski G, Funch-Jensen P, Arendt-Nielsen L. Is the pain in chronic pancreatitis of neuropathic origin? Support from EEG studies during experimental pain. World J Gastroenterol 14: 4020–4027, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eilers H, Cattaruzza F, Nassini R, Materazzi S, Andre E, Chu C, Cottrell GS, Schumacher M, Geppetti P, Bunnett NW. Pungent general anesthetics activate transient receptor potential-A1 to produce hyperalgesia and neurogenic bronchoconstriction. Anesthesiology 112: 1452–1463, 2010 [DOI] [PubMed] [Google Scholar]
- 20. Etemad B, Whitcomb DC. Chronic pancreatitis: diagnosis, classification, and new genetic developments. Gastroenterology 120: 682–707, 2001 [DOI] [PubMed] [Google Scholar]
- 21. Fitzsimmons D, Kahl S, Butturini G, van Wyk M, Bornman P, Bassi C, Malfertheiner P, George SL, Johnson CD. Symptoms and quality of life in chronic pancreatitis assessed by structured interview and the EORTC QLQ-C30 and QLQ-PAN26. Am J Gastroenterol 100: 918–926, 2005 [DOI] [PubMed] [Google Scholar]
- 22. Friess H, Shrikhande S, Shrikhande M, Martignoni M, Kulli C, Zimmermann A, Kappeler A, Ramesh H, Buchler M. Neural alterations in surgical stage chronic pancreatitis are independent of the underlying aetiology. Gut 50: 682–686, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fujita F, Uchida K, Moriyama T, Shima A, Shibasaki K, Inada H, Sokabe T, Tominaga M. Intracellular alkalization causes pain sensation through activation of TRPA1 in mice. J Clin Invest 118: 4049–4057, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gardner TB, Vege SS, Chari ST, Petersen BT, Topazian MD, Clain JE, Pearson RK, Levy MJ, Sarr MG. Faster rate of initial fluid resuscitation in severe acute pancreatitis diminishes in-hospital mortality. Pancreatology 9: 770–776, 2009 [DOI] [PubMed] [Google Scholar]
- 25. Glasbrenner B, Adler G. Evaluating pain and the quality of life in chronic pancreatitis. Int J Pancreatol 22: 163–170, 1997 [DOI] [PubMed] [Google Scholar]
- 26. Haber PS, Keogh GW, Apte MV, Moran CS, Stewart NL, Crawford DH, Pirola RC, McCaughan GW, Ramm GA, Wilson JS. Activation of pancreatic stellate cells in human and experimental pancreatic fibrosis. Am J Pathol 155: 1087–1095, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hofer MA, Brunelli SA, Shair HN. Ultrasonic vocalization responses of rat pups to acute separation and contact comfort do not depend on maternal thermal cues. Dev Psychobiol 26: 81–95, 1993 [DOI] [PubMed] [Google Scholar]
- 28. Hoogerwerf WA, Gondesen K, Xiao SY, Winston JH, Willis WD, Pasricha PJ. The role of mast cells in the pathogenesis of pain in chronic pancreatitis. BMC Gastroenterol 5: 8, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kloppel G, Detlefsen S, Feyerabend B. Fibrosis of the pancreas: the initial tissue damage and the resulting pattern. Virchows Arch 445: 1–8, 2004 [DOI] [PubMed] [Google Scholar]
- 30. Ko SW, Chatila T, Zhuo M. Contribution of CaMKIV to injury and fear-induced ultrasonic vocalizations in adult mice. Mol Pain 1: 10, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krug HE, Frizelle S, McGarraugh P, Mahowald ML. Pain behavior measures to quantitate joint pain and response to neurotoxin treatment in murine models of arthritis. Pain Med 10: 1218–1228, 2009 [DOI] [PubMed] [Google Scholar]
- 32. Lindsay TH, Jonas BM, Sevcik MA, Kubota K, Halvorson KG, Ghilardi JR, Kuskowski MA, Stelow EB, Mukherjee P, Gendler SJ, Wong GY, Mantyh PW. Pancreatic cancer pain and its correlation with changes in tumor vasculature, macrophage infiltration, neuronal innervation, body weight and disease progression. Pain 119: 233–246, 2005 [DOI] [PubMed] [Google Scholar]
- 33. Macpherson LJ, Xiao B, Kwan KY, Petrus MJ, Dubin AE, Hwang S, Cravatt B, Corey DP, Patapoutian A. An ion channel essential for sensing chemical damage. J Neurosci 27: 11412–11415, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mews P, Phillips P, Fahmy R, Korsten M, Pirola R, Wilson J, Apte M. Pancreatic stellate cells respond to inflammatory cytokines: potential role in chronic pancreatitis. Gut 50: 535–541, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miyamoto T, Nakamura H, Nagashio Y, Asaumi H, Harada M, Otsuki M. Overexpression of Smad6 exacerbates pancreatic fibrosis in murine caerulein-induced chronic pancreatic injuries. Pancreas 39: 385–391, 2010 [DOI] [PubMed] [Google Scholar]
- 36. Navaneethan U, Venkataraman J. Recent advancements in the pathogenesis of pain in chronic pancreatitis: the argument continues. Minerva Gastroenterol Dietol 56: 55–63, 2010 [PubMed] [Google Scholar]
- 37. Neuschwander-Tetri BA, Burton FR, Presti ME, Britton RS, Janney CG, Garvin PR, Brunt EM, Galvin NJ, Poulos JE. Repetitive self-limited acute pancreatitis induces pancreatic fibrogenesis in the mouse. Dig Dis Sci 45: 665–674, 2000 [DOI] [PubMed] [Google Scholar]
- 38. Obideen K, Paul Y, Mohammad W. Pancreatitis, Chronic (Online). http://emedicine.medscape.com/article/181554-overview [2008].
- 39. Olesen SS, Brock C, Krarup AL, Funch-Jensen P, Arendt-Nielsen L, Wilder-Smith OH, Drewes AM. Descending inhibitory pain modulation is impaired in patients with chronic pancreatitis. Clin Gastroenterol Hepatol 8: 724–730, 2010 [DOI] [PubMed] [Google Scholar]
- 40. Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest 117: 50–59, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pasricha PJ. Unraveling the mystery of pain in chronic pancreatitis. Nat Rev Gastroenterol Hepatol 9: 140–151, 2012 [DOI] [PubMed] [Google Scholar]
- 42. Puig-Divi V, Molero X, Salas A, Guarner F, Guarner L, Malagelada JR. Induction of chronic pancreatic disease by trinitrobenzene sulfonic acid infusion into rat pancreatic ducts. Pancreas 13: 417–424, 1996 [DOI] [PubMed] [Google Scholar]
- 43. Rich L, Whittaker P. Collagen and picrosirius red staining: a polarized light assessment of fibrillar hue and spatial distribution. Braz J Morphol Sci 22: 97–104, 2005 [Google Scholar]
- 44. Romac JM, Ohmuraya M, Bittner C, Majeed MF, Vigna SR, Que J, Fee BE, Wartmann T, Yamamura K, Liddle RA. Transgenic expression of pancreatic secretory trypsin inhibitor-1 rescues SPINK3-deficient mice and restores a normal pancreatic phenotype. Am J Physiol Gastrointest Liver Physiol 298: G518–G524, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schwartz ES, Christianson JA, Chen X, La JH, Davis BM, Albers KM, Gebhart GF. Synergistic role of TRPV1 and TRPA1 in pancreatic pain and inflammation. Gastroenterology 140: 1283–1291, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Staruschenko A, Jeske NA, Akopian AN. Contribution of TRPV1-TRPA1 interaction to the single channel properties of the TRPA1 channel. J Biol Chem 285: 15167–15177, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Trevisani M, Siemens J, Materazzi S, Bautista DM, Nassini R, Campi B, Imamachi N, Andre E, Patacchini R, Cottrell GS, Gatti R, Basbaum AI, Bunnett NW, Julius D, Geppetti P. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci USA 104: 13519–13524, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vera-Portocarrero LP, Lu Y, Westlund KN. Nociception in persistent pancreatitis in rats: effects of morphine and neuropeptide alterations. Anesthesiology 98: 474–484, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wakabayashi A. Serum amylase isozyme changes in chronic pancreatitis and their clinical significance. Gastroenterol Jpn 11: 196–202, 1976 [DOI] [PubMed] [Google Scholar]
- 50. Wallace VC, Norbury TA, Rice AS. Ultrasound vocalisation by rodents does not correlate with behavioural measures of persistent pain. Eur J Pain 9: 445–452, 2005 [DOI] [PubMed] [Google Scholar]
- 51. Williams WO, Riskin DK, Mott AK. Ultrasonic sound as an indicator of acute pain in laboratory mice. J Am Assoc Lab Anim Sci 47: 8–10, 2008 [PMC free article] [PubMed] [Google Scholar]
- 52. Winston JH, He ZJ, Shenoy M, Xiao SY, Pasricha PJ. Molecular and behavioral changes in nociception in a novel rat model of chronic pancreatitis for the study of pain. Pain 117: 214–222, 2005 [DOI] [PubMed] [Google Scholar]
- 53. Winston JH, Toma H, Shenoy M, He ZJ, Zou L, Xiao SY, Micci MA, Pasricha PJ. Acute pancreatitis results in referred mechanical hypersensitivity and neuropeptide up-regulation that can be suppressed by the protein kinase inhibitor k252a. J Pain 4: 329–337, 2003 [DOI] [PubMed] [Google Scholar]
- 54. Witt H, Luck W, Hennies HC, Classen M, Kage A, Lass U, Landt O, Becker M. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet 25: 213–216, 2000 [DOI] [PubMed] [Google Scholar]
- 55. Xu GY, Winston JH, Shenoy M, Yin H, Pendyala S, Pasricha PJ. Transient receptor potential vanilloid 1 mediates hyperalgesia and is up-regulated in rats with chronic pancreatitis. Gastroenterology 133: 1282–1292, 2007 [DOI] [PubMed] [Google Scholar]
- 56. Yang L, Shen J, He S, Hu G, Wang F, Xu L, Dai W, Xiong J, Ni J, Guo C, Wan R, Wang X. l-Cysteine administration attenuates pancreatic fibrosis induced by TNBS in rats by inhibiting the activation of pancreatic stellate cell. PLos One 7: e31807, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Young DM, Schenk AK, Yang SB, Jan YN, Jan LY. Altered ultrasonic vocalizations in a tuberous sclerosis mouse model of autism. Proc Natl Acad Sci USA 107: 11074–11079, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]