

Abstract
Brain damage due to severe hypoglycemia occurs in insulin-treated people with diabetes. This study tests the hypothesis that chronic insulin therapy that normalizes elevated blood glucose in diabetic rats would be neuroprotective against brain damage induced by an acute episode of severe hypoglycemia. Male Sprague-Dawley rats were split into three groups: 1) control, non-diabetic; 2) STZ-diabetic; and 3) insulin-treated STZ-diabetic. After 3 wk of chronic treatment, unrestrained awake rats underwent acute hyperinsulinemic severe hypoglycemic (10–15 mg/dl) clamps for 1 h. Rats were subsequently analyzed for brain damage and cognitive function. Severe hypoglycemia induced 15-fold more neuronal damage in STZ-diabetic rats compared with nondiabetic rats. Chronic insulin treatment of diabetic rats, which nearly normalized glucose levels, markedly reduced neuronal damage induced by severe hypoglycemia. Fortunately, no cognitive defects associated with the hypoglycemia-induced brain damage were observed in any group. In conclusion, antecedent blood glucose control represents a major modifiable therapeutic intervention that can afford diabetic subjects neuroprotection against severe hypoglycemia-induced brain damage.
Keywords: brain damage, diabetes, severe hypoglycemia
severe hypoglycemia is a side effect of insulin therapy affecting ∼40% of insulin-treated diabetics (7). Insulin-treated diabetic patients experience an average of two symptomatic hypoglycemic episodes per week, whereas severe hypoglycemia is experienced approximately once a year (7). By depriving the brain of glucose, severe hypoglycemia can lead to brain damage. The cortex and hippocampus are especially sensitive to the brain-damaging effects of severe hypoglycemia (5). Deficits in learning and memory have been shown to be a direct consequence of severe hypoglycemia-induced hippocampal neuronal damage (2, 22, 23). Clinical research has shown that severe hypoglycemia induces cognitive deficits in some (13, 15, 19, 28) but not all studies (3, 11, 12). Animal studies have shown that rats exposed to severe hypoglycemia perform worse in a Morris water maze (17), indicating that brain damage is functionally related to poorer cognitive performance. Since uncontrolled diabetes magnifies the extent of severe hypoglycemia-induced brain damage (5), it was hypothesized that chronic hyperglycemia associated with chronic insulin deficiency induces a maladaptive process that predisposes the brain to more extensive hypoglycemia-induced damage. Insulin has been shown to protect against ischemia-induced neuronal death both in vitro (8) and in vivo (10), but the potential neuroprotective effects of long-term insulin therapy for diabetic animals have yet to be examined in response to severe hypoglycemia-induced brain damage.
Therefore, the aim of this study was to determine whether chronic insulin therapy resulting in the near-normalization of glycemia could protect diabetic rats from hippocampal and cortical brain damage induced by a single episode of severe hypoglycemia. It was also determined whether this neuroprotective effect would also safeguard against hypoglycemia-induced deficits in learning and memory.
MATERIALS AND METHODS
Animals and surgery.
Nine-week-old male Sprague-Dawley rats (Charles River Laboratories) were housed individually in temperature- and light-controlled environments and fed ad libitum. All studies were done in accordance with and approved by the Animal Studies Committee at Washington University School of Medicine. In rats destined to undergo severe hypoglycemia, indwelling catheters were implanted under anesthesia in the left carotid artery and right jugular vein, as described previously (5) (Fig. 1). In rats not destined to undergo hypoglycemia, the left carotid artery and right jugular vein were ligated under anesthesia.
Fig. 1.
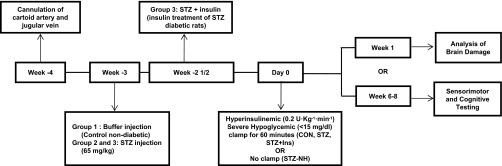
Experimental protocol. After recovery from arterial and venous cannulation, rats were injected with streptozotocin (STZ) or vehicle (CON; n = 18). Three days later, insulin treatment was started in a subset of the STZ-diabetic rats (STZ + Ins; n = 15) by implanting an insulin pellet and starting insulin injections as needed to maintain blood glucose <250 mg/dl. About 3 wk later, hyperinsulinemic (0.2 U·kg−1·min−1) severe hypoglycemic (10–15 mg/dl) clamps were performed in all rats, except for a group of diabetic rats that were not subjected to the severe hypoglycemic clamp (STZ-NH). After the episode of severe hypoglycemia, either neuronal damage was assessed by Flouro-Jade B and hematoxylin-eosin staining 1 wk later or rats were studied 6–8 wk later with sensorimotor and cognitive testing. Brain damage study: CON (n = 6), STZ (n = 6), and STZ + Ins (n = 6); cognitive testing study: CON (n = 12), STZ (n = 13), STZ + Ins (n = 12), and STZ-NH (n = 10).
Induction of diabetes.
Three days postsurgery, rats received intraperitoneal injections of either streptozotocin (STZ) (65 mg/kg, n = 37; Sigma) to induce diabetes or vehicle 0.1 mM sodium citrate buffer (Fisher Scientific) for nondiabetic controls (CON; n = 18). Some rats were injected a second time with STZ to ensure random glucose of >300 mg/dl (Ascensia Contour BG monitors; Bayer HealthCare, Mishawaka, IN) (Fig. 2A).
Fig. 2.

Glucose levels during the protocol. A: blood glucose levels throughout the experiment. The nondiabetic control (CON, n = 18; ●) rats had glucose levels of ∼100 mg/dl. After STZ injection, STZ-diabetic rats (n = 19; ○) and STZ-diabetic rats not exposed to hypoglycemia (STZ-NH, n = 10; △) had glucose levels of >450 mg/dl. The group of diabetic rats chronically treated with insulin (STZ + Ins, n = 18; ▼) had markedly improved blood glucose levels (160 ± 11 mg/dl) throughout the experimental timeline. B: glucose levels during the hyperinsulinemic hypoglycemic clamp. Insulin infusion decreased glucose levels over a 4-h period in the CON (n = 18), STZ (n = 19), and STZ + Ins (n = 18) groups until glucose reached 15 mg/dl. Glucose was clamped between 10 and 15 mg/dl for 1 h, and then hypoglycemia was terminated with increased glucose infusion throughout a 4-h recovery period. Note: time axis in A is not to scale. Data shown as means ± SE. HYPO, day of severe hypoglycemic clamp.
Insulin treatment.
Eighteen of the diabetic rats were treated with subcutaneous insulin pellets (STZ + Ins; Linplant, ∼2 U/day; Lin Shin, Toronto, ON, Canada) to achieve blood glucose levels of 100–250 mg/dl. If these rats were noted to have tail vein blood glucose >250 mg/dl, insulin (Lantus, 0–2.5 U/day; Sanofi-Aventis, Bridgewater, NJ) was injected subcutaneously once daily. Monitoring the adequacy of insulin treatment was determined by tail vein-obtained blood glucose levels that were assessed one to three times per day. One rat whose blood glucose values were <50 mg/dl for 3 days was excluded from the study.
Hyperinsulinemic hypoglycemic clamp.
Three weeks after STZ or vehicle injections, hyperinsulinemic (0.2 U·kg−1·min−1) hypoglycemic (10–15 mg/dl) clamps were performed in overnight-fasted, awake, unrestrained, nondiabetic CON, uncontrolled STZ-diabetic, and chronically insulin-treated STZ rats, as described previously (5, 17). At the start of insulin infusion (Humulin R; Eli Lilly, Indianapolis, IN), all clamped rats received intravenous glucose (50% dextrose; Hospira, Lake Forest, IL) at an adjustable infusion rate so that all rats reached severe hypoglycemia by 4 h. All groups were then precisely matched for duration (1 h) and depth of hypoglycemia (10–15 mg/dl). This depth and duration of hypoglycemia has previously been shown to be necessary to induce brain damage in this model (2, 5). After 1 h of severe hypoglycemia, insulin infusion was discontinued, and glucose was given to end hypoglycemia. Rats were monitored for 4 h during glucose reperfusion and then returned to their cages for recovery. Rats subjected to severe hypoglycemia were examined for either brain damage or behavioral testing (Fig. 1).
To serve as a negative control, a fourth group of diabetic rats [STZ-no hypoglycemia (STZ-NH); n = 10] was not subjected to an episode of hypoglycemia.
Fluoro-Jade B and hematoxylin-eosin staining.
One week after the severe hypoglycemic clamps, anesthetized rats were perfused intracardially with 0.01 mol/l PBS (Sigma), followed by 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) (5, 17). Brains were immersed in 4% paraformaldehyde overnight and then cryoprotected in 30% sucrose. Beginning at 2.8 mm posterior to the bregma, four coronal cryostat sections (20 μm), 120 μm apart, were analyzed for neuronal damage by Fluoro-Jade B (Chemicon International) and hematoxylin-eosin (Sigma) staining. Fluoro-Jade B is a well-characterized stain for degenerating neurons and was performed as described by Schmued and Hopkins (20). Fluorescent cells (Fluoro-Jade-positive cells) were quantified in both hemispheres of the cortex and of the hippocampal structures CA1 and dentate gyrus. For each region of interest, data are expressed as the average number of Fluoro-Jade B-positive cells per section.
Behavioral testing.
Consistent with other protocol designs (22, 24, 25), histopathological outcomes were assessed 1 wk following the hypoglycemic neuronal insult, whereas cognitive studies were performed 6–8 wk later in a separate group of similarly treated rats. This later assessment of cognitive function is a more useful measurement of clinical outcome and a better functional index of neuroprotection because it allows for a complete and integrated evaluation of ongoing damage and possible recovery (6). After a 6- to 8-wk recovery from the severe hypoglycemic clamp, CON (n = 12), STZ (n = 11), and STZ + Ins (n = 12) rats, as well as a fourth group of sham-operated negative control diabetic rats not subjected to hypoglycemia (STZ-NH; n = 10), were evaluated on a series of behavioral tests. Rats were first assessed on a 1-h locomotor activity test and then on a battery of sensorimotor measurements the following day. Testing on the cued condition in the Morris water maze began the next day for 2 consecutive days. After a 2-day respite, spatial learning and memory (place and probe conditions) were tested in the water maze over 5 consecutive days as described (17).
One-hour locomotor activity test and sensorimotor battery.
General locomotor activity and exploratory behavior were evaluated for 1 h in an open field (41 × 41 × 38.5 cm high) constructed of Pleixglas and containing computerized photobeam instrumentation consisting of a 16 × 16 matrix of photocell pairs (MotorMonitor; Kinder Scientific) to quantify ambulations (whole body movements) and vertical rearing frequency. As described previously (29), the ledge, platform, 90° inclined screen, and walking initiation tests were conducted to measure balance, strength, coordination, and initiation of movement.
Cognitive testing in the Morris water maze.
Spatial learning and memory were assessed for the CON (n = 12), STZ (n = 11), STZ + Ins (n = 12), and STZ-NH (n = 10) groups using the Morris water maze test, similar to previously published methods (29). Cued (visible platform, variable location) and place (submerged, hidden platform, constant location) trials were conducted to train rats to navigate to a submerged platform (1.5 cm below the surface of opaque water) in a round pool (118 cm in diameter). A computerized tracking system (ANY-maze; Stoelting, Wood Dale, IL) was used to quantify escape path length, latency, and swimming speeds, which served as dependent variables. Rats first received cued trials to determine whether nonassociative dysfunctions (sensorimotor or visual disturbances or alterations in motivation) were likely to affect performance in subsequent place trials. The cued condition involved conducting two sessions of three trials each (60-s maximum/trial) per day for 2 consecutive days, during which the rats were trained to swim to the submerged platform marked (cued) by a visible pole, with 3 h intervening between sessions. The data were analyzed in blocks of three trials each for a total of four blocks. Three days later, the spatial learning capabilities of the rats were tested using the place condition in the water maze. This is a reference memory-based task where the rats were trained to learn the position of a submerged and hidden (since pole is removed) platform, which remained in the same location across all trials. The place trials protocol involved conducting two sessions of three trials each (60-s maximum/trial) per day for 5 consecutive days, using a 3-h interval between sessions. The data were analyzed in blocks of six trials each (2 daily sessions) for a total of five blocks. On days 3 and 5, a probe trial was conducted 1 h after the last place trial, which involved removing the platform from the pool and quantifying the rats' search behaviors in the four pool quadrants for 30 s to evaluate retention of the platform location. Probe trial performance variables included the number of times a rat passed directly over the platform location (platform crossings), the time spent in the target quadrant, and spatial bias that involved comparing the time spent in the target quadrant with time spent in each of the other quadrants.
Statistics.
All data are represented as means ± SE. Data were analyzed by one-way (ANOVA) or repeated-measures analysis of variance (rmANOVA). The spatial learning data were analyzed using an ANOVA model containing one between-subjects variable (treatment: CON, STZ, STZ + Ins, and STZ-NH) and one within-subjects (repeated measures) variable, such as blocks of trials. For rmANOVAs, the Huynh-Feldt correction was used for all within-subjects effects containing more than two levels to help protect against violations of sphericity/compound symmetry assumptions underlying the rmANOVA model. In addition, Bonferroni correction was used to help maintain prescribed α-levels (e.g., 0.05) when multiple comparisons were conducted.
RESULTS
Chronic insulin treatment nearly normalizes glucose level of diabetic rats.
Rats that received STZ displayed elevated blood glucose levels 2 days postinjection and remained diabetic throughout the experiment compared with nondiabetic controls (CON: 100 ± 1 mg/dl vs. STZ: 484 ± 10 mg/dl; Fig. 2A). Insulin treatment reduced blood glucose levels to nearly normal (STZ + Ins: 160 ± 11 mg/dl). Sham-operated diabetic rats that did not undergo severe hypoglycemia (STZ-NH) had glucose levels similar to the STZ group (STZ-NH: 467 ± 12 mg/dl). Prior to randomization, body weight did not differ between groups (CON: 302 ± 10 g; STZ: 308 ± 5 g; STZ + Ins: 302 ± 7 g; STZ-NH: 310 ± 9 g). On the day of the severe hypoglycemic clamp, the nondiabetic and insulin-treated diabetic rats had increases in body weight to 408 ± 37 and 387 ± 14 g, respectively, but 3 wk of uncontrolled diabetes in the STZ and STZ-NH rats resulted in a failure to increase body weight (329 ± 22 and 339 ± 10 g, respectively; P = not significant compared with pretreatment).
Hyperinsulinemic hypoglycemic clamp.
All rats experienced the same degree of severe hypoglycemia (CON: 12 ± 0.3 mg/dl; STZ: 13 ± 0.7 mg/dl; STZ + Ins: 12 ± 0.3 mg/dl; Fig. 2B). During severe hypoglycemia, animals lost their righting reflex, displayed seizure-like activity, and became minimally responsive. After 1 h of severe hypoglycemia, glucose reperfusion was started so that animals reached euglycemia within 30 min (CON: 159 ± 48 mg/dl; STZ: 181 ± 31 mg/dl; STZ + Ins: 172 ± 48 mg/dl). Almost immediately, animals regained consciousness, and their righting reflex and seizure-like activity ceased. The animals were allowed to recover for 4 h and then returned to their cages. The following day, blood glucose levels were back to their respective values (CON: 101 ± 6 mg/dl; STZ: 418 ± 49 mg/dl; STZ + Ins: 82 ± 13 mg/dl).
Overall mortality associated with the episode of severe hypoglycemia was 29%. Control rats had a mortality of 22%, whereas STZ and STZ + Ins rats tended to have increased mortality to 36 and 42%, respectively, but this did not reach significance (P < 0.25, Fisher exact test, Freeman Halton extension).
Chronic insulin treatment decreases severe hypoglycemia-induced brain damage.
One week following the acute severe hypoglycemic episode, rats were euthanized, and brains were sectioned for neuronal damage using the marker Flouro-Jade B, which stains for degenerating neurons. Despite equal depth and duration of hypoglycemia, diabetic rats had markedly increased (15-fold) brain damage in the hippocampus and cortex compared with nondiabetic rats (Fig. 3, A and B). In particular, within the hippocampus, the CA1, CA3, and dentate gyrus regions had fivefold, 15-fold, and 45-fold increases in damage, respectively. Chronic antecedent insulin treatment reduced neuronal damage in diabetic rats (STZ + INS, P < 0.05, ANOVA) compared with uncontrolled diabetic rats (STZ) in all regions to levels not significantly different from that of nondiabetic rats (Fig. 3A, B).
Fig. 3.
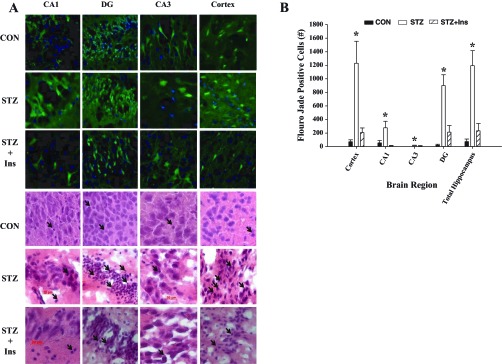
Severe hypoglycemia induced brain damage. A: representative Fluoro-Jade B (green) and DAPI (blue) (top 3 rows) and hematoxylin-eosin (bottom 3 rows) staining of the cortex and hippocampal structures, CA1, CA3, and the dentate gyrus (DG) 1 wk following an episode of severe hypoglycemia in CON, STZ, and STZ + Ins rats. Neuronal damage is indicated with Fluoro-Jade B-positive cells (green fluorescence) or by pyknotic cells (hematoxylin-eosin staining; black arrows). B: brain damage was markedly elevated in the cortex and hippocampus in STZ (open bars) rats compared with controls (black bars). STZ + Ins (diagonally striped bars) reduced this damage in both regions. *P < 0.05 ANOVA; n = 6/group. Data are shown as means ± SE.
Severe hypoglycemia does not disrupt behavior.
Severe hypoglycemia did not significantly impair locomotor or sensorimotor functions (Fig. 4, A and B) when tested 6–8 wk posttreatment (severe hypoglycemic clamp). An rmANOVA did not reveal any significant effects involving treatment on total ambulations (whole body movements) measured during the 1-h locomotor activity test (Fig. 4A), nor were any differences observed in vertical rearing frequency (not shown). Similarly, no performance differences between treatment groups were observed with regard to any measurement on the battery of sensorimotor tests. For example, although the CON group tended to perform not as well as the diabetic groups in terms of being able to remain on a small elevated platform, a one-way ANOVA showed no significant differences among groups on this sensorimotor test (Fig. 4B) or on other sensorimotor measurements of balance (ledge), strength (90° inclined screen), or initiation of movement (walking initiation) out of a small delineated space (not shown).
Fig. 4.
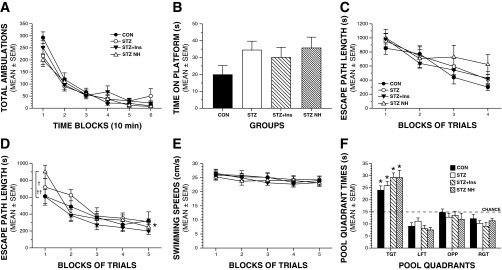
Severe hypoglycemia does not produce impairment in behavioral and cognitive functions. A: there were no significant effects involving treatment (severe hypoglycemia) on general activity, as measured by total ambulations (whole body movements) during a 1-h locomotor activity test. B: although the CON group tended to perform not as well as the diabetic groups in terms of being able to remain on a small elevated platform, there were no significant differences among groups on this sensorimotor test. C: groups did not differ in escape path length during the cued (visible platform location) trials. D: a repeated-measures ANOVA conducted on the path length data during the place (spatial learning) trials yielded a significant group (treatment) by blocks of trials interaction (*P = 0.011). This was due mostly to increased path length in the STZ-NH group vs. the STZ + Ins (†P = 0.036) or the CON (††P = 0.0369) groups during the first block of trials only, but not thereafter. E: no differences among groups were observed in swimming speeds during the place trials. F: groups performed similarly in terms of time in the target quadrant (TGT) during probe trial 1 (mid-acquisition). In addition, all groups demonstrated significant spatial bias for the TGT that had contained the hidden platform, as evidenced by each group spending significantly more time in the TGT compared with times spent in each of the other quadrants (*P < 0.01). LFT, left quadrant; OPP, opposite quadrant; RGT, right quadrant.
Interestingly, we also found no general performance differences between treatment groups on the learning and memory indices quantified during testing in the Morris water maze. Specifically, no effects involving treatment were found following rmANOVAs conducted on the escape path length (Fig. 4C), latency, or swimming speed data (not shown) during the cued (visible platform location) trials. These results suggest that there were no visual, sensorimotor, or motivational disturbances in any of the groups that would compromise swimming performance and thus confound interpretation of the subsequent place trials data. With regard to the acquisition (spatial learning) performance of the treatment groups during the place trials (Fig. 4D), an rmANOVA conducted on the path length data yielded a significant treatment by blocks of trials interaction, (F12,64 = 2.54, P = 0.011). Subsequent comparisons showed that this was due mostly to increased path length in the STZ-NH group vs. the STZ-Ins (P = 0.036) or the CON (P = 0.037) groups during the first block of trials only, but not thereafter. A significant treatment by blocks of trials interaction (F12,64 = 2.32; P = 0.016) was also found for escape latency (not shown), although only the comparison between the STZ-NH vs. STZ + Ins groups was significantly different (P = 0.017). Note that although the comparisons cited here for block 1 revealed certain large (P < 0.05) differences between treatment groups, none of the P values were less than the Bonferroni corrected level (P = 0.05/4 = 0.0125). Thus, it is appropriate to conclude that the performance of the treatment groups did not differ at any time with regard to path length or latency during acquisition (place trials). Consistent with the general lack of differences between treatment groups during the place trials, comparable performance levels were observed among groups during the probe trials. For example, one-way ANOVAs revealed nonsignificant treatment effects with regard to time in the target quadrant during probe trial 1 (midacquisition) (Fig. 4F) and platform crossings (not shown). In addition, all groups demonstrated significant spatial bias for the target quadrant that had contained the hidden platform, as evidenced by each group spending significantly more time in the target quadrant compared with times spent in each of the other quadrants (P < 0.01). Similar results were found for the same variables during the second probe trial conducted 1 h after the last place trial on day 5 (not shown).
DISCUSSION
Studies investigating the potential neuroprotective role of insulin often do not differentiate the acute vs. chronic effects of insulin therapy. The present findings show that although acute insulin-induced hypoglycemia can cause brain damage, chronic insulin treatment in diabetic rats to restore normoglycemia is markedly neuroprotective and reduces the extent of severe hypoglycemia-induced brain damage significantly.
The Morris maze has consistently been shown to be a sensitive test to quantify impaired cognitive function following a single episode of hypoglycemia-induced brain damage (17, 22, 23). Therefore, it was hypothesized that the uncontrolled diabetic rats that displayed markedly elevated hypoglycemia-induced brain damage would perform much worse than control nondiabetic rats tested and that the marked neuroprotective effects of chronic insulin therapy would mitigate against demonstrable deficits in cognitive performance. Therefore, it was surprising that Morris maze testing did not demonstrate differences in cognitive performances between the groups. This indicates that either 1) the STZ-diabetic rats were able to develop adequate cognitive compensatory mechanisms in the 6- to 8-wk recovery period or 2) the extent of hypoglycemia-induced brain damage was not of a sufficient magnitude to produce observable behavioral deficits. The latter interpretation is favored based on the following. 1) Although the extent of hippocampal neuronal regeneration may contribute to cognitive recovery after severe hypoglycemia injury (24), it is unlikely that the uncontrolled STZ-diabetic rats had markedly elevated neuronal recovery following the documented degree of increased hypoglycemia-induced brain damage; 2) the brain damaged diabetic rats performed as well as the diabetic rats not exposed to hypoglycemia (Fig. 4); 3) our previous Morris maze studies in nonhypoglycemic normal rats (17) showed learning and memory testing profiles very similar to the currently tested groups of rats, suggesting that all of the current groups of rats performed equally as well as (historical control) rats not exposed to hypoglycemia; and 4) previous experiments in our laboratory that induced hypoglycemia of longer duration, causing up to sixfold more brain damage than observed in the current study, seemed to be necessary to induce demonstrable deficits in cognitive testing by the Morris maze (17). Therefore, it is suspected that the extent of brain damage achieved in the current studies did not reach a high enough threshold to cause demonstrable deficits in learning/memory performances, as determined by Morris maze testing. Although considered, the relatively high mortality in the current series of rats precluded hypoglycemia experiments of longer duration. Interestingly, a similar dissociation between severe hypoglycemia-induced brain damage and detectable cognitive deficits is consistent with clinical studies. Specifically, severe hypoglycemia can significantly alter brain structure (16), yet the ability to demonstrate cognitive deficits due to severe hypoglycemia varies, with cognitive deficits noted in some (16, 19) but not all (11) studies.
Consistent with a previous report from our laboratory revealing that diabetic rats are more susceptible to severe hypoglycemia-induced death (18), mortality due to severe hypoglycemia tended to be increased in both the uncontrolled diabetic (STZ) and insulin-treated diabetic (STZ + Ins) rats compared with controls. However, since the difference was not significant, future research is focusing on how diabetes per se might increase the risk of severe hypoglycemia-induced death. Although the cause of death in the current experiments is unknown, it is hypothesized that severe hypoglycemia induces fatal cardiac arrhythmias (18).
Hypoglycemia has been shown to induce neuronal cell death via a cascade of molecular events, including increased release of glutamate, calcium, zinc, and reactive oxygen species, leading to cell death (1). The mechanism by which hypoglycemia-induced neuronal damage was markedly augmented in diabetic rats was not assessed directly in these studies, although many potential mechanisms exist. 1) The greater net drop in glycemia experienced in the diabetic rats (i.e., from 484 to 13 mg/dl) may have exacerbated brain damage; 2) the decreased number of brain glucose transporters (26) and altered glucose transport and metabolism (9) may have led to the increased susceptibility to brain damage in the diabetic rats; 3) an upregulation of glutamate leading to calcium overload may have enhanced neuronal death in diabetic rats (27); 4) STZ-diabetic rats show a downregulation of cortical NMDA receptors (4), which may be particularly important given the role of excess glutamate in mediating hypoglycemia-induced neuronal damage (21); and 5) following the hypoglycemic episode, the effects of chronic hyperglycemia may have been toxic to vulnerable neurons, resulting in increased brain damage (14).
In the absence of severe hypoglycemia, uncontrolled diabetes (induced by STZ) does not result in brain damage, as shown by a previous report (5). Therefore, in the current studies, diabetic rats not subjected to severe hypoglycemia (STZ-NH) were not analyzed for brain damage.
The mechanism by which chronic insulin therapy afforded neuroprotection against brain damage cannot be precisely determined in this model. It was hypothesized that chronic insulin deficiency may predispose the diabetic brain to greater hypoglycemia-induced damage. Studies in ischemic brain injury models are consistent with the notion that insulin acts directly in the brain to afford neuroprotection (8, 10). Alternatively, since the downregulation of brain glucose transporters by chronic hyperglycemia (26) may predispose the brain to profound neuroglycopenia in response to acute severe hypoglycemia, it may be that the correction of chronic hyperglycemia alone could be neuroprotective. Another possible mechanism for the neuroprotective effect of chronic insulin therapy could be related to the preconditioning effect of recurrent hypoglycemia (17). Although rats that displayed recurrent hypoglycemia were specifically excluded from the study, we cannot rule out the possibility that, in an attempt to achieve near-normalization of blood glucose levels, some of the insulin-treated diabetic rats might have had episodes of undetected antecedent hypoglycemia that could have preconditioned the brain and protected it against the subsequent severe hypoglycemic episode (17). Regardless of the mechanism, the effect was profound. The 15-fold reduction in hypoglycemia-induced brain damage indicates that antecedent blood glucose achieved with chronic insulin therapy is a major beneficial therapeutic intervention that provided the diabetic rats with significant neuroprotection.
For patients and clinicians, the choice to pursue an intensive insulin regime as a therapeutic approach to realize the established long-term microvascular benefits associated with tight glycemic control has to be weighed against the increased risk of severe hypoglycemia and associated adverse outcomes, including brain damage. In this study, chronic insulin treatment in diabetic rats that led to a near normalization of glucose levels markedly reduced the extent of brain damage induced by severe hypoglycemia. These intriguing findings indicate that chronic insulin therapy that improved glycemic control paradoxically rendered the diabetic rats more prone to, but less vulnerable to, an episode of severe hypoglycemia. In conclusion, antecedent blood glucose control represents a major modifiable therapeutic intervention that can afford diabetic subjects neuroprotection against a subsequent episode of severe hypoglycemia-induced brain damage.
GRANTS
Research support from the National Institutes of Health (NS-070235) and core grant support from the Neuroscience Blueprint Center (NS-057105) and Washington University's Diabetes Research and Training Center (DK-020579) and Clinical Nutrition Research Unit (DK-056341) are gratefully acknowledged.
DISCLOSURES
The authors have no conflicts of interest, financial or otherwise, relative to this article to report.
AUTHOR CONTRIBUTIONS
C.M.R., T.T., D.F.W., and S.J.F. contributed to the conception and design of the research; C.M.R., T.T., A.B., D.D.-I., C.C., and S.E.M. performed the experiments; C.M.R., T.T., A.B., D.D.-I., C.C., S.E.M., D.F.W., and S.J.F. analyzed the data; C.M.R., T.T., A.B., S.E.M., D.F.W., and S.J.F. interpreted the results of the experiments; C.M.R., D.D.-I., S.E.M., and D.F.W. prepared the figures; C.M.R. and D.F.W. drafted the manuscript; C.M.R., D.F.W., and S.J.F. edited and revised the manuscript; C.M.R., D.F.W., and S.J.F. approved the final version of the manuscript.
REFERENCES
- 1. Alano CC, Ying W, Swanson RA. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem 279: 18895–18902, 2004 [DOI] [PubMed] [Google Scholar]
- 2. Auer RN. Hypoglycemic brain damage. Forensic Sci Int 146: 105–110, 2004 [DOI] [PubMed] [Google Scholar]
- 3. Austin EJ, Deary IJ. Effects of repeated hypoglycemia on cognitive function: a psychometrically validated reanalysis of the Diabetes Control and Complications Trial data. Diabetes Care 22: 1273–1277, 1999 [DOI] [PubMed] [Google Scholar]
- 4. Bean L, Zheng H, Patel KP, Monaghan DT. Regional variations in NMDA receptor downregulation in streptozotocin-diabetic rat brain. Brain Res 1115: 217–222, 2006 [DOI] [PubMed] [Google Scholar]
- 5. Bree AJ, Puente EC, Daphna-Iken D, Fisher SJ. Diabetes increases brain damage caused by severe hypoglycemia. Am J Physiol Endocrinol Metab 297: E194–E201, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Corbett D, Nurse S. The problem of assessing effective neuroprotection in experimental cerebral ischemia. Prog Neurobiol 54: 531–548, 1998 [DOI] [PubMed] [Google Scholar]
- 7. Cryer PE, Davis SN, Shamoon H. Hypoglycemia in diabetes. Diabetes Care 26: 1902–1912, 2003 [DOI] [PubMed] [Google Scholar]
- 8. Duarte AI, Proenca T, Oliveira CR, Santos MS, Rego AC. Insulin restores metabolic function in cultured cortical neurons subjected to oxidative stress. Diabetes 55: 2863–2870, 2006 [DOI] [PubMed] [Google Scholar]
- 9. Gutniak M, Blomqvist G, Widén L, Stone-Elander S, Hamberger B, Grill V. d-[U-11C]glucose uptake and metabolism in the brain of insulin-dependent diabetic subjects. Am J Physiol Endocrinol Metab 258: E805–E812, 1990 [DOI] [PubMed] [Google Scholar]
- 10. Hui L, Pei DS, Zhang QG, Guan QH, Zhang GY. The neuroprotection of insulin on ischemic brain injury in rat hippocampus through negative regulation of JNK signaling pathway by PI3K/Akt activation. Brain Res 1052: 1–9, 2005 [DOI] [PubMed] [Google Scholar]
- 11. Jacobson AM, Musen G, Ryan CM, Silvers N, Cleary P, Waberski B, Burwood A, Weinger K, Bayless M, Dahms W, Harth J. Long-term effect of diabetes and its treatment on cognitive function. N Engl J Med 356: 1842–1852, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kramer L, Fasching P, Madl C, Schneider B, Damjancic P, Waldhausl W, Irsigler K, Grimm G. Previous episodes of hypoglycemic coma are not associated with permanent cognitive brain dysfunction in IDDM patients on intensive insulin treatment. Diabetes 47: 1909–1914, 1998 [DOI] [PubMed] [Google Scholar]
- 13. Langan SJ, Deary IJ, Hepburn DA, Frier BM. Cumulative cognitive impairment following recurrent severe hypoglycaemia in adult patients with insulin-treated diabetes mellitus. Diabetologia 34: 337–344, 1991 [DOI] [PubMed] [Google Scholar]
- 14. Moreira T, Cebers G, Pickering C, Ostenson CG, Efendic S, Liljequist S. Diabetic Goto-Kakizaki rats display pronounced hyperglycemia and longer-lasting cognitive impairments following ischemia induced by cortical compression. Neuroscience 144: 1169–1185, 2007 [DOI] [PubMed] [Google Scholar]
- 15. Musen G, Lyoo IK, Sparks CR, Weinger K, Hwang J, Ryan CM, Jimerson DC, Hennen J, Renshaw PF, Jacobson AM. Effects of type 1 diabetes on gray matter density as measured by voxel-based morphometry. Diabetes 55: 326–333, 2006 [DOI] [PubMed] [Google Scholar]
- 16. Northam EA, Rankins D, Lin A, Wellard RM, Pell GS, Finch SJ, Werther GA, Cameron FJ. Central nervous system function in youth with type 1 diabetes 12 years after disease onset. Diabetes Care 32: 445–450, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Puente EC, Silverstein J, Bree AJ, Musikantow DR, Wozniak DF, Maloney S, Daphna-Iken D, Fisher SJ. Recurrent moderate hypoglycemia ameliorates brain damage and cognitive dysfunction induced by severe hypoglycemia. Diabetes 59: 1055–1062, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reno CM, Tanoli T, Puente EC, Bree AJ, Cui C, Silverstein J, Daphna-Iken D, Fisher SJ. Deaths due to severe hypoglycemia are exacerbated by diabetes and ameliorated by hypoglycemic preconditioning (Abstract). Diabetes 60: A81, 2011 [Google Scholar]
- 19. Rovet JF, Ehrlich RM. The effect of hypoglycemic seizures on cognitive function in children with diabetes: a 7-year prospective study. J Pediatr 134: 503–506, 1999 [DOI] [PubMed] [Google Scholar]
- 20. Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 874: 123–130, 2000 [DOI] [PubMed] [Google Scholar]
- 21. Silverstein JM, Musikantow D, Puente EC, Daphna-Iken D, Bree AJ, Fisher SJ. Pharmacologic amelioration of severe hypoglycemia-induced neuronal damage. Neurosci Lett 492: 23–28, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suh SW, Aoyama K, Chen Y, Garnier P, Matsumori Y, Gum E, Liu J, Swanson RA. Hypoglycemic neuronal death and cognitive impairment are prevented by poly(ADP-ribose) polymerase inhibitors administered after hypoglycemia. J Neurosci 23: 10681–10690, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suh SW, Aoyama K, Matsumori Y, Liu J, Swanson RA. Pyruvate administered after severe hypoglycemia reduces neuronal death and cognitive impairment. Diabetes 54: 1452–1458, 2005 [DOI] [PubMed] [Google Scholar]
- 24. Suh SW, Fan Y, Hong SM, Liu Z, Matsumori Y, Weinstein PR, Swanson RA, Liu J. Hypoglycemia induces transient neurogenesis and subsequent progenitor cell loss in the rat hippocampus. Diabetes 54: 500–509, 2005 [DOI] [PubMed] [Google Scholar]
- 25. Suh SW, Gum ET, Hamby AM, Chan PH, Swanson RA. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J Clin Invest 117: 910–918, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vannucci SJ, Koehler-Stec EM, Li K, Reynolds TH, Clark R, Simpson IA. GLUT4 glucose transporter expression in rodent brain: effect of diabetes. Brain Res 797: 1–11, 1998 [DOI] [PubMed] [Google Scholar]
- 27. Vizi ES. Role of high-affinity receptors and membrane transporters in nonsynaptic communication and drug action in the central nervous system. Pharmacol Rev 52: 63–89, 2000 [PubMed] [Google Scholar]
- 28. Whitmer RA, Karter AJ, Yaffe K, Quesenberry CP, Jr, Selby JV. Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus. JAMA 301: 1565–1572, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wong M, Wozniak DF, Yamada KA. An animal model of generalized nonconvulsive status epilepticus: immediate characteristics and long-term effects. Exp Neurol 183: 87–99, 2003 [DOI] [PubMed] [Google Scholar]