

Abstract
Diabetes manifests from a loss in functional β-cell mass, which is regulated by a dynamic balance of various cellular processes, including β-cell growth, proliferation, and death as well as secretory function. The cell cycle machinery comprised of cyclins, kinases, and inhibitors regulates proliferation. However, their involvement during β-cell stress during the development of diabetes is not well understood. Interestingly, in a screen of multiple cell cycle inhibitors, p21 was dramatically upregulated in INS-1-derived 832/13 cells and rodent islets by two pharmacological inducers of β-cell stress, dexamethasone and thapsigargin. We hypothesized that β-cell stress upregulates p21 to activate the apoptotic pathway and suppress cell survival signaling. To this end, p21 was adenovirally overexpressed in pancreatic rat islets and 832/13 cells. As expected, p21 overexpression resulted in decreased [3H]thymidine incorporation. Flow cytometry analysis in p21-transduced 832/13 cells verified lower replication, as indicated by a decreased cell population in the S phase and a block in G2/M transition. The sub-G0 cell population was higher with p21 overexpression and was attributable to apoptosis, as demonstrated by increased annexin-positive stained cells and cleaved caspase-3 protein. p21-mediated caspase-3 cleavage was inhibited by either overexpression of the antiapoptotic mitochondrial protein Bcl-2 or siRNA-mediated suppression of the proapoptotic proteins Bax and Bak. Therefore, an intact intrinsic apoptotic pathway is central for p21-mediated cell death. In summary, our findings indicate that β-cell apoptosis can be triggered by p21 during stress and is thus a potential target to inhibit for protection of functional β-cell mass.
Keywords: cyclin-dependent kinase inhibitor-1a, dexamethasone, thapsigargin, islet, stress
insulin-secreting β-cells, along with glucagon-secreting α-cells, are the primary physiological regulators of glucose homeostasis. Diabetes is a metabolic disease characterized by a decline in functional β-cell mass that is attributed to either impaired insulin secretion or an imbalance in the proliferation to death ratio of β-cells. In the adult pancreas, β-cell proliferation in islets is minuscule (7, 9, 20, 29). However, in diseased states there could be a decrease in the already low rate of proliferation in addition to an increase in death of existing β-cells. Progress toward understanding the events that lead to β-cell death or inhibition of proliferative potential are necessary to identify novel therapeutic targets for diabetes. Specifically, mechanisms to inhibit cell death and release the inhibition on proliferation are essential to protect and/or restore functional β-cell mass in the setting of diabetes.
The cellular mechanisms regulating β-cell proliferation have been studied extensively and reviewed (10). Like other cells, the β-cell expresses cell cycle regulators, including cyclins, cyclin-dependent kinases (Cdk), and Cdk inhibitors separated into two family subtypes, Cdk-interacting protein/kinase inhibitory protein (CIP/KIP) and inhibitors of CDK4 (INK4/Arf). Cyclin-dependent kinase inhibitor-1a (CDKN1a), also known as CDKN1a/p21Cip1/WAF1 (hereafter referred to as p21), is a CIP/KIP member that has been suggested to function as a molecular brake during β-cell mass expansion and is upregulated in pregnancy and mitogen stimulation [e.g., hepatocyte growth factor (HGF) and placental lactogen] to avoid excessive proliferation (10, 18, 23).
In addition to its role in the β-cell, p21 has been studied in cancer biology, and its expression is increased after DNA damage and cell stress (15, 38). This rise in p21 occurs to halt proliferation and permit cell repair. However, if a cell becomes too damaged, its ultimate fate becomes apoptosis, a highly regulated form of cell death mediated through two distinct pathways, the extrinsic or intrinsic pathway, that converge upon activation of the effector caspase-3. Similarly, during the pathogenesis of diabetes, increased cellular stress, such as endoplasmic reticulum (ER) stress due to the increased secretory demand placed on the β-cell, could ultimately lead to apoptosis. Whereas p21 clearly plays a major role in halting proliferation in pancreatic β-cells, whether upregulation of p21 in β-cells contributes directly to the apoptotic death pathway in the development of diabetes remains to be determined.
Here, we discovered that p21 is the only cell cycle inhibitor to be increased dramatically with two independent pharmacological inducers of β-cell stress. The aim of this study was to gain more insight on the importance and function for the p21 upregulation during these processes. To this end, a p21-overexpressing adenovirus was used to characterize the role of p21 on apoptosis and cell survival signaling within rodent β-cell lines and isolated pancreatic islets.
MATERIALS AND METHODS
Cell culture and rodent islet isolation.
INS-1-derived 832/13 and 828/33 rat insulinoma cell lines were maintained in complete RPMI 1640 medium with l-glutamine and 11.2 mM glucose supplemented with 50 U/ml penicillin, 50 μg/ml streptomycin, 10 mM HEPES buffer, 10% fetal bovine serum (FBS), and an INS-1 supplement, as described previously (17, 30). Hepatocellular carcinoma HepG2 cells were maintained in DMEM containing 4.5 g/l d-glucose, 2 mM l-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin, 1 mM sodium pyruvate, and 10% FBS. Pancreatic islets were isolated from male Wistar rats weighing 200–250 g, using a protocol that was approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee (22, 25). Islets were cultured in RPMI medium with l-glutamine and supplemented with 8 mM glucose, 10% FBS, 50 U/ml penicillin, and 50 μg/ml streptomycin. Freshly isolated islets were hand-picked into separate treatment groups.
Drug treatment groups and adenoviral transduction.
β-Cell lines were treated with complete medium containing dexamethasone (100 nM for 16 h; BioVision, Mountain View, CA) or thapsigargin (1 μM for 6 h; Sigma, St. Louis, MO) and compared with DMSO (Sigma) as a control. Isolated primary islets were treated with 1 μM dexamethasone or 10 μM thapsigargin overnight in complete RPMI medium. Dose- and time-dependent experiments were also performed with thapsigargin treatment. For dose-dependent experiments, 832/13 cells were treated with DMSO or 250, 500, or 1,000 nM for 6 h. For time-dependent experiments, 832/13 cells were treated with DMSO or 1,000 nM thapsigargin for 0.5, 1, 2, 4, or 6 h. At the end of the experiments, cells were harvested for collection of either mRNA for quantitative real-time PCR or protein for immunoblotting.
For gene overexpression, 832/13 cells were transduced for 48 h, with purified adenoviruses expressing green fluorescent protein under the control of the cytomegalovirus (CMV) promoter (AdCMV-GFP), human p21 under control of the CMV promoter (AdCMV-p21; Vector Biolabs), or human Bcl-2 under the control of the CMV promoter (AdCMV-Bcl2, a kind gift from Dr. David Curiel at Washington University) (2–4), with 200 multiplicities of infection. Rat islets were transduced immediately following isolation for 72 h with 1,000 multiplicities of infection (assuming an islet contains 1,000 cells). Cells and islets were cultured in adenoviral-containing medium only for the first 24 h and subsequently switched into fresh medium for the remainder of the experiment before harvesting.
[3H]thymidine incorporation.
Proliferation was assessed via [3H]methyl-thymidine incorporation into genomic DNA by incubating 832/13 cells or isolated rat islets in 1 μCi/ml for 4 or 16 h, respectively. Islets that underwent drug treatments or adenovirus transduction were separated into triplicate groups of 30, as described previously (13). Cold 10% trichloroacetic acid was used to precipitate DNA from each sample and subsequently solubilized with 0.3 N NaOH. The total amount of [3H]thymidine incorporation was measured using liquid scintillation counting and normalized to cellular protein content.
Quantitative real-time polymerase chain reaction.
To isolate RNA, 832/13 cells and ∼50 isolated rat islets were lysed in RLT buffer containing 1% β-mercaptoethanol and harvested using the RNeasy mini or micro kit, respectively (Qiagen, Valencia, CA). Next, cDNA was synthesized using the High Capacity Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Samples underwent real-time PCR using Taqman Gene Expression Master Mix Reagents and Gene Expression assay probes (Applied Biosystems). The threshold cycle (CT) method (24) was used to calculate relative quantities of mRNA products; each sample was performed in triplicate and normalized by the CT value of GAPDH, an internal control within each reading.
Flow cytometry.
After adenoviral transduction, the media from cultured 832/13 cells were collected and spun at 1900 rpm at 4°C for 3 min in clear polyethylene tubes. Cultured cells were then washed with PBS and trypsinized until cells detached from the plate. Cells were gently collected and spun together with the apoptotic cells (i.e., those collected from the media) at 1,000 rpm for 3 min at 4°C. Supernatants were removed, and cells were washed with PBS, vortexed, and spun at 1,900 rpm for 10 min at 4°C. Duplicate samples were processed for either cell cycle or apoptosis analysis. For cell cycle analysis, samples were resuspended and stained with propidium iodide using the Guava cell cycle reagent (Millipore, Billerica, MA). For apoptosis analysis, samples were resuspended and costained with Annexin V-APC (Invitrogen, Carlsbad, CA) and propidium iodide (BioVision) in annexin-binding buffer (BioVision). Cells were sorted on a BD FACSCalibur APC instrument using the CellQuest Pro software (BD Biosciences, San Jose, CA). Cell cycle populations were analyzed with the ModFit LT Application.
Immunoblot analysis.
Cells or ∼150 isolated islets were washed and lysed with radioimmunoprecipitation buffer (Santa Cruz Biotechnology, Santa Cruz, CA) supplemented with protease inhibitors (Santa Cruz Biotechnology) and phosphatase inhibitor cocktails 2 and 3 (Sigma). Total cellular protein content was quantified using the BCA assay kit (Pierce, Rockford, IL). Protein samples (30 μg) were heated with NuPage reducing agent and resolved on NuPage 10% Tris-Bis gels (Invitrogen). Proteins were transferred onto Immobulin-FL membranes (Millipore), and membranes were incubated overnight at 4°C with the following primary antibodies that were diluted 1:1,000 in polyvinylpyrrolidone (16): caspase-3 (no. 9662S; Cell Signaling Technology, Beverly, MA), phosphorylated-Akt (Thr308) (no. 4056S; Cell Signaling Technology), Akt (no. 2920S; Cell Signaling Technology), caspase-8 (no. 9746S; Cell Signaling Technology), phosphorylated eukaryotic initiation factor-2α (eIF-2α) (no. 3398S; Cell Signaling Technology), eIF-2α (no. 2103S; Cell Signaling Technology), p21 (sc-397; Santa Cruz Biotechnology), Bcl-2 (sc-7382; Santa Cruz Biotechnology), Bax (06-499; Millipore), Bak (06-536; Millipore), and phosphorylated cdk1 (pTpY14/15) (44-686G; Biosource). The loading control for all immunoblots was actin (1:5,000, no. 691001; MP Biomedical, Solon, OH). Primary antibodies were detected by incubating membranes with IRDye 700- or 800-conjugated secondary antibodies for 1 h; fluorophore-labeled protein bands were detected on the Odyssey System (LI-COR, Lincoln, NE) and were quantified using ImageJ software (National Institutes of Health, Bethesda, MD).
siRNA transfection.
For siRNA-mediated suppression of Bax and Bak, preannealed siRNA duplexes from Ambion (ID nos. 49750 and 190465, respectively) were utilized and compared with a scrambled control siRNA, as described previously (8). 832/13 cells were incubated for 24 h with complete medium containing a final concentration of 0.1 μM siRNA in Dharmafect transfection reagents (ThermoScientific, Waltham, MA). Subsequently, adenovirus transduction occurred for the remaining 48 h of culture before harvesting for protein and mRNA analysis.
Statistical methods.
For analysis between two groups, Student's t-test was used, and differences were considered significant when P < 0.05. Comparisons between GFP- and p21-overexpressing groups in the cell lines were performed using a two-tailed Student's t-test; islet experiments were analyzed using a paired two-tailed t-test. One-way analysis of variance (ANOVA) was used in experiments that had three or more groups. Differences within ANOVA were determined using Tukey's post hoc tests, with significance determined when P < 0.05. All data are reported as means ± SE.
RESULTS
Dexamethasone and thapsigargin suppress proliferation and preferentially increase p21 transcription.
Both dexamethasone and thapsigargin decreased proliferation in 832/13 cells, as indicated by a decrease in thymidine incorporation (Fig. 1A). To determine the cell cycle inhibitor(s) responsible for the decrease in proliferation, a quantitative PCR screen of the CIP/KIP and INK4/Arf family members was performed in 832/13 cells (Fig. 1B). Unexpectedly, p21 was the only cell cycle inhibitor markedly upregulated by both dexamethasone and thapsigargin. Whereas p27 was increased significantly, 2.7-fold with thapsigargin treatment, it was not induced by dexamethasone. These findings were verified in isolated rat islets and resulted in a nearly threefold increase in p21 transcripts with overnight treatment of dexamethasone or thapsigargin (Fig. 1C). Transcripts for p15, p16, and p57 were undetectable in 832/13 cells.
Fig. 1.

Dexamethasone (Dex) and thapsigargin (Tg) halt proliferation and preferentially increase p21 transcription. 832/13 cells were treated with the vehicle DMSO (control), Dex (100 nM for 16 h), or Tg (1 μM for 6 h). A: cell proliferation was assessed by [3H]thymidine incorporation of triplicate samples. B: mRNA levels of cell cycle inhibitors were measured by quantitative PCR. C: p21 mRNA was measured in isolated rat islets treated overnight with vehicle, 1 μM Dex, or 10 μM Tg. Data are normalized to the DMSO-treated control group and are presented as means ± SE; n = 3–5. *Significance vs. control using a 1-way ANOVA test; P < 0.05. Cdk, cyclin-dependent kinase.
To examine the dose- and time-dependent induction of p21 during the initiation of β-cell stress by thapsigargin, we treated 832/13 cells with increasing concentrations of thapsigargin and over a time course (Fig. 2). We used cleavage of caspase-3 as a readout of the development of β-cell stress-mediated cell death. The induction of p21 with increasing doses of thapsigargin largely mirrored that of caspase-3 activation/cleavage. Interestingly, the time-dependent induction of p21 by thapsigargin also coincided with the activation/cleavage of caspase-3.
Fig. 2.

Dose- and time-dependent upregulation of p21 transcription with Tg. A–C: 832/13 cells were treated with the vehicle DMSO (control) or increasing doses of Tg (250, 500, or 1,000 nM) for 6 h. D–F: 832/13 cells were treated with Tg (1,000 nM) for 0.5, 1, 2, 4, or 6 h. A and D: mRNA expression of p21 transcripts was measured by quantitative PCR and expressed relative to control. B and E: representative Western blot of cleaved caspase-3 protein (Cl casp-3) with each treatment. C and F: quantified results normalized to actin expression. Data are presented as means ± SE; n = 3–4. *Significance vs. control using a 1-way ANOVA test; P < 0.05.
p21 overexpression decreases β-cell proliferation and arrests the cell cycle at G1/S and G2/M transitions.
To further investigate the role for p21 in β-cells, an adenovirus that overexpressed human p21, thus inhibiting Cdk activation (Fig. 3, A and B), was used to transduce 832/13 cells and isolated rat islets. Expression of p21 was adenovirally increased approximately seven- and threefold in 832/13 cells and rat islets, respectively (832/13 cells: 0.30 ± 0.01 vs. 2.21 ± 0.31 p21/actin, P < 0.05; rat islets: 0.86 ± 0.25 vs. 2.34 ± 0.45 p21/actin, P < 0.05). In both 832/13 cells and rat islets, p21 overexpression decreased proliferation, as indicated by tritiated-thymidine incorporation assays (Fig. 3, C and D). In addition, flow cytometry analysis revealed a significant decrease in the S phase cell population but an increase in the G2 cell population (Fig. 3, E and F), consequently demonstrating cell cycle arrest at the G1/S and G2/M transitions. Unexpectedly, an increase in the sub-G0 cell population was also evident (Fig. 3G) and led us to further investigate the role of p21 in apoptosis.
Fig. 3.
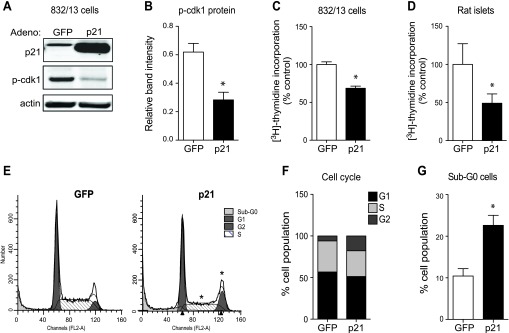
p21-overexpressing adenovirus inhibits Cdk activation and entry into S and M phase. A: representative Western blot of 832/13 cells transduced with a control green fluorescent protein (GFP)- or p21-overexpressing adenovirus and probed for p21 and phosphorylated Cdk 1 (p-Cdk1). B: quantification of phosphorylated Cdk1 normalized to actin. *Signifcance vs. GFP; n = 3 independent experiments. 832/13 cells (C) and pooled rat islets (D) of 3 animals were treated with a control GFP- or p21-overexpressing adenovirus, and [3H]thymidine incorporation was measured to assess proliferation. Data are normalized to the control GFP group for [3H]thymidine incorporation and presented as means ± SE; n = 3–4 independent experiments in triplicate. *Significance vs. GFP in an unpaired or paired 1-tailed t-test for 832/13 cells and islets, respectively. 832/13-transduced cells underwent flow cytometry and were analyzed by ModFit LT to acquire representative cell cycle peaks for each treatment (E) and to measure the percentage of cells at each cell cycle phase (F). G: the percentage of sub-G0 cells was also acquired during this process. Data for cell populations are presented as means ± SE; n = 4–5 independent experiments with duplicate samples. *Significance vs. GFP in an unpaired 2-tailed t-test; P < 0.05.
p21 directly activates apoptosis in β-cells.
Using propidium iodide and annexin costaining to sort apoptotic cells by flow cytometry (Fig. 4A), p21 overexpression decreased the percentage of viable cells (Fig. 4B). Additionally, the number of annexin- and propidium iodide-positive cells was sevenfold higher with p21 overexpression, indicating a significant increase in the number of apoptotic cells (Fig. 4C). Furthermore, protein analysis of 832/13 cells transduced with p21-overexpressing adenovirus demonstrated an increase in the hallmark apoptotic marker cleaved caspase-3 (Fig. 5, A and C). More importantly, caspase-3 cleavage in isolated primary rat islets nearly tripled with p21 overexpression (Fig. 5, D and F). To determine whether p21 was disrupting cell survival signaling, levels of phosphorylated Akt were assessed. Although there was a substantial decrease in phosphorylated Akt after p21 overexpression in 832/13 cells (Fig. 5, A and B), this result did not translate to isolated islets under the experimental conditions used (Fig. 5, D and E).
Fig. 4.

p21 overexpression increases the number of apoptotic β-cells. A: annexin-V and propidium iodide were used to costain GFP- or p21-transduced 832/13 cells, and cells were analyzed with flow cytometry. B: annexin- and propidium iodide-negative cells are represented as viable cells. C: annexin- and propidium iodide-positive cells are represented as apoptotic cells. Data are presented as means ± SE; n = 3 independent experiments with duplicate samples. *Significance vs. GFP in unpaired 2-tailed t-tests; P < 0.05.
Fig. 5.
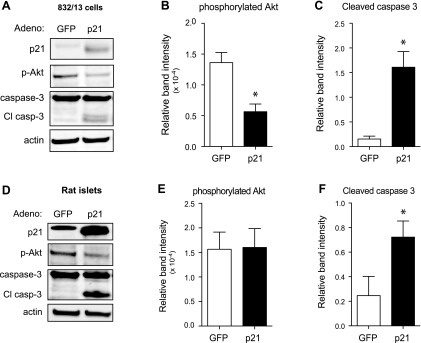
p21 overexpression activates caspase-3 and decreases cell survival. Representative Western blot images from whole cell lysates of 832/13 cells transduced with GFP- or p21-overexpressing adenovirus for 48 h (A) or rat islets transduced with the same adenoviruses for 72 h (D). Quantification of phospho-Akt normalized to total Akt (B and E) and Cl casp-3 (C and F), both normalized to actin protein, for lysates from 832/13 cells (B and C) and rat islets (E and F). Data are represented as mean intensities ± SE; n = 3 experiments. *Significance vs. GFP in an unpaired 2-tailed t-test for 832/13 cells and in a paired 2-tailed t-test for islets; P < 0.05.
To determine whether the induction of p21 during stress and p21's ability to trigger apoptosis were novel phenomena in β-cells, we performed complementary experiments in HepG2 cells, a hepatocyte cell line. Thapsigargin but not dexamethasone induced p21 in HepG2 cells (Fig. 6A). Interestingly, adenoviral overexpression of p21 did not stimulate apoptosis in HepG2 cells; as a positive control to establish that HepG2 cells are capable of undergoing apoptosis, HepG2 cells were treated with the topoisomerase inhibitor etoposide, which also induces p21, and this treatment increased caspase-3 cleavage, indicating apoptosis (Fig. 6B). Thus, p21's ability to induce apoptosis is dependent on the cellular context.
Fig. 6.

p21 upregulation does not activate caspase-3 in HepG2 cells. A: HepG2 cells were treated with Dex or Tg (1 μM for 24 h), and mRNA levels for p21 were measured by quantitative PCR. B: representative Western blot of HepG2 cells transduced with GFP- or p21-overexpressing adenovirus or treated with etoposide (100 μM for 6 or 24 h) as a positive control. Data are normalized to the DMSO-treated control group and are presented as means ± SE; n = 3–4. *Significance vs. control using 1-way ANOVA test; P < 0.05.
p21-induced apoptosis is mediated through the intrinsic mitochondrial death pathway.
The next objective was to determine whether p21 was activating apoptosis through the extrinsic or intrinsic pathway. Protein analysis of caspase-8, an intermediate of the extrinsic pathway, indicated no change with p21 overexpression (Fig. 7A). mRNA expression of mitochondrial pro- and antiapoptotic Bcl-2 family members that are regulated in the intrinsic pathway was not significantly different with p21 overexpression (Fig. 7B). However, when 828/33 cells, a Bcl-2-overexpressing β-cell line, were utilized, p21-mediated caspase-3 cleavage was blocked (Fig. 8, A and B). Similarly, thapsigargin-induced caspase-3 cleavage was also blocked in 828/33 cells despite persistent ER stress, as noted by the increase in phosphorylated eIF-2α (Fig. 8, C and D). Furthermore, acute overexpression of Bcl-2 with a recombinant adenovirus attenuated p21-mediated caspase-3 cleavage (Fig. 8, E and F). In addition, siRNA-mediated suppression of the proapoptotic Bax and Bak proteins also inhibited p21-mediated cell death, as indicated by a decrease in caspase-3 cleavage (Fig. 9). The promotion of caspase-3 cleavage by p21 was mediated by both Bax and Bak, as siRNA-mediated suppression of either protein significantly reduced caspase-3 cleavage following p21 overexpression, and when both proteins were suppressed simultaneously, there was a further reduction in caspase-3 cleavage. These data suggest that p21-induced apoptosis is mediated through the intrinsic mitochondrial death pathway.
Fig. 7.

p21-mediated apoptosis is not regulated through the extrinsic mitochondrial death pathway or by a change in Bcl-2 family member expression. A: Western blot analysis of caspase-8 (Cl casp-8) protein levels in whole cell lysates from 832/13 cells transduced with GFP- or p21-overexpressing adenovirus for 48 h. B: these cells also underwent a quantitative PCR screen of various pro- and antiapoptotic Bcl-2 family transcripts; n = 3 independent experiments.
Fig. 8.
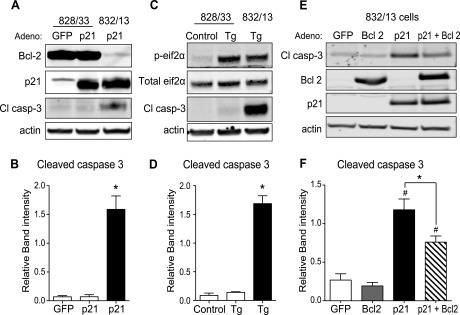
p21- or ER stress-mediated apoptosis is blocked by Bcl-2 overexpression. A: 828/33 cells, which stably overexpress Bcl-2, were transduced with GFP- or p21-overexpressing adenovirus, and as a positive control 832/13 cells were transduced with p21-overexpressing adenovirus. Whole cell lysates underwent Western blot analysis for Bcl-2, p21, Cl casp-3, and actin as a loading control. B: quantification of Cl casp-3 normalized to actin. Data are represented as mean intensities ± SE; n = 3 independent experiments. *Significance vs. 828/33 cells transduced with GFP or p21 adenovirus in a 1-way ANOVA; P < 0.05. C: representative Western blot of 828/33 and 832/13 cells treated with Tg for 6 h and probed for total and phosphorylated eukaryotic initiation factor-2α (eIF-2α) to indicate ER stress, Cl casp-3 to indicate ER stress-induced apoptosis, and actin as a loading control. D: quantification of Cl casp-3 normalized to actin. Data are represented as mean intensities ± SE; n = 3 independent experiments. *Significance vs. 828/33 cells treated with vehicle or Tg in a 1-way ANOVA; P < 0.05. E and F: 832/13 cells were transduced with GFP-, Bcl2-, or p21-overexpressing adenovirus for 48 h (E), and caspase-3 activation was observed and quantified via Western blot analysis (F). Data are represented as mean intensities ± SE; n = 4 independent experiments. *Significance between p21- and p21 + Bcl-2 adenovirus-treated groups by ANOVA; P < 0.05. #Significance vs. GFP- and p21 adenovirus-treated groups by ANOVA; P < 0.05.
Fig. 9.
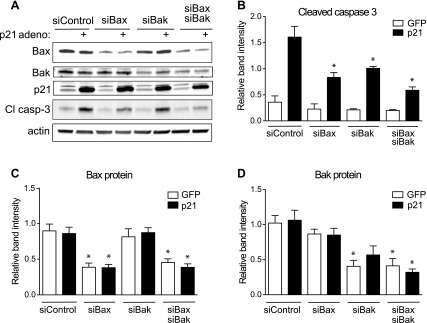
p21-mediated apoptosis is blocked by siRNA-mediated suppression of Bax and/or Bak. A: 832/13 cells were transfected with a scrambled control siRNA (siControl) or siRNAs directed against Bax (siBax), Bak (siBak), or the combination (siBax siBak) for 72 h and transduced with GFP- or p21-overexpressing adenovirus for the last 48 h of transfection for Western blot analysis and quantification for Cl casp 3 (B), Bax (C), and Bak (D) protein normalized to actin. Data are represented as mean intensities ± SE; n = 4 independent experiments. *Significance vs. siControl + GFP in a 1-way ANOVA test using Tukey's post hoc, P < 0.05.
DISCUSSION
During the development of type 2 diabetes, cellular stress impairs β-cell proliferation and function, promotes apoptosis, and ultimately triggers the demise of functional β-cell mass. Therefore, preservation of functional β-cell mass is essential to maintain euglycemia and prevent the transition from glucose intolerance/insulin resistance to frank diabetes. Various stressors known to influence functional β-cell mass during the progression to diabetes include inflammation, ER stress, free fatty acids, and glucotoxicity, to name a few (11). However, the precise molecular events linking cellular stress to β-cell impairment and destruction are not fully understood.
In an attempt to determine how a selection of stressors modulates functional β-cell mass and whether independent stressors converge on a uniform pathway, we focused initially on factors regulating cellular proliferation. Thus, we examined the inhibitory proteins of the cell cycle machinery during exposure to the synthetic glucocorticoid agonist dexamethasone, described previously as a β-cell stressor (34), and a pharmacological inducer of ER stress, thapsigargin. Both dexamethasone and thapsigargin reduced β-cell proliferation, and we speculated that the induction of p21 mediates this response, as it was the only cell cycle inhibitory protein induced by both stressors. Using p21 overexpression in isolated primary rat islets and β-cell lines, we demonstrated that p21 is sufficient to inhibit proliferation by preventing the transition between the G1/S and G2/M phases of the cell cycle. The ability of p21 to prevent cell cycle transitions has been well established given its ability to directly inhibit the activity of several cyclins and Cdks, such as Cdk1, that are necessary for cell cycle progression (1).
Several physiological and pathophysiological processes seem to converge on p21 as a mechanism to restrain β-cell growth. Previous work has implicated p21 as a molecular brake for β-cell proliferation during stimulation with mitogens such as HGF and placental lactogen to avoid excessive proliferation (10). In addition to mitogen stimulation, ER stress models within β-cells increase p21 levels to inhibit proliferation (36). Finally, treatment with glucocorticoids shown here also induces p21 and presumably limits β-cell proliferation. Although it is counterintuitive that growth factors would induce the expression of a proliferation inhibitor, namely p21, a likely explanation is that these mechanisms are initiated to keep proliferation in check. Therefore, it is possible that negative feedback mechanisms of cell growth, proliferation, and survival signaling pathways are activated simultaneously. This example of negative feedback by a cell cycle inhibitor in the β-cell is not exclusive of p21. Pascoe et al. (27) had demonstrated previously that free fatty acids, which themselves can drive β-cell replication, induce both p16 and p18, thereby blocking glucose-stimulated β-cell proliferation. Together, these findings suggest that the targeting of cell cycle inhibitors may be warranted to maximize β-cell expansion since they serve as molecular brakes to growth-promoting stimuli.
While the role of p21 in proliferation was being characterized, sub-G0 cells were notably higher, leading us to further investigate p21 in β-cell death. Our findings conclude that p21 initiates apoptosis through the intrinsic apoptotic pathway, and the interplay between pro- and antiapoptotic mitochondrial proteins is able to modulate the amount of p21-induced caspase-3 cleavage, a hallmark of apoptosis. Few studies have investigated the role of p21 in β-cell death. Interestingly, in a rat model of diabetes, isolated islets of 12-wk-old Zucker diabetic fatty rats had increased p21 mRNA transcripts that corresponded to a decrease in insulin mRNA (19). Although this study correlated decreased β-cells with an increase in p21, our experiments were able to directly establish that p21 itself is capable of acutely inducing apoptosis through the intrinsic pathway in β-cells, as we demonstrated increased caspase-3 cleavage with p21 overexpression, which could be inhibited by Bcl-2 overexpression or siRNA-mediated suppression of Bax or Bak. In support of our findings, chronically induced p21-overexpressing mice exhibit hyperglycemia accompanied by increased apoptotic β-cells (37). Furthermore, Blandino-Rosano et al. (5) demonstrated that when a constitutively active form of Akt was overexpressed in vivo, which robustly increased β-cell replication and induced β-cell apoptosis, as measured by TUNEL staining, deletion of a single p21 allele attenuated the apoptosis. Thus, chronic elevation of p21 leads to β-cell apoptosis, and decreased levels of p21 are able to prevent it. Beyond its level of expression, p21 localization is central to its function, since triggering p21 localization into the cytoplasm allows for its interaction with other molecules that are involved in stimulating apoptosis, such as JNK and even caspase-3 itself (26, 28). Despite its proapoptotic function in the β-cell, p21 has had a conflicting role in apoptosis, and it has been suggested to also have antiapoptotic actions depending on its subcellular localization (14, 21, 31–33). An explanation for these contradictory assertions could be that p21's actions are cell type specific. Support for this idea is gained by our observation that p21 overexpression does not trigger apoptosis in a hepatocyte cell line, unlike its ability to do so in β-cells.
One well-known cell survival signaling molecule is protein kinase B/Akt. Here, we provide evidence that p21 overexpression inhibits phosphorylation of Akt in 832/13 β-cells. Previous studies have shown that Akt stabilizes p21 protein expression when upregulated and that it is capable of binding to p21 (5, 12, 18, 35, 39). However, our findings suggest that p21 is able to negatively modulate Akt, indicating a possible feedback regulation. Further studies are necessary to determine the direct mechanisms for how p21 is able to attenuate Akt activation. Aside from reducing proliferation and cell survival signaling, our findings suggest that p21 is also mediating the death process that occurs as a result of excessive β-cell stress.
In the β-cell, we have determined that p21 is transcriptionally upregulated under stress conditions. p21 overexpression in turn negatively regulates cell proliferation and survival signaling while simultaneously inducing intrinsic apoptotic pathways in β-cells. Undoubtedly, an increase in p21-mediated apoptosis in the pancreatic islet would be damaging to β-cell function and the integrated insulin secretory capacity. Decreasing the numbers of β-cells during states of insulin resistance or type 2 diabetes would be detrimental to maintaining glucose homeostasis by compromising the ability to secrete insulin. These findings suggest that inhibiting p21's apoptotic role could be a useful therapeutic target to prevent β-cell death, although experiments to directly test this possibility remain to be completed. Additionally, further studies are necessary to determine the exact mechanism by which p21 induces the death pathway in β-cells. Nevertheless, this work suggests a new mechanism for β-cell destruction during stress.
GRANTS
This work was supported by a William Townsend Porter Predoctoral Fellowship from the American Physiological Society (to A. M. Hernandez), National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-078732 (to P. T. Fueger), and a Showalter Research Trust Award from Indiana University School of Medicine (to P. T. Fueger).
DISCLOSURES
The authors have nothing to disclose.
AUTHOR CONTRIBUTIONS
A.M.H. and P.T.F. contributed to the conception and design of the research; A.M.H., E.S.C., Y.-C.C., S.L.G., and L.E.E. performed the experiments; A.M.H., E.S.C., Y.-C.C., S.L.G., L.E.E., and P.T.F. analyzed the data; A.M.H., E.S.C., Y.-C.C., S.L.G., L.E.E., and P.T.F. interpreted the results of the experiments; A.M.H. prepared the figures; A.M.H. drafted the manuscript; A.M.H., E.S.C., Y.-C.C., and P.T.F. edited and revised the manuscript; A.M.H., E.S.C., Y.-C.C., S.L.G., L.E.E., and P.T.F. approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Natalie Stull for technical assistance with rodent islet isolations, Susan Rice and the Indiana University Simon Cancer Center Flow Cytometry Resource Facility for support with flow cytometry, and Dr. David Curiel at Washington University for kindly providing the adenovirus expressing human Bcl-2.
REFERENCES
- 1. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 9: 400–414, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bilbao G, Contreras JL, Eckhoff DE, Mikheeva G, Krasnykh V, Douglas JT, Thomas FT, Thomas JM, Curiel DT. Reduction of ischemia-reperfusion injury of the liver by in vivo adenovirus-mediated gene transfer of the antiapoptotic Bcl-2 gene. Ann Surg 230: 185–193, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bilbao G, Contreras JL, Gómez-Navarro J, Eckhoff DE, Mikheeva G, Krasnykh V, Hynes T, Thomas FT, Thomas JM, Curiel DT. Genetic modification of liver grafts with an adenoviral vector encoding the Bcl-2 gene improves organ preservation. Transplantation 67: 775–783, 1999 [DOI] [PubMed] [Google Scholar]
- 4. Bilbao G, Contreras JL, Mikheeva G, Krasnykh V, Eckhoff DE, Thomas FT, Thomas J, Curiel DT. Genetic cytoprotection of human endothelial cells during preservation time with an adenoviral vector encoding the anti-apoptotic human Bcl-2 gene. Transplant Proc 31: 1012–1015, 1999 [DOI] [PubMed] [Google Scholar]
- 5. Blandino-Rosano M, Alejandro EU, Sathyamurthy A, Scheys JO, Gregg B, Chen AY, Rachdi L, Weiss A, Barker DJ, Gould AP, Elghazi L, Bernal-Mizrachi E. Enhanced beta cell proliferation in mice overexpressing a constitutively active form of Akt and one allele of p21Cip. Diabetologia 55: 1380–1389, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52: 102–110, 2003 [DOI] [PubMed] [Google Scholar]
- 8. Collier JJ, Fueger PT, Hohmeier HE, Newgard CB. Pro- and antiapoptotic proteins regulate apoptosis but do not protect against cytokine-mediated cytotoxicity in rat islets and beta-cell lines. Diabetes 55: 1398–1406, 2006 [DOI] [PubMed] [Google Scholar]
- 9. Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, Takane KK, Garcia-Ocana A, Vasavada R, Stewart AF. Molecular control of cell cycle progression in the pancreatic beta-cell. Endocr Rev 27: 356–370, 2006 [DOI] [PubMed] [Google Scholar]
- 10. Cozar-Castellano I, Weinstock M, Haught M, Velázquez-Garcia S, Sipula D, Stewart AF. Evaluation of beta-cell replication in mice transgenic for hepatocyte growth factor and placental lactogen: comprehensive characterization of the G1/S regulatory proteins reveals unique involvement of p21cip. Diabetes 55: 70–77, 2006 [PubMed] [Google Scholar]
- 11. Donath MY, Ehses JA, Maedler K, Schumann DM, Ellingsgaard H, Eppler E, Reinecke M. Mechanisms of beta-cell death in type 2 diabetes. Diabetes 54, Suppl 2: S108–S113, 2005 [DOI] [PubMed] [Google Scholar]
- 12. Fatrai S, Elghazi L, Balcazar N, Cras-Meneur C, Krits I, Kiyokawa H, Bernal-Mizrachi E. Akt induces beta-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes 55: 318–325, 2006 [DOI] [PubMed] [Google Scholar]
- 13. Fueger PT, Hernandez AM, Chen YC, Colvin ES. Assessing replication and beta cell function in adenovirally-transduced isolated rodent islets. J Vis Exp. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gartel AL. The conflicting roles of the cdk inhibitor p21(CIP1/WAF1) in apoptosis. Leuk Res 29: 1237–1238, 2005 [DOI] [PubMed] [Google Scholar]
- 15. Gudas J, Nguyen H, Li T, Hill D, Cowan KH. Effects of cell cycle, wild-type p53 and DNA damage on p21CIP1/Waf1 expression in human breast epithelial cells. Oncogene 11: 253–261, 1995 [PubMed] [Google Scholar]
- 16. Haycock JW. Polyvinylpyrrolidone as a blocking agent in immunochemical studies. Anal Biochem 208: 397–399, 1993 [DOI] [PubMed] [Google Scholar]
- 17. Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49: 424–430, 2000 [DOI] [PubMed] [Google Scholar]
- 18. Hughes E, Huang C. Participation of Akt, menin, and p21 in pregnancy-induced beta-cell proliferation. Endocrinology 152: 847–855, 2011 [DOI] [PubMed] [Google Scholar]
- 19. Kaneto H, Kajimoto Y, Fujitani Y, Matsuoka T, Sakamoto K, Matsuhisa M, Yamasaki Y, Hori M. Oxidative stress induces p21 expression in pancreatic islet cells: possible implication in beta-cell dysfunction. Diabetologia 42: 1093–1097, 1999 [DOI] [PubMed] [Google Scholar]
- 20. Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B. Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes 49: 1325–1333, 2000 [DOI] [PubMed] [Google Scholar]
- 21. Koster R, di Pietro A, Timmer-Bosscha H, Gibcus JH, van den Berg A, Suurmeijer AJ, Bischoff R, Gietema JA, de Jong S. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J Clin Invest 120: 3594–3605, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li DS, Yuan YH, Tu HJ, Liang QL, Dai LJ. A protocol for islet isolation from mouse pancreas. Nat Protoc 4: 1649–1652, 2009 [DOI] [PubMed] [Google Scholar]
- 23. Liu Y, Martindale JL, Gorospe M, Holbrook NJ. Regulation of p21WAF1/CIP1 expression through mitogen-activated protein kinase signaling pathway. Cancer Res 56: 31–35, 1996 [PubMed] [Google Scholar]
- 24. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[−Delta Delta C(T)] Method. Methods 25: 402–408, 2001 [DOI] [PubMed] [Google Scholar]
- 25. Milburn JL, Jr, Hirose H, Lee YH, Nagasawa Y, Ogawa A, Ohneda M, BeltrandelRio H, Newgard CB, Johnson JH, Unger RH. Pancreatic beta-cells in obesity. Evidence for induction of functional, morphologic, and metabolic abnormalities by increased long chain fatty acids. J Biol Chem 270: 1295–1299, 1995 [DOI] [PubMed] [Google Scholar]
- 26. Park JA, Kim KW, Kim SI, Lee SK. Caspase 3 specifically cleaves p21WAF1/CIP1 in the earlier stage of apoptosis in SK-HEP-1 human hepatoma cells. Eur J Biochem 257: 242–248, 1998 [DOI] [PubMed] [Google Scholar]
- 27. Pascoe J, Hollern D, Stamateris R, Abbasi M, Romano LC, Zou B, O'Donnell CP, Garcia-Ocana A, Alonso LC. Free fatty acids block glucose-induced beta-cell proliferation in mice by inducing cell cycle inhibitors p16 and p18. Diabetes 61: 632–641, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ranta F, Leveringhaus J, Theilig D, Schulz-Raffelt G, Hennige AM, Hildebrand DG, Handrick R, Jendrossek V, Bosch F, Schulze-Osthoff K, Haring HU, Ullrich S. Protein kinase C delta (PKCdelta) affects proliferation of insulin-secreting cells by promoting nuclear extrusion of the cell cycle inhibitor p21Cip1/WAF1. PLoS One 6: e28828, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes 54: 2557–2567, 2005 [DOI] [PubMed] [Google Scholar]
- 30. Tran VV, Chen G, Newgard CB, Hohmeier HE. Discrete and complementary mechanisms of protection of beta-cells against cytokine-induced and oxidative damage achieved by bcl-2 overexpression and a cytokine selection strategy. Diabetes 52: 1423–1432, 2003 [DOI] [PubMed] [Google Scholar]
- 31. Vitiello PF, Staversky RJ, Keng PC, O'Reilly MA. PUMA inactivation protects against oxidative stress through p21/Bcl-XL inhibition of bax death. Free Radic Biol Med 44: 367–374, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vitiello PF, Wu YC, Staversky RJ, O'Reilly MA. p21(Cip1) protects against oxidative stress by suppressing ER-dependent activation of mitochondrial death pathways. Free Radic Biol Med 46: 33–41, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Warfel NA, El-Deiry WS. p21WAF1 and tumourigenesis: 20 years after. Curr Opin Oncol 25: 52–58, 2013 [DOI] [PubMed] [Google Scholar]
- 34. Weinhaus AJ, Bhagroo NV, Brelje TC, Sorenson RL. Dexamethasone counteracts the effect of prolactin on islet function: implications for islet regulation in late pregnancy. Endocrinology 141: 1384–1393, 2000 [DOI] [PubMed] [Google Scholar]
- 35. Wu DD, Feng C, Xu XY, Xiao JY, Liu C, Meng J, Wang EH, Yu BZ. Protein kinase B/Akt may regulate G2/M transition in the fertilized mouse egg by changing the localization of p21(Cip1/WAF1). Cell Biochem Funct 29: 265–271, 2011 [DOI] [PubMed] [Google Scholar]
- 36. Yamada T, Ishihara H, Tamura A, Takahashi R, Yamaguchi S, Takei D, Tokita A, Satake C, Tashiro F, Katagiri H, Aburatani H, Miyazaki J, Oka Y. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic beta-cells. Hum Mol Genet 15: 1600–1609, 2006 [DOI] [PubMed] [Google Scholar]
- 37. Yang J, Zhang W, Jiang W, Sun X, Han Y, Ding M, Shi Y, Deng H. P21cip-overexpression in the mouse beta cells leads to the improved recovery from streptozotocin-induced diabetes. PLoS One 4: e8344, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhan Q, Eldeiry W, Bae I, Alamo I, Kastan M, Vogelstein B, Fornace A. Similarity of the DNA-damage responsiveness and growth-suppressive properties of waf1/cip1 and gadd45. Int J Oncol 6: 937–946, 1995 [DOI] [PubMed] [Google Scholar]
- 39. Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol 3: 245–252, 2001 [DOI] [PubMed] [Google Scholar]