

Abstract
Accumulating evidence suggests that insulin acts within the hypothalamus to alter sympathetic nerve activity (SNA) and baroreflex function. Although insulin receptors are widely expressed across the hypothalamus, recent evidence suggests that neurons of the arcuate nucleus (ARC) play an important role in the sympathoexcitatory response to insulin. The purpose of the present study was to determine whether circulating insulin acts directly in the ARC to elevate SNA. In anesthetized male Sprague-Dawley rats (275–425 g), the action of insulin was neutralized by microinjection of an anti-insulin affibody (1 ng/40 nl). To verify the efficacy of the affibody, ARC pretreatment with injection of the anti-insulin affibody completely prevented the increase in lumbar SNA produced by ARC injection of insulin. Next, ARC pretreatment with the anti-insulin affibody attenuated the lumbar sympathoexcitatory response to intracerebroventricular injection of insulin. Third, a hyperinsulinemic-euglycemic clamp increased lumbar, but not renal, SNA in animals that received ARC injection of a control affibody. However, this sympathoexcitatory response was absent in animals pretreated with the anti-insulin affibody in the ARC. Injection of the anti-insulin affibody in the adjacent ventromedial hypothalamus did not alter the sympathoexcitatory response to insulin. The ability of the anti-insulin affibody to prevent the sympathetic effects of insulin cannot be attributed to a general inactivation or nonspecific effect on ARC neurons as the affibody did not alter the sympathoexcitatory response to ARC disinhibition by gabazine. Collectively, these findings suggest that circulating insulin acts within the ARC to increase SNA.
Keywords: obesity, blood pressure, sympathetic nerve activity, lumbar, renal
insulin acts within the central nervous system to increase sympathetic nerve activity (SNA) and alter baroreflex function. This notion is supported by experimental studies that demonstrated that a hyperinsulinemic-euglycemic clamp elevated muscle or lumbar SNA in humans (3, 40) or rodents (4, 28, 47), respectively. Systemic hyperinsulinemia has also been reported to increase baroreflex gain in both humans and rodents (8, 32, 37, 50). These neural effects are attributed to a central action as intracerebroventricular injection of insulin produces similar responses (4, 31, 47). Furthermore, the sympathoexcitatory effects of insulin are prevented or attenuated by a number of central manipulations, including lesion of the anteroventral third ventricle region (30) or intracerebroventricular administration of a phosphoinositol 3-kinase (PI3K) inhibitor (39). Despite these observations, little is known regarding where circulating insulin is detected in the brain to initiate downstream changes in sympathetic outflow and baroreflex function.
Insulin receptors are expressed throughout the central nervous system with notably higher levels reported in the hypothalamus, including structures such as the hypothalamic paraventricular nucleus (PVH), ventromedial hypothalamus (VMH), and arcuate nucleus (ARC) (26, 48). A hypothalamus-mediated site of action for insulin is supported by studies in rats in which administration of insulin into the lateral, but not fourth, ventricle increased lumbar SNA (37). Within the hypothalamus, previous studies have demonstrated that inhibition of PVH or ARC neurons reversed the sympathoexcitatory response to insulin (8, 47); however, microinjection of insulin into the ARC, but not PVH, increased lumbar SNA (8, 47). Anatomic studies have indicated that ARC neurons densely innervate several hypothalamic structures, including the PVH (9, 11). The ARC contains an abundance of proopiomelanocortin neurons (20, 34). Blockade of melanocortin 3/4 receptors within the PVH attenuates the sympathoexcitatory and pressor response to ARC stimulation (19) and also reverses the sympathoexcitatory response to intracerebroventricular insulin and a systemic hyperinsulinemic-euglycemic clamp (47). Collectively, the results of these studies suggest that ARC neurons may detect changes in circulating insulin to initiate changes in the activity of downstream circuits through the PVH to alter sympathetic outflow (4, 28, 47).
The above studies were limited by the absence of any experimental manipulation to directly interrupt insulin-mediated actions within the ARC and definitively address whether circulating insulin acts on ARC neurons. Therefore, the purpose of the present study was to determine whether systemic insulin acts locally within the ARC to increase lumbar SNA. In these experiments, the actions of insulin were neutralized by microinjection of an anti-insulin affibody. Affibody molecules have a smaller molecular weight versus traditional antibodies. The anti-insulin affibody was recently characterized to prevent the phosphorylation of Akt in differentiated adipocytes by insulin or within VMH neurons in vivo during insulin-induced hypoglycemia (35).
MATERIALS AND METHODS
Animals
All experimental procedures conformed with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Pennsylvania State University College of Medicine. Adult male Sprague-Dawley rats (250–400 g, Charles River Laboratories) were housed in a temperature-controlled room (22–23°C) and maintained on a 12:12-h light-dark cycle for at least 1 wk before experiments. Animals had ad libitium access to laboratory chow (Harlan Teklad Global Diet no. 2018) and deionized water.
General Experimental Procedures
Rats were anesthetized with isoflurane (2–4% in 100% O2) and prepared for recordings of arterial blood pressure (ABP), lumbar SNA, and/or renal SNA as previously described (1, 4, 47). Lumbar SNA was measured in every experiment since previous studies across multiple laboratories have reported that insulin consistently raises lumbar SNA (4, 8, 28, 47). Briefly, the lumbar sympathetic nerve was exposed through a ventral midline incision. The venae cava distal to the renal vessels was gently retracted. The lumbar sympathetic nerve was isolated, placed on a bipolar stainless steel electrode, and insulated with KWIK-SIL (World Precision Instruments). The incision was closed with staples. After the animal had been placed into a stereotaxic frame in the prone position, the left renal sympathetic nerve was isolated through a retroperitoneal incision, placed on bipolar stainless steel electrodes, and insulated with KWIK-SIL. The incision was closed with staples. Nerve signals were amplified (10 K) and filtered (100–1,000 Hz) using a model 1700 differential alternating current amplifier (AM Systems). Signals were digitized at 5 kHz, rectified, and integrated (2-s time constant) using a Micro1401 and Spike 2 software (Cambridge Electronic Design). Animals were artificially ventilated with O2-enriched room air. End-tidal CO2 was maintained between 4.0% and 4.5% using a MicroCapstar End-Tidal CO2 monitor (CWE). Body temperature was maintained at 37.0 ± 0.5°C with a rectal temperature probe (Sable Systems) and a water circulating blanket. Finally, a craniotomy was performed to remove bone overlying the cortex to gain access to the ARC and other brain structures.
After completion of the surgical procedures, isoflurane anesthesia was replaced by α-chloralose (experiments 1 and 2) or Inactin (experiments 3 and 4). α-Chloralose was initially administered as a bolus (50 mg/kg iv) followed by a continuous infusion (25 mg·kg−1·h−1 at 0.25–0.5 ml/h iv). α-Chloralose was dissolved in 2% sodium borate. Inactin was administered as a bolus (120 mg/kg iv) followed by a continuous infusion (5 mg·kg−1·h−1 at 0.5 ml/h). Inactin instead of α-chloralose was used in experiments 3 and 4 as preliminary data in our laboratory indicated that α-chloralose may interfere with the analysis of plasma insulin levels via ELISA. Both Inactin and α-chloralose solutions were dissolved in 0.45% NaCl to maintain electrolytes (Na+ and Cl−) at baseline values. The level of anesthesia was assessed by the absence of a withdrawal reflex to a toe pinch. Animals were allowed to stabilize for a minimum of 60 min before experimental protocols began.
Experimental Protocols
Experiment 1: validation of the anti-insulin affibody in the ARC.
Initial experiments were performed to test the ability of the anti-insulin affibody to prevent sympathetic responses to insulin directly injected into the ARC. Baseline values were recorded for 20 min. Rats then received a bilateral injection of the anti-insulin affibody (1 ng·40 nl−1·side−1, ab31906, Abcam). Control injections were performed using an anti-IgG affibody molecule (1 ng·40 nl−1·side−1, ab31900, Abcam). As mentioned above, the dose of the anti-insulin affibody has been previously used to prevent the phosphorylation of Akt in differentiated adipocytes by insulin or within VMH neurons in vivo during insulin-induced hypoglycemia (35). Ten minutes later, insulin (4 μU·40 nl−1·side−1) was injected bilaterally into the ARC. Variables were recorded for 2 h after the insulin injection. Blood glucose levels were measured from a drop of arterial blood every 30 min using a standard glucometer (One Touch Ultra). ARC microinjections were performed over 5–10 s using single-barrel glass pipettes (outer diameter: 20–40 μm) connected to a pneumatic picopump and lowered into the ARC using the following coordinates in reference to the bregma: 2.4–2.9 mm caudal, 9.7–9.9 mm ventral, and 0.5–0.7 mm lateral. Bilateral injections were performed using a glass pipette for each side. Pipettes remained in the ARC for 5 min after the injections, were raised and rinsed four times with artificial cerebrospinal fluid (aCSF), and then lowered into the ARC for the second injection.
Experiment 2: gabazine in the ARC after pretreatment with the anti-insulin affibody.
A second set of experiments was performed to determine whether injection of the anti-insulin affibody produced a general inhibition or had a nonspecific action on ARC neurons. Baseline values were recorded for 20 min. Rats then received a unilateral injection of the anti-insulin affibody (1 ng·40 nl−1·side−1, ab31906, Abcam). Control injections were performed using an anti-IgG affibody molecule (1 ng·40 nl−1·side−1, ab31900, Abcam). Ten minutes later, the GABAA receptor antagonist gabazine (1 mM·20 nl−1·side−1) was injected unilaterally into the ipsilateral ARC. Injections of the anti-insulin and anti-IgG affibodies were performed in the same animal but contralateral ARC separated by a minimum of 60 min when variables returned and stabilized at baseline values. The order was randomized.
Experiment 3: ARC anti-insulin affibody pretreatment before intracerebroventricular insulin.
A third set of experiments was performed to determine whether ARC neurons mediate the sympathetic response to intracerebroventricular insulin. Inactin-anesthetized rats received a bilateral injection of the anti-insulin affibody (1 ng·40 nl−1·side−1, ab31906, Abcam). Control injections were performed using aCSF or an anti-IgG affibody molecule (1 ng·40 nl−1·side−1, ab31900, Abcam). Ten minutes later, insulin (1 mU/1 μl) was administered into the lateral ventricle using a glass micropipette with the following coordinates in reference to the bregma: −1.5 mm caudal, 1.6 mm lateral, and 4.6 mm ventral. Variables were recorded for 2 h after the injection of insulin. Blood glucose was measured every 30 min. Blood samples (0.2 ml) were collected from an arterial line into microcentrifuge tubes containing EDTA (5mM) at baseline and 30, 60, and 120 min after injection of insulin. Samples were centrifuged (10,000 g, 1 min), and the plasma was stored at −80°C until insulin levels were determined via ELISA. At the end of experiments, Evan's blue dye (0.5%, 1 μl) was injected into the lateral ventricle to verify the intracerebroventricular injection site.
Experiment 4: ARC anti-insulin affibody pretreatment during hyperinsulinemic-euglycemic clamp.
A final set of experiments was performed to determine whether ARC neurons mediate the sympathetic response to a hyperinsulinemic-euglycemic clamp. Inactin-anesthetized rats received a bilateral injection of the anti-insulin affibody (1 ng·40 nl−1·side−1, ab31906, Abcam). Control injections were performed using aCSF or an anti-IgG affibody molecule (1 ng·40 nl−1·side−1, ab31900, Abcam). Ten minutes later, insulin (4.0 mU·kg−1·min−1 iv, Humulin R) and 50% dextrose solution (0.25–2.0 ml/h iv) were infused for 120 min. Blood glucose was measured every 10 min through a brachial arterial line. The dextrose infusion rate was adjusted to maintain euglycemia. Blood samples (0.2 ml) were collected at baseline and 30, 60, and 120 min after the start of the insulin infusion to determine plasma insulin levels. Dextrose was dissolved in 0.45% NaCl to avoid increases in the plasma Na+ concentration. Plasma electrolytes were measured by an i-STAT-1 analyzer and 6+ cartridges (Abbott Laboratories). Control animals were infused with equal volumes of 0.45% saline.
To determine whether the anti-insulin affibody diffused through the pipette track dorsally to the VMH or dorsomedial hypothalamus (DMH), another set of animals was infused with insulin and dextrose as described above. However, the anti-insulin affibody was injected dorsally using the following coordinates in reference to the bregma: 2.4–2.9 mm caudal, 9.0 mm ventral, and 0.5–0.7 mm lateral.
Histology
All injection sites were marked by the addition of rhodamine beads (0.1%) to all solutions. In experiments that required a second injection, FITC beads (0.1%) were added to the second solution. Intracerebroventricular injection sites were marked by an injection of 2% Evan's blue dye (1 μl) after recordings were complete. At the end of the experiments, animals were perfused with 4% paraformaldehyde. Brains were removed, postfixed in 4% paraformaldehyde, and sectioned at 50–100 μm. Adjacent sections were counterstained with cresyl violet. Injection sites were visualized using a Nikon 90i microscope with the appropriate filters. For experiments 1, 3, and 4 with bilateral injections, data are reported for animals with both left and right injections localized to the ARC (or outside the ARC for anatomic controls). Animals with one injection in the ARC but the contralateral injection outside the ARC were omitted.
Determination of Plasma Insulin Levels
Plasma insulin levels were measured by a rat insulin ELISA (EZRMI-13K, Millipore). For purposes of comparison, insulin levels were also analyzed from plasma samples of a rodent model of diet-induced obesity previously described in our laboratory (43). Briefly, male Sprague-Dawley rats (200–250 g, Charles River Laboratories) were fed a low-fat diet (LF group; 10% kcal from fat, D12489B, Research Diets) or a moderately high-fat diet (32% kcal from fat, D12266B, Research Diets) for 13 wk. Those on the moderately high-fat diet segregated into obesity-resistant (OR) and obesity-prone (OP) groups. After 13 wk, these rats were anesthetized with isoflurane and prepared for recordings of SNA, and isoflurane anesthesia was replaced with Inactin as described above. These animals did not receive ARC injections but were used for other experiments not described here. However, plasma insulin values of these animals are reported to verify that the plasma insulin concentrations of animals receiving an intravenous hyperinsulinemic-euglycemic clamp were physiological. The inter- and intraassay coefficients of variance were 5.2% and 7.4%, respectively.
Data Analysis
All data are expressed as means ± SE. Changes in SNA were calculated by subtracting background noise after hexamethonium (30 mg/kg iv) or physically crushing the nerve. For all variables, data were averaged over 5 min and compared with three 5-min baseline periods (before any injection). All data were analyzed by one- or two-way ANOVA with repeated measures when appropriate. All post hoc tests were performed using independent or paired t-tests with a layered Bonferroni correction. P values of <0.05 were considered statistically significant.
RESULTS
Pretreatment With Anti-Insulin Affibody Prevents the Sympathoexcitatory Response to Insulin Injection in the ARC
Initial experiments tested whether pretreatment with the anti-insulin affibody prevented the sympathoexcitatory response to local application of insulin in the ARC. As previously reported (8), ARC injection of insulin significantly increased lumbar SNA at 60, 90, and 120 min (Fig. 1). While values tended to increase at 30 min, this was not statistically significant (P = 0.061). However, pretreatment with the anti-insulin affibody eliminated the sympathetic response to ARC insulin (Fig. 1). In fact, lumbar SNA did not change from baseline values and was significantly lower than animals injected with the control affibody at 60, 90, and 120 min. Injection of the control or anti-insulin affibody alone did not alter any variable (Table 1). Mean ABP and heart rate (control antibody: 420 ± 10 beats/min, anti-insulin affibody: 422 ± 8 beats/min) did not change from baseline values at any time. Figure 1D shows the injection sites for each experiment. In every case, injection of the affibodies overlapped with injection of insulin as shown by the location of red and green beads. Therefore, the injection site for each animal is shown as one injection site.
Fig. 1.

Anti-insulin affibody prevents the sympathoexcitatory response to insulin in the arcuate nucleus (ARC). A and B: arterial blood pressure (ABP) and mean ABP (gray line) (top), integrated (∫) lumbar sympathetic nerve activity (SNA; middle), and raw lumbar SNA (bottom; 1-s segments) of rats that received an injection of the control antibody (A; n = 6) or anti-insulin affibody (B; n = 4) into the ARC 10 min before ARC injection of insulin. C: mean ± SE values of mean ABP and lumbar SNA. *P < 0.05, control vs. anti-insulin affibody. Arrows denote ARC injection. D: schematic illustration and photomicrographs of ARC injection sites. Coordinates are rostrocaudal levels in reference to the bregma using the atlas of Paxinos and Watson. (36). The photomicrograph shows the injection site for red (i) and green (ii) beads for the same animal, although sections are 80 μm apart. Scale bars = 200 μm. DMH, dorsomedial hypothalamus; VMH, ventromedial hypothalamus; 3V, third ventricle.
Table 1.
Changes in mean ABP and SNA after injection of control or anti-insulin affibody into the arcuate nucleus in Inactin-anesthetized rats that received an infusion of 0.45% NaCl (0.2–2.0 ml/h iv)
| Time |
|||||
|---|---|---|---|---|---|
| −20 min | 0 min | 30 min | 60 min | 120 min | |
| Control affibody | |||||
| Mean ABP, mmHg | 94 ± 3 | 93 ± 2 | 95 ± 4 | 94 ± 3 | 93 ± 2 |
| Lumbar SNA, % | 100 ± 1 | 101 ± 3 | 102 ± 3 | 104 ± 4 | 104 ± 6 |
| Renal SNA, % | 100 ± 1 | 99 ± 2 | 99 ± 4 | 98 ± 5 | 97 ± 5 |
| Anti-insulin affibody | |||||
| Mean ABP, mmHg | 95 ± 2 | 94 ± 4 | 93 ± 4 | 92 ± 6 | 92 ± 5 |
| Lumbar SNA, % | 100 ± 1 | 100 ± 4 | 101 ± 6 | 102 ± 7 | 102 ± 6 |
| Renal SNA, % | 100 ± 1 | 100 ± 2 | 101 ± 4 | 102 ± 6 | 103 ± 7 |
Values are means ± SE; n = 3 animals injected with control affibody and 4 animals injected with anti-insulin affibody. ABP, arterial blood pressure; SNA, sympathetic nerve activity.
Anti-Insulin Affibody Did Not Alter the Sympathoexcitatory Response to ARC Injection of Gabazine
To test whether the anti-insulin affibody produced a general nonspecific inactivation of ARC neurons, the GABAA antagonist gabazine was injected into the ARC 10 min after the injection of the control or anti-insulin affibody. Unilateral injection of gabazine significantly increased mean ABP, lumbar SNA, and renal SNA (Fig. 2). Injection of the anti-insulin affibody did not significantly alter these responses. In both groups, gabazine significantly increased heart rate (0 min: 425 ± 12 beats/min vs. 20 min: 535 ± 19 beats/min for all groups, P < 0.01), end-tidal CO2 (0 min: 2.6 ± 0.1% vs. 20 min: 3.4 ± 0.1% for all groups, P < 0.01), and core body temperature (0 min: 37.2 ± 0.1°C vs. 30 min: 37.8 ± 0.1°C for all groups, P < 0.01). In these experiments, expired CO2 and core temperature were permitted to change based on preliminary experiments showing that ARC disinhibition altered these variables. Figure 2B shows the injection sites for each experiment. In every case, injection of the affibodies overlapped with injection of gabazine. Therefore, the injection site for each animal is shown as one injection site.
Fig. 2.

A: mean ABP, integrated lumbar SNA, and integrated renal SNA of rats injected with gabazine 10 min after injection of control or anti-insulin affibodies in the ARC. Gabazine significantly increased all variables, but the magnitudes were not different between groups. Values are means ± SE. Arrows denote ARC injection. B: schematic illustration of ARC injection sites.
ARC Pretreatment With Anti-Insulin Affibody Attenuates the Sympathoexcitatory Response to Intracerebroventricular Insulin
To test whether intracerebroventricular insulin increased SNA through insulin-dependent actions on ARC neurons, the control or anti-insulin affibody was injected 10 min before an injection of insulin into the lateral ventricle. As shown in Fig. 3, intracerebroventricular injection of insulin significantly increased lumbar, but not renal, SNA over 120 min. Although intracerebroventricular insulin also increased lumbar SNA in animals pretreated with the anti-insulin affibody in the ARC, the magnitude of the effect was significantly attenuated at every time point (Fig. 3). Renal SNA, mean ABP, and heart rate did not change significantly from baseline values in any group.
Fig. 3.

A: mean ABP, integrated and raw lumbar SNA, and integrated and raw renal SNA of rats injected with control or anti-insulin affibodies at 10 min before an intracerebroventricular injection of insulin. In animals injected with the control affibody, intracerebroventricular injection of insulin significantly increased lumbar, but not renal, SNA. This lumbar sympathoexcitatory effect was attenuated in animals pretreated with ARC anti-insulin affibody. B: mean ± SE values of mean ABP, lumbar SNA, and renal SNA in both groups. Arrows denote ARC injection. *P < 0.05, control vs. anti-insulin affibody.
ARC Pretreatment with Anti-Insulin Affibody Prevents the Sympathoexcitatory Response to Hyperinsulinemic-Euglycemic Clamp
The major goal of this study was to determine whether circulating insulin acts within the ARC to increase SNA. Figure 4 shows examples of raw traces. Figure 5 shows summary data. As expected (4, 47), the hyperinsulinemic-euglycemic clamp significantly increased lumbar, but not renal, SNA over 120 min in animals pretreated with the control affibody in the ARC. In marked contrast, ARC pretreatment with the anti-insulin affibody eliminated the sympathoexcitatory response to insulin. In fact, values of lumbar SNA were not different from those of animals that received an ARC injection but were infused with saline intravenously (Fig. 5 and Table 1). As noted above, a number of hypothalamic nuclei contain insulin receptors, including those located dorsal to the ARC, including the VMH and DMH. To control for diffusion of the anti-insulin affibody dorsally through the pipette track to the VMH or DMH, a hyperinsulinemic-euglycemic clamp was performed in a final set of animals pretreated with the anti-insulin affibody in the VMH. As shown in Figs. 4 and 5, insulin still produced a lumbar sympathoexcitatory response in rats pretreated with the anti-insulin affibody in the VMH. Figure 6 shows the injection sites for all three groups.
Fig. 4.

A–C: examples of mean ABP, integrated and raw lumbar SNA, and integrated and raw renal SNA of rats that received a hyperinsulinemic-euglycemic clamp after pretreatment with control affibody in the ARC (A), anti-insulin affibody in the ARC (B), or anti-insulin affibody in the VMH (C). Summary data are shown in Fig. 5, and histology is shown in Fig. 6. Infusion of insulin increased lumbar SNA in rats that received a control antibody injection in the ARC or an anti-insulin injection in the VMH. However, the lumbar sympathoexcitatory response was absent in rats pretreated with the anti-insulin affibody in the ARC. Arrows denote ARC injection.
Fig. 5.

Mean ± SE values of mean ABP, lumbar SNA, renal SNA, heart rate [in beats/min (bpm)], and blood glucose of rats that received a hyperinsulinemic-euglycemic clamp after pretreatment with the control affibody in the ARC, anti-insulin affibody in the ARC, or anti-insulin affibody in the VMH. Injection sites are shown in Fig. 6. Infusion of insulin increased lumbar SNA in rats that received a control antibody injection in the ARC or anti-insulin injection in the VMH. However, the lumbar sympathoexcitatory response was absent in rats pretreated with the anti-insulin affibody in the ARC. Arrows denote ARC injection. *P < 0.05, ARC control affibody vs. ARC anti-insulin affibody; †P < 0.05, VMH anti-insulin vs. ARC anti-insulin.
Fig. 6.
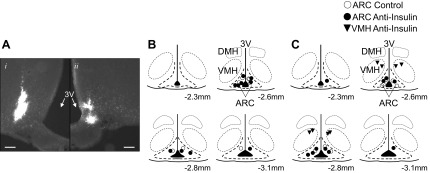
A: photomicrograph of a bilateral ARC injection site with left (i) and right (ii) sides. In this animal, the injection sites were 120 μm apart in the rostral-caudal plane. Scale bars = 100 μm. B and C: schematic illustration of ARC or VMH injection sites for animals injected with intracerebroventricular insulin (B) or that received a hyperinsulinemic-euglycemic clamp (C).
An interesting observation was the significant difference in mean ABP at 120 min between rats pretreated with the ARC anti-insulin affibody versus the ARC control affibody or VMH anti-insulin affibody. The latter two groups had a significantly higher mean ABP than the former. Heart rate did not differ across groups.
Plasma insulin values are shown in Fig. 7. The differences in the sympathoexcitatory response to insulin between animals pretreated with the anti-insulin antibody in the ARC versus those pretreated with the anti-insulin antibody in the VMH or control affibody in the ARC cannot be explained by differences in plasma insulin values. Injection of the control or anti-insulin affibody into the ARC alone did not alter plasma insulin levels (data not shown). Furthermore, the resultant plasma insulin values during the hyperinsulinemic-euglycemic clamps were not statistically different from those of OP animals fed a moderately high-fat diet for 13 wk. OP animals had significantly higher plasma insulin values than LF or OR animals. As expected (43), OP animals versus LF or OR animals had higher body weight (OP: 775 ± 16 g vs. LF: 610 ± 9 g vs. OR: 601 ± 7 g, n = 8 animals/group, P < 0.05) and adiposity index (OP: 7.7 ± 0.3% vs. LF: 4.8 ± 0.2% vs. OR: 4.7 ± 0.2%, n = 8 animals/group, P < 0.01).
Fig. 7.

Left: mean ± SE values of plasma insulin levels in rats treated with a hyperinsulinemic-euglycemic clamp plus pretreatment with a control or an anti-insulin affibody in the ARC or VMH. As expected, plasma insulin levels increased from baseline values (P < 0.01). Right: mean ± SE values of plasma insulin levels of low-fat (LF), obesity-resistant (OR), or obesity-prone (OP) rats. OP rats had a significantly higher plasma insulin values than LF or OR rats (n = 8 animals/group). *P < 0.05. Plasma insulin values of OP rats did not significantly differ from those of normal rats infused with insulin.
DISCUSSION
Despite the ability of insulin to act within the hypothalamus to alter cardiovascular function, the primary site responsible for detecting circulating insulin and initiating downstream changes in sympathetic outflow was previously unknown. The present findings provide several novel observations: 1) microinjection of the anti-insulin affibody prevented the sympathetic response to injection of insulin in the ARC, 2) ARC pretreatment with the anti-insulin affibody attenuated the sympathetic response to intracerebroventricular insulin, 3) ARC injection of the anti-insulin affibody prevented the elevated lumbar SNA during a hyperinsulinemic-euglycemic clamp, and 4) VMH injection of the anti-insulin affibody did not alter the sympathoexcitatory response to insulin. Taken together, these findings suggest that circulating insulin acts on ARC neurons to increase sympathetic outflow.
The ARC has been hypothesized as a primary site for the cardiovascular actions of insulin due to the abundance of insulin receptors, neurochemistry, and anatomic connections. Although insulin receptor binding or mRNA is present at high levels within several hypothalamic nuclei, including in the ARC and VMH (26, 48), anatomic studies using injection of transneuronal viruses into sympathetic vasomotor tissues have repeatedly reported labeled cell bodies in the ARC but not VMH (6, 7). Second, the ARC contains proopiomelanocortin neurons that densely innervate the PVH (9–11, 34). Activation of ARC neurons by N-methyl-d-aspartate increases SNA and ABP (19, 20), and this sympathoexcitatory response was attenuated or prevented by blockade of glutamate or melanocortin 3/4 receptors in the PVH (19, 20). Interestingly, direct injection of insulin into the ARC increases lumbar SNA (8), and the sympathoexcitatory effects of insulin are prevented in melanocortin 4 receptor-deficient mice (38) or by PVH injection of the melanocortin 3/4 antagonist SHU-9119 (47). Finally, inhibition of ARC neurons with injection of the GABAA agonist muscimol reversed the sympathoexcitatory response to systemic insulin (8). Taken together, these observations suggest that insulin raises SNA through a pathway originating from the ARC to the PVH and rostral ventrolateral medulla; however, these studies were limited by the absence of any direct experimental manipulation of insulin actions within the ARC.
The present study used an anti-insulin affibody to locally neutralize the actions of insulin within a specific brain region. The efficacy of this affibody has been previously reported to block the phosphorylation of Akt in differentiated adipocytes by insulin or within VMH neurons in vivo during insulin-induced hypoglycemia (35). While we did not perform similar measures within the ARC, our initial experiments validated the use of the affibody as ARC injection of the anti-insulin affibody prevented the sympathoexcitatory response to local ARC application of insulin and partially attenuated the response to intracerebroventricular insulin. These effects cannot be attributed to a nonspecific action of the anti-insulin affibody as local ARC injection did not affect the sympathoexcitatory response to ARC-injected gabazine. Although there were differences in the baseline mean ABP of animals injected with ARC insulin, this difference is not likely to explain the absence of a sympathetic response in the anti-insulin affibody-treated group. The discrepancy between a complete prevention of the sympathetic response to local ARC insulin versus a partial attenuation to intracerebroventricular insulin can be attributed to one of two explanations: 1) intracerebroventricular insulin can also act outside of the ARC to increase SNA or 2) the dose of intracerebroventricular insulin exceeded the neutralizing ability of the anti-insulin affibody. Indeed, the amount of insulin administered intracerebroventricularly was high. On the other hand, it remains plausible that insulin administered intracerebroventricularly may act on other structures to increase SNA as insulin receptors are expressed widely throughout the hypothalamus (26, 48). To the extent that it has been tested, insulin injection into these other hypothalamic areas, such as the PVH, does not increase SNA (4, 8, 47).
The main goal of the present study was to use the anti-insulin affibody to directly test whether circulating insulin acts within the ARC to increase SNA. As previously reported by a number of laboratories (4, 28, 47), a hyperinsulinemic-euglycemic clamp significantly increased lumbar SNA. ARC pretreatment with the anti-insulin affibody prevented the sympathoexcitatory response. Since nuclei adjacent to the ARC also express insulin receptors (26, 48), these effects may be attributed to diffusion of the antibody through the pipette track and outside the ARC. However, injection of the anti-insulin affibody into the VMH did not significantly alter the lumbar SNA response to systemic insulin administration. This observation is also consistent with the lack of positively labeled cell bodies in the VMH after injection of transneuronal viruses into sympathetically innervated vasomotor tissues such as the kidney, spleen, adrenal gland, or skeletal muscle (6, 7, 17, 21). Altogether, these data demonstrate that circulating insulin acts within the ARC to increase SNA.
Insulin has been well documented across several laboratories to increase lumbar SNA with variable effects on ABP including a decrease (4), no change (4, 8, 28, 30, 37, 47), or increase (47) in mean ABP. In the present study, the route of insulin administration and perhaps different anesthetics may have influenced the effect of insulin on mean ABP. Local ARC injection of insulin tended to decrease mean ABP in α-chloralose-anesthetized rats (Fig. 1), intracerebroventricular insulin did not alter mean ABP in Inactin-anesthetized rats, and an intravenous hyperinsulinemic-euglycemic clamp produced a small increase in mean ABP of Inactin-anesthetized rats. As mentioned above, we have found that mean ABP is more stable over time in Inactin- versus α-chloralose-anesthetized rodents (see Table 1). This observation coupled with the increase in lumbar SNA and blood volume due to the intravenous clamp may have produced the increase in mean ABP. Furthermore, the lack of a change in mean ABP during an increase in lumbar SNA could be attributed to several factors. First, it is not clear whether the increase in lumbar SNA significantly reduced muscle blood flow during insulin injection into ARC, intracerebroventricularly, or intravenously. Second, we did not observe an increase in renal SNA after intracerebroventricular insulin or a hyperinsulinemic-euglycemic clamp. This was a consistent observation in our laboratory over a 2-h period (4, 47). A small change in the resistance of muscle arterioles would likely have little impact on total peripheral resistance in an anesthetized animal. Therefore, these factors may explain the lack of a change in ABP during a selective increase in lumbar SNA in response to ARC insulin or intracerebroventricular insulin.
The absence of any change in renal SNA during insulin administration is in marked contrast to the findings of Rahmouni and colleagues (29, 38, 39). These studies reported that large doses of insulin administered intracerebroventricularly increased renal SNA of rats or mice at 3–6 h after insulin injection. In these studies, renal and lumbar SNA increased 100–200% without any change in mean ABP (29, 38, 39). Second, the time course of the renal sympathoexcitatory response to intracerebroventricular insulin appears variable with a statistical increase at 120 or 240 min. To address whether intravenous insulin influences renal SNA over longer time periods, we did extend the hyperinsulinemic-euglycemic clamps to 4 h in a subset of animals that received an ARC injection of the control or anti-insulin affibody. In control affibody-injected animals, lumbar SNA continued to increase (2 h: 145 ± 10% vs. 4 h: 161 ± 14%, n = 4) but renal SNA did not change (2 h: 96 ± 8% vs. 4 h: 102 ± 11, n = 4). However, the ARC anti-insulin affibody injection still prevented the increase in lumbar SNA (2 h: 107 ± 3% vs. 4 h: 110 ± 5, n = 3) with no change in renal SNA (2 h: 103 ± 4% vs. 4 h: 109 ± 6%, n = 3). To our knowledge, there are limited data on the impact of intravenous hyperinsulinemic-euglyemic clamps on renal SNA (4, 28, 47), and all of these studies examined the renal SNA response over 2 h. In humans, a hyperinsulinemic-euglycemic clamp has been reported to increase muscle SNA but failed to significantly alter renal norepinephrine spillover (15). Collectively, the above observations indicate that intravenous insulin administered acutely at physiological concentrations has limited impact on renal SNA.
An interesting observation in the present study was that the injection of gabazine increased lumbar SNA, renal SNA, and ABP, but also increased end-tidal CO2 and body temperature. Although the SNA and ABP responses to ARC gabazine have been previously reported (19), the latter effect may be attributed to increased metabolism via elevated brown adipose tissue SNA. Indeed, unpublished data in our laboratory indicate that gabazine microinjection into the ARC increases brown adipose tissue SNA. Since a source of GABA in the ARC originates from local neuropeptide Y neurons projecting onto proopiomelanocortin neurons (19, 20), gabazine may block this inhibitory input to produce the sympathetic and cardiovascular responses. In addition, recent reports (23, 44) have suggested that GABAergic ARC neurons are important to mediate the thermogenic actions of leptin. Given the differences in the hemodynamic and thermogenic responses between insulin and gabazine in the ARC, it is likely that blockade of GABAA receptors targets a much larger population of neurons than those affected by insulin.
Within the ARC nucleus, in vitro patch-clamp studies (16, 49) have reported that insulin hyperpolarized and leptin depolarized distinct populations of proopiomelanocortin neurons. In both instances, the electrophysiological responses were prevented by inhibition of PI3K (16, 49). The role of PI3K as a key mediator of insulin's actions is supported by the results of an in vivo study (39) in which intracerebroventricular administration of the PI3K inhibitors LY-294002 or wortmannin attenuated the lumbar sympathoexcitatory response to intracerebroventricular insulin in rats. While these studies underscore the potential importance of PI3K in leptin and insulin's actions, it also questions how PI3K can have two diametric actions within ARC proopiomelanocortin neurons. Furthermore, there is no evidence regarding the electrophysiological responses of ARC neurons to insulin or leptin during synaptic blockade or to leptin and insulin under much longer timeframes (minutes to hours) that may relate to its effects on feeding and sympathetic regulation.
Numerous laboratories have reported that insulin acutely increases SNA and enhances baroreflex gain (3, 4, 8, 28, 32, 37, 40, 47, 50). Yet, the role of insulin in cardiovascular dysfunction in diabetes or obesity remains controversial. Obesity is characterized by elevated SNA to the kidney and muscle (12, 45), with the latter consistent with a sympathoexcitatory role for insulin. However, baroreflexes are attenuated (14, 42, 51). Whereas some studies (24, 27, 41, 46) have reported a direct correlation between obesity, plasma insulin levels, and blood pressure or muscle SNA, others (2, 13, 45) have failed to do so. One complicating factor is that insulin works centrally to exert its effects on SNA and baroreflex function. Therefore, the interpretation of these studies is potentially confounded by changes in insulin transport across the blood-brain barrier in obese animals/subjects (5, 18, 22, 33). These latter studies have clearly indicated that insulin is transported into the brain via a saturable transporter, but pathological disease states (such as obesity) decrease insulin delivery into the central nervous system. Furthermore, there is little information regarding the insulin responsiveness or sensitivity of ARC neurons regulating sympathetic outflow in obesity. Interestingly, a recent study (25) in high-fat diet-fed rabbits reported that acute intracerebroventricular administration of an insulin receptor antagonist lowered ABP but not renal SNA. Baroreflex function was not assessed. Clearly, future studies are needed to address the contribution of brain insulin and ARC neurons to altered SNA and baroreflex function in diabetes and/or obesity.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant R01-HL-090826 (to S. D. Stocker), an American Heart Association (AHA) Established Investigator Grant (to S. D. Stocker), and an AHA-Great Rivers Predoctoral Fellowship (to L. Wolfgang). J. L. Frielle was supported, in part, by an undergraduate fellowship from the American Physiological Society and AHA Undergraduate Fellowship 10UFEL3900000.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: B.S.L., J.L.F., L.W., and S.D.S. conception and design of research; B.S.L., J.L.F., L.W., and S.D.S. performed experiments; B.S.L., J.L.F., and S.D.S. analyzed data; B.S.L., J.L.F., and S.D.S. interpreted results of experiments; B.S.L., J.L.F., L.W., and S.D.S. edited and revised manuscript; B.S.L., J.L.F., L.W., and S.D.S. approved final version of manuscript; S.D.S. prepared figures; S.D.S. drafted manuscript.
ACKNOWLEDGMENTS
The authors thank Shane Miller for technical assistance.
Present address of B. Luckett: Integrative Physiology and Health Sciences, Alma College, 614 W. Superior St., Alma, MI 48801.
REFERENCES
- 1.Adams JM, Madden CJ, Sved AF, Stocker SD. Increased dietary salt enhances sympathoexcitatory and sympathoinhibitory responses from the rostral ventrolateral medulla. Hypertension 50: 354–359, 2007 [DOI] [PubMed] [Google Scholar]
- 2.Alvarez GE, Beske SD, Ballard TP, Davy KP. Sympathetic neural activation in visceral obesity. Circulation 106: 2533–2536, 2002 [DOI] [PubMed] [Google Scholar]
- 3.Anderson EA, Hoffman RP, Balon TW, Sinkey CA, Mark AL. Hyperinsulinemia produces both sympathetic neural activation and vasodilation in normal humans. J Clin Invest 87: 2246–2252, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bardgett ME, McCarthy JJ, Stocker SD. Glutamatergic receptor activation in the rostral ventrolateral medulla mediates the sympathoexcitatory response to hyperinsulinemia. Hypertension 55: 284–290, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baura GD, Foster DM, Porte D, Jr, Kahn SE, Bergman RN, Cobelli C, Schwartz MW. Saturable transport of insulin from plasma into the central nervous system of dogs in vivo. A mechanism for regulated insulin delivery to the brain. J Clin Invest 92: 1824–1830, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cano G, Card JP, Sved AF. Dual viral transneuronal tracing of central autonomic circuits involved in the innervation of the two kidneys in rat. J Comp Neurol 471: 462–481, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Cano G, Sved AF, Rinaman L, Rabin BS, Card JP. Characterization of the central nervous system innervation of the rat spleen using viral transneuronal tracing. J Comp Neurol 439: 1–18, 2001 [DOI] [PubMed] [Google Scholar]
- 8.Cassaglia PA, Hermes SM, Aicher SA, Brooks VL. Insulin acts in the arcuate nucleus to increase lumbar sympathetic nerve activity and baroreflex function in rats. J Physiol 589: 1643–1662, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron 23: 775–786, 1999 [DOI] [PubMed] [Google Scholar]
- 10.Elias CF, Kelly JF, Lee CE, Ahima RS, Drucker DJ, Saper CB, Elmquist JK. Chemical characterization of leptin-activated neurons in the rat brain. J Comp Neurol 423: 261–281, 2000 [PubMed] [Google Scholar]
- 11.Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, Tatro JB, Hoffman GE, Ollmann MM, Barsh GS, Sakurai T, Yanagisawa M, Elmquist JK. Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J Comp Neurol 402: 442–459, 1998 [PubMed] [Google Scholar]
- 12.Esler M, Straznicky N, Eikelis N, Masuo K, Lambert G, Lambert E. Mechanisms of sympathetic activation in obesity-related hypertension. Hypertension 48: 787–796, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Gentile CL, Orr JS, Davy BM, Davy KP. Modest weight gain is associated with sympathetic neural activation in nonobese humans. Am J Physiol Regul Integr Comp Physiol 292: R1834–R1838, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Grassi G, Seravalle G, Colombo M, Bolla G, Cattaneo BM, Cavagnini F, Mancia G. Body weight reduction, sympathetic nerve traffic, and arterial baroreflex in obese normotensive humans. Circulation 97: 2037–2042, 1998 [DOI] [PubMed] [Google Scholar]
- 15.Gudbjornsdottir S, Friberg P, Elam M, Attvall S, Lonnroth P, Wallin BG. The effect of metformin and insulin on sympathetic nerve activity, norepinephrine spillover and blood pressure in obese, insulin resistant, normoglycemic, hypertensive men. Blood Press 3: 394–403, 1994 [DOI] [PubMed] [Google Scholar]
- 16.Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, Cowley MA, Cantley LC, Lowell BB, Elmquist JK. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest 118: 1796–1805, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jansen AS, Hoffman JL, Loewy AD. CNS sites involved in sympathetic and parasympathetic control of the pancreas: a viral tracing study. Brain Res 766: 29–38, 1997 [DOI] [PubMed] [Google Scholar]
- 18.Kaiyala KJ, Prigeon RL, Kahn SE, Woods SC, Schwartz MW. Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes 49: 1525–1533, 2000 [DOI] [PubMed] [Google Scholar]
- 19.Kawabe T, Kawabe K, Sapru HN. Cardiovascular responses to chemical stimulation of the hypothalamic arcuate nucleus in the rat: role of the hypothalamic paraventricular nucleus. PLos One 7: e45180, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawabe T, Kawabe K, Sapru HN. Effect of barodenervation on cardiovascular responses elicited from the hypothalamic arcuate nucleus of the rat. PLos One 7: e53111, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kerman IA, Akil H, Watson SJ. Rostral elements of sympatho-motor circuitry: a virally mediated transsynaptic tracing study. J Neurosci 26: 3423–3433, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kern W, Benedict C, Schultes B, Plohr F, Moser A, Born J, Fehm HL, Hallschmid M. Low cerebrospinal fluid insulin levels in obese humans. Diabetologia 49: 2790–2792, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Kong D, Tong Q, Ye C, Koda S, Fuller PM, Krashes MJ, Vong L, Ray RS, Olson DP, Lowell BB. GABAergic RIP-Cre neurons in the arcuate nucleus selectively regulate energy expenditure. Cell 151: 645–657, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landsberg L. Insulin-mediated sympathetic stimulation: role in the pathogenesis of obesity-related hypertension (or, how insulin affects blood pressure, and why). J Hypertens 19: 523–528, 2001 [DOI] [PubMed] [Google Scholar]
- 25.Lim K, Burke SL, Head GA. Obesity-related hypertension and the role of insulin and leptin in high-fat-fed rabbits. Hypertension 61: 628–634, 2013 [DOI] [PubMed] [Google Scholar]
- 26.Marks JL, Porte D, Jr, Stahl WL, Baskin DG. Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology 127: 3234–3236, 1990 [DOI] [PubMed] [Google Scholar]
- 27.Monroe MB, Van Pelt RE, Schiller BC, Seals DR, Jones PP. Relation of leptin and insulin to adiposity-associated elevations in sympathetic activity with age in humans. Int J Obes Relat Metab Disord 24: 1183–1187, 2000 [DOI] [PubMed] [Google Scholar]
- 28.Morgan DA, Balon TW, Ginsberg BH, Mark AL. Nonuniform regional sympathetic nerve responses to hyperinsulinemia in rats. Am J Physiol Regul Integr Comp Physiol 264: R423–R427, 1993 [DOI] [PubMed] [Google Scholar]
- 29.Morgan DA, Rahmouni K. Differential effects of insulin on sympathetic nerve activity in agouti obese mice. J Hypertens 28: 1913–1919, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muntzel M, Beltz T, Mark AL, Johnson AK. Anteroventral third ventricle lesions abolish lumbar sympathetic responses to insulin. Hypertension 23: 1059–1062, 1994 [DOI] [PubMed] [Google Scholar]
- 31.Muntzel MS, Morgan DA, Mark AL, Johnson AK. Intracerebroventricular insulin produces nonuniform regional increases in sympathetic nerve activity. Am J Physiol Regul Integr Comp Physiol 267: R1350–R1355, 1994 [DOI] [PubMed] [Google Scholar]
- 32.Okada M, Bunag RD. Insulin acts centrally to enhance reflex tachycardia in conscious rats. Am J Physiol Regul Integr Comp Physiol 266: R481–R486, 1994 [DOI] [PubMed] [Google Scholar]
- 33.Owen OE, Reichard GA, Jr, Boden G, Shuman C. Comparative measurements of glucose, beta-hydroxybutyrate, acetoacetate, and insulin in blood and cerebrospinal fluid during starvation. Metabolism 23: 7–14, 1974 [DOI] [PubMed] [Google Scholar]
- 34.Palkovits M, Mezey E, Eskay RL. Pro-opiomelanocortin-derived peptides (ACTH/beta-endorphin/alpha-MSH) in brainstem baroreceptor areas of the rat. Brain Res 436: 323–338, 1987 [DOI] [PubMed] [Google Scholar]
- 35.Paranjape SA, Chan O, Zhu W, Horblitt AM, McNay EC, Cresswell JA, Bogan JS, McCrimmon RJ, Sherwin RS. Influence of insulin in the ventromedial hypothalamus on pancreatic glucagon secretion in vivo. Diabetes 59: 1521–1527, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates(4th ed CD-ROM).San Diego, CA: Academic, 1998 [Google Scholar]
- 37.Pricher MP, Freeman KL, Brooks VL. Insulin in the brain increases gain of baroreflex control of heart rate and lumbar sympathetic nerve activity. Hypertension 51: 514–520, 2008 [DOI] [PubMed] [Google Scholar]
- 38.Rahmouni K, Haynes WG, Morgan DA, Mark AL. Role of melanocortin-4 receptors in mediating renal sympathoactivation to leptin and insulin. J Neurosci 23: 5998–6004, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rahmouni K, Morgan DA, Morgan GM, Liu X, Sigmund CD, Mark AL, Haynes WG. Hypothalamic PI3K and MAPK differentially mediate regional sympathetic activation to insulin. J Clin Invest 114: 652–658, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rowe JW, Young JB, Minaker KL, Stevens AL, Pallotta J, Landsberg L. Effect of insulin and glucose infusions on sympathetic nervous system activity in normal man. Diabetes 30: 219–225, 1981 [DOI] [PubMed] [Google Scholar]
- 41.Scherrer U, Randin D, Tappy L, Vollenweider P, Jequier E, Nicod P. Body fat and sympathetic nerve activity in healthy subjects. Circulation 89: 2634–2640, 1994 [DOI] [PubMed] [Google Scholar]
- 42.Schreihofer AM, Mandel DA, Mobley SC, Stepp DW. Impairment of sympathetic baroreceptor reflexes in obese Zucker rats. Am J Physiol Heart Circ Physiol 293: H2543–H2549, 2007 [DOI] [PubMed] [Google Scholar]
- 43.Stocker SD, Meador R, Adams JM. Neurons of the rostral ventrolateral medulla contribute to obesity-induced hypertension in rats. Hypertension 49: 640–646, 2007 [DOI] [PubMed] [Google Scholar]
- 44.Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci 11: 998–1000, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vaz M, Jennings G, Turner A, Cox H, Lambert G, Esler M. Regional sympathetic nervous activity and oxygen consumption in obese normotensive human subjects. Circulation 96: 3423–3429, 1997 [DOI] [PubMed] [Google Scholar]
- 46.Ward KD, Sparrow D, Landsberg L, Young JB, Vokonas PS, Weiss ST. Influence of insulin, sympathetic nervous system activity, and obesity on blood pressure: the Normative Aging Study. J Hypertens 14: 301–308, 1996 [DOI] [PubMed] [Google Scholar]
- 47.Ward KR, Bardgett JF, Wolfgang L, Stocker SD. Sympathetic response to insulin is mediated by melanocortin 3/4 receptors in the hypothalamic paraventricular nucleus. Hypertension 57: 435–441, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Werther GA, Hogg A, Oldfield BJ, McKinley MJ, Figdor R, Allen AM, Mendelsohn FA. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology 121: 1562–1570, 1987 [DOI] [PubMed] [Google Scholar]
- 49.Williams KW, Margatho LO, Lee CE, Choi M, Lee S, Scott MM, Elias CF, Elmquist JK. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci 30: 2472–2479, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Young CN, Deo SH, Chaudhary K, Thyfault JP, Fadel PJ. Insulin enhances the gain of arterial baroreflex control of muscle sympathetic nerve activity in humans. J Physiol 588: 3593–3603, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao D, McCully BH, Brooks VL. Rosiglitazone improves insulin sensitivity and baroreflex gain in rats with diet-induced obesity. J Pharmacol Exp Ther 343: 206–213, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]