

Abstract
Heat shock protein 90 (Hsp90) inhibitors were initially developed as anticancer agents; however, it is becoming increasing clear that they also possess potent anti-inflammatory properties. Posttranslational modifications of Hsp90 have been reported in tumors and have been hypothesized to affect client protein- and inhibitor-binding activities. In the present study we investigated the posttranslational modification of Hsp90 in inflammation. LPS, a prototypical inflammatory agent, induced concentration- and time-dependent tyrosine (Y) phosphorylation of Hsp90α and Hsp90β in bovine pulmonary arterial and human lung microvascular endothelial cells (HLMVEC). Mass spectrometry identified Y309 as a major site of Y phosphorylation on Hsp90α (Y300 of Hsp90β). LPS-induced Hsp90 phosphorylation was prevented by the Hsp90 inhibitor 17-allyl-amino-demethoxy-geldanamycin (17-AAG) in vitro as well as in lungs from LPS-treated mice, in vivo. Furthermore, 17-AAG prevented LPS-induced pp60src activation. LPS-induced Hsp90 phosphorylation was also prevented by the pp60src inhibitor PP2. Additionally, Hsp90 phosphorylation was induced by infecting cells with a constitutively active pp60src adenovirus, whereas either a dominant-negative pp60src adenovirus or reduced expression of pp60src by a specific siRNA prevented the LPS-induced Y phosphorylation of Hsp90. Transfection of HLMVEC with the nonphosphorylatable Hsp90β Y300F mutant prevented LPS-induced Hsp90β tyrosine phosphorylation but not pp60src activation. Furthermore, the Hsp90β Y300F mutant showed a reduced ability to bind the Hsp90 client proteins eNOS and pp60src and HLMVEC transfected with the mutant exhibited reduced LPS-induced barrier dysfunction. We conclude that inflammatory stimuli cause posttranslational modifications of Hsp90 that are Hsp90-inhibitor sensitive and may be important to the proinflammatory actions of Hsp90.
Keywords: endothelial cells, Hsp90, human, LPS, posttranslational modifications
molecular chaperones aid in the precise polypeptide folding as well as the protection of protein aggregation during cell metabolism. Heat shock protein 90 (Hsp90) is a highly conserved cellular chaperone and one of the most abundant proteins in eukaryotic cells (20). It represents 1–2% of total cellular proteins and participates in the stabilization and activation of more than 200 “client” proteins (48). Hsp90 forms multichaperone complexes with a variety of cochaperones and proinflammatory client proteins. These complexes cycle between an open and a closed conformation of the dimeric Hsp90. Hsp90 inhibitors, such as 17-allyl-amino-demethoxy-geldanamycin (17-AAG), lock the complex in the open state, leading to client protein deactivation, destabilization, and proteosomal degradation (33).
Hsp90 is subjected to posttranslational modifications that modulate its activity, such as acetylation, S-nitrosylation, and phosphorylation (41). Phosphorylation occurs at both serine/threonine and tyrosine sites with different functional consequences. Phosphorylation of Hsp90 in response to serine/threonine phosphatase inhibition leads to reduced association with pp60v-src in NIH3T3 cells (26). Protein phosphatase 5 dephosphorylates Hsp90 in vitro and inhibits Hsp90 function in yeast cells (49). The tyrosine kinase WEE1, a client protein of Hsp90, phosphorylates a tyrosine residue of Hsp90 and promotes its ability to chaperone client protein kinases, such as pp60c-src, RAF1, CDK4, and WEE itself (29). In endothelial cells, phosphorylation of the Tyr 300 residue of Hsp90 by pp60c-src is crucial for VEGF2-induced angiogenesis (14). However, inflammation-induced activation of Hsp90 remains unexplored.
Lipopolysaccharide (LPS) is the major component of the outer wall of gram-negative bacteria. It activates macrophages, neutrophils, dendritic, and other cells that induce inflammation and produce reactive oxygen and nitrogen species and endothelial damage (7). Exposure to LPS leads to endothelial barrier dysfunction, to increased endothelial permeability, and to the development of acute lung injury and acute respiratory distress syndrome (2). Recent studies have revealed a potent anti-inflammatory action of Hsp90 inhibitors in vascular tissues, but it is still not known whether these actions involve modulation of Hsp90 activation (18, 20). In this study we have investigated the LPS-induced activation of Hsp90, its mechanism of action, and its functional role.
MATERIALS AND METHODS
Reagents.
17-AAG was obtained from the National Cancer Institute (Bethesda, MD). Anti-Hsp90 (cat. no. 624088) and anti-eNOS (cat. no. 610297) mouse monoclonal antibodies were purchased from BD Biosciences Transduction Laboratories (San Jose, CA). Phospho-tyrosine (PY99) (cat. no. sc-7020) mouse monoclonal antibody, pp60c-src (cat. no. sc-5266) mouse monoclonal antibody, p Yes (cat. no. sc-130182) and p Fyn (cat. no. sc-16848) rabbit polyclonal antibodies, protein A/G Plus agarose beads, siRNA against pp60c-src, and control siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Hsp90α (cat. no. ab59459) and Hsp90β (cat. no. ab82395) mouse monoclonal antibodies were purchased from Abcam (Cambridge, MA). pY416 pp60c-src (cat. no. 2101) monoclonal rabbit antibody, Lyn (cat. no. 4576) mouse monoclonal antibody, and Yes (cat. no. 3201), Fyn (cat. no. 4023), and pLyn (cat. no. 2731) rabbit polyclonal antibodies were obtained from Cell Signaling (Danvers, MA). β-Actin (cat. no. A5441), c-myc (cat. no. M4439) mouse monoclonal antibodies, and the CelyticM Lysis Reagent were purchased from Sigma-Aldrich (St. Louis, MO). Secondary anti-mouse (cat. no. NA931V) and rabbit (cat. no. NA934V) antibodies, as well as the Pierce BCA protein assay were obtained from Fisher Scientific (Pittsburgh, PA). Lipofectamine and Opti MEM 1 reduced serum were purchased from Invitrogen (Carlsbad, CA). Effectene Transfection Reagent was purchased from Qiagen (Valencia, CA). Nitrocellulose membranes were bought from Bio-Rad (Hercules, CA). Myc-WT-Hsp90β and Myc-Y300F-Hsp90β plasmids were generously donated by Dr. Jean-Philippe Gratton (McGill University, Montreal, QC, Canada) and their construction has been previously reported (14). Escherichia coli LPS (cat. no. L4130) was purchased from Sigma and exhibited an activity of 6,000,000 endotoxin units (EU)/mg.
Animals.
Male C57BL/6 mice, 7–8 wk old, from Harlan Laboratories were used in all experiments. Mice were maintained under pathogen-free conditions in a 12:12-h light-dark cycle. All animal care and experimental procedures were approved by the Animal Care Committee of the Georgia Health Sciences University and were in line with the principles of humane animal care adopted by the American Physiological Society.
Cell culture.
In-house harvested bovine pulmonary arterial endothelial cells (BPAEC) were subcultured from primary cultures and used at an early passage (<6). Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2-95% air in medium M199 supplemented with 10% fetal bovine serum (FBS), 5% iron-supplemented calf serum, 20 mg/ml l-glutamine, 1× MEM amino acid and vitamin solutions, 0.6 mg/ml thymidine, 500 IU/ml penicillin, and 500 mg/ml streptomycin (7). In-house harvested human lung microvascular endothelial cells (HLMVEC) were maintained in M199 medium supplemented with 10% FBS and antibiotics/antimycotics, as described previously (5).
Measurement of endothelial barrier function.
Barrier function of endothelial cell monolayers grown on electrode arrays (8W10E+) was estimated by the electric cell-substrate impedance sensing (ECIS), as previously described (47) using an ECIS model 1600R from Applied BioPhysics. Experiments were conducted on wells that achieved >700 Ω of baseline, steady-state resistance.
Immunoprecipitation.
Confluent cell culture dishes were placed on ice and washed three times with ice-cold PBS. PBS was removed and ice-cold lysis buffer was added. Adherent cells were scraped and the cell suspension was transferred to a cold microcentrifuge tube. Constant agitation was maintained for 30 min at 4°C and lysates were centrifuged for 30 min at 4°C. The protein concentration in the supernatant was calculated by the BCA protein assay, and 2.5 μg of appropriate antibody was added to lysates containing 250 μg protein and incubated overnight under agitation. The next day, 50λ A/G agarose beads were added to the lysates and the mixture was incubated at 4°C under rotary agitation. The mixture was then washed four times with lysis buffer, and the last supernatant was removed and replaced with 1× sample buffer. Proteins were denatured and separated from the beads by 5 min boiling at 100°C. The lysates where then subjected to SDS-PAGE electrophoresis.
Protein isolation and Western blot analysis.
Proteins were isolated from cells or tissues by using CelyticM Lysis Reagent or RIPA buffer and concentration was determined by the BCA method according to manufacturer's instructions. Protein-matched samples (40 μg per lane) were separated by electrophoresis through 12% sodium dodecyl sulfate (SDS-PAGE) Tris·HCl gels. Wet transfer was used to transfer the proteins onto nitrocellulose membranes. The membranes were incubated for 1 h at room temperature in 5% nonfat dry milk in Tris-buffered saline-0.1% (vol/vol) Tween 20. The blots were then incubated at 4°C overnight with the appropriate antibody. The signal for the immunoreactive proteins was developed by using the appropriate peroxidase-conjugated secondary antibody and visualized by exposure to chemiluminescence substrate. β-Actin was used as a loading control.
Transfections.
siRNA against human pp60c-src was used to knock down the expression of pp60c-src. An RNA that does not lead to any specific degradation of any known cellular mRNA was used as control. Seventy percent confluent cells were used. For transfection, the siRNA was diluted in Opti-MEM I Reduced Serum Medium, and Lipofectamine 2000 was diluted in an equal volume of Opti-MEM I and incubated for 30 min at room temperature. The oligomer-Lipofectamine complexes were added to the cells, which were cultured in media free of antibiotics. The medium was changed 8 h after transfection. Cells were incubated at 37°C in an atmosphere of 5% CO2 and 95% air for 72 h after transfection and then assayed by Western blotting. Myc-WT-Hsp90β and Myc-Y300F-Hsp90β plasmids were transfected into the cells by using the Effectene Transfection Reagent according to manufacturer's instructions.
In vivo experiments.
Stock solutions of LPS were prepared in saline. Mice received either vehicle (10% DMSO in saline) or Hsp90 inhibitor dissolved in 10% DMSO intraperitoneally (10 μg/g body wt) 24 h before LPS (2 μg/g body wt dissolved in saline, intratracheally). Mice were euthanized 24 h after LPS by cervical dislocation and the lungs were flushed with 1 ml ice-cold PBS (5 mM EDTA), excised, dipped in saline, blotted dry, snap frozen in liquid nitrogen, crushed to powder in a prechilled mortar, and stored at −80°C.
MALDI-TOF mass spectrometry.
The peptide calibration standards were from Applied Biosystems (Carlsbad, CA). All spectra were taken on an ABSciex 5800 matrix-assisted laser desorption ionization-time-of-flight/time-of-flight (MALDI-TOF/TOF) mass spectrometer in positive reflector mode (10 kV) with a matrix of α-cyano-4-hydroxycinnamic acid (CHCA). At least 1,000 laser shots were averaged to obtain each spectrum. Masses were calibrated to known peptide standards, and 5-μl aliquots of Hsp90 tryptic digest (from the band in the Hsp90α immunoprecipitant) were cleaned on a C18 ZipTip column (Millipore, Bedford, MA) as per manufacturer's instructions. Bound peptides were desalted with two 5-μl washes of 0.1% trifluoroacetic acid (TFA) and then eluted with 2.5 μl aqueous, acidic acetonitrile (75% CH3CN, 0.1% TFA). The eluant was mixed with 2.5 μl freshly prepared CHCA stock solution (20 mg/ml CHCA in aqueous acetonitrile, as above); 1.5 μl portions of this mixture were spotted onto a MALDI sample plate for air drying; and 1.5 μl crude peptides were additionally mixed with 1.5 μl CHCA and spotted. MS and MS/MS spectra were analyzed in Protein Pilot 3.0, Mascot Distiller, and PEAKS software packages.
Adenovirus construction.
Wild-type (c-SrcWT), chicken constitutively active (c-SrcY527F), and mouse dominant-negative (c-SrcK296R/Y528F) pp60Src coding sequences in pUSEamp vector were purchased from Upstate Biotechnology. An adenoviral vector expressing wild-type or mutant pp60Src was constructed with the pAd/pENTR System (Invitrogen). Briefly, a pAd/cytomegalovirus plasmid containing wild-type or constitutively active or dominant-negative pp60Src cDNA was created and transfected into 293A cells. A selected clone was propagated and viral lysate was collected and used for further experiments. Infections on endothelial cells were performed for 48 h with virus diluted in normal DMEM to the desired multiplicity of infection. Verification of expression was performed by Western blot analysis by using 20 μg of protein extracts probed with pp60src antibody (1:1,000) and normalized using β-actin antibody (1:1,000).
Densitometry/statistical analysis.
Image J software (National Institute of Health) was used to perform densitometry of immunoblots. All data are expressed as means ± SE. Student's t-test was performed to determine the significance in the difference between two mean values. A value of P < 0.05 was considered significant. GraphPad Prism 4 (version 4.03, Graph Pad Software) was used for data analysis.
RESULTS
LPS increases tyrosine phosphorylation of Hsp90 in BPAEC.
BPAEC were exposed to 1 EU/ml LPS for 5 to 60 min. LPS induced tyrosine phosphorylation of Hsp90 in a time-dependent manner. The maximum increase in phosphorylation occurred 60 min after exposure to LPS (Fig. 1A).
Fig. 1.
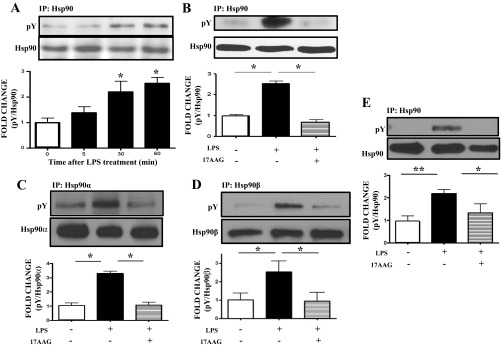
Modulation of pY levels of heat shock protein 90 (Hsp90) by LPS and 17-AAG. A: Western blot analysis of the expression levels of tyrosine-phosphorylated Hsp90 after 5-, 30-, or 60-min treatment with LPS in bovine pulmonary arterial endothelial cells (BPAEC). B: Western blot analysis of tyrosine-phosphorylated Hsp90 levels in BPAEC treated with LPS or vehicle and pretreated with PP2 or vehicle. C: Western blot analysis of tyrosine-phosphorylated Hsp90α levels in human lung microvascular endothelial cells (HLMVEC) treated with LPS or vehicle and pretreated with 17-AAG or vehicle. D: Western blot analysis of tyrosine-phosphorylated Hsp90β levels in HLMVEC treated with LPS or vehicle and pretreated with 17-AAG or vehicle. E: Western blot analysis of tyrosine-phosphorylated Hsp90 levels in lungs from mice treated with LPS or vehicle and pretreated with 17-AAG or vehicle. In all panels, Western blotting was performed on samples immunoprecipitated with antibodies against Hsp90 (A, B, E), Hsp90α (C), or Hsp90β (D), and each blot shown is representative of 3 separate experiments per group. Band intensity was analyzed by densitometry and normalized to Hsp90. *P < 0.05 vs. control, **P < 0.01 vs. control. Bars represent SE.
17-AAG suppresses LPS-mediated tyrosine phosphorylation of Hsp90 in BPAEC.
Confluent BPAEC were treated for 4 h with 1 EU/ml LPS, following 1 h pretreatment with either vehicle or 1 μg/ml 17-AAG. LPS-induced tyrosine phosphorylation of Hsp90 was completely suppressed by 17-AAG (Fig. 1B).
17-AAG suppresses LPS-mediated phosphotyrosine levels of Hsp90α and HSP90β in HLMVEC.
HLMVEC were treated for 4 h with 10 EU/ml LPS, following 1 h pretreatment with either vehicle or 1 μg/ml 17-AAG. LPS induced robust tyrosine phosphorylation of Hsp90α and Hsp90β, which was completely suppressed by 17-AAG (Fig. 1, C and D).
17-AAG suppresses LPS-mediated phosphotyrosine levels of Hsp90 in mice lungs.
Mice received either vehicle or Hsp90 inhibitor dissolved in 10% DMSO intraperitoneally 24 h before LPS. Animals were euthanized 12 h after LPS. 17-AAG suppressed the LPS-induced tyrosine phosphorylation of Hsp90, in vivo (Fig. 1E).
LPS increases activation of pp60src and the association between active pp60src and Hsp90 in BPAEC.
BPAEC were treated with either vehicle or 1 EU/ml LPS for 4 h. pY416 pp60src expression levels were increased in LPS-treated cells compared with controls (Fig. 2A). LPS also increased the association between pY416 pp60src and Hsp90, without affecting the expression levels of pp60src (Fig. 2B).
Fig. 2.

LPS activates pp60src and promotes its association with Hsp90 in BPAEC. A: Western blot analysis of the expression of active (pY416) pp60src in BPAEC after treatment with LPS or vehicle. Band density of pY416pp60src was analyzed by densitometry and normalized to pp60src. *P < 0.05 vs. control. The blot shown represents 1 of 3 independent experiments. Bars represent SE. B: Western blot analysis of the expression of pY416 (active) pp60src (left) or total pp60src (right) associated with Hsp90 in BPAEC after treatment with LPS or vehicle. Band density of pY416pp60src and pp60src was analyzed by densitometry and normalized to Hsp90. *P < 0.05 vs. control. Each blot shown represents 1 of 3 independent experiments. Bars represent SE.
The pp60src inhibitor PP2 blocks LPS-induced tyrosine phosphorylation of Hsp90 without affecting the expression of pp60src in BPAEC.
BPAEC were treated for 4 h with 1 EU/ml LPS following 2 h pretreatment with either vehicle or 10 μM PP2. PP2 suppressed the LPS-induced phosphotyrosine levels of Hsp90 (Fig. 3A) but did not affect the expression levels of pp60src (Fig. 3B).
Fig. 3.

The pp60src inhibitor PP2 blocks LPS-induced Y phosphorylation of Hsp90, whereas constitutively active ad-pp60src increases Hsp90 Y phosphorylation in BPAEC. A: Western blot analysis of pY Hsp90 levels in BPAEC treated with LPS or vehicle and pretreated with PP2 or vehicle. Band density of pY was analyzed by densitometry and normalized to Hsp90 expression. *P < 0.05 vs. control. Western blotting was performed on samples immunoprecipitated with antibodies against Hsp90. B: Western blot analysis of pp60src expression in BPAEC treated with LPS or vehicle and pretreated with PP2 or vehicle. Band density of pp60src was analyzed by densitometry and normalized to β-actin. C: Western blot analysis of the expression of pY in BPAEC transduced with adenoviruses containing GFP (ad-GFP), wild-type pp60src (ad-wt-pp60src), dominant-negative pp60src (ad-DN-pp60src), or constitutively active pp60src (ad-CA-pp60src). Band density of pY was analyzed by densitometry and normalized to Hsp90. *P < 0.05 vs. ad-GFP. Western blotting was performed on samples immunoprecipitated with antibodies against Hsp90 (top). Western blot analysis of the expression of pp60src and Hsp90 in BPAEC transduced with ad-GFP, ad-wt-pp60src, ad-DN-pp60src and ad-CA-pp60src. Band density of pp60src was analyzed by densitometry and normalized to Hsp90. *P < 0.05 vs. ad-GFP (right). In all cases, each blot shown represents 1 of 3 independent experiments. Bars represent SE.
Constitutively active ad-pp60src increases Hsp90 phosphorylation in BPAEC.
BPAEC were exposed for 48 h to GFP adenovirus, wild-type pp60src adenovirus, constitutively active pp60src adenovirus, or dominant-negative pp60src adenovirus. Cells transduced with the dominant-negative pp60src adenovirus did not exhibit any tyrosine-phosphorylated Hsp90. The wild-type pp60src-transduced cells showed high levels of tyrosine-phosphorylated Hsp90, and those transduced with constitutively active pp60src adenovirus showed the highest phosphorylation compared with the GFP-transduced cells (Fig. 3C).
Effects of the SFK inhibitor PP2 on LPS-induced SFK activation in HLMVEC.
HLMVEC were treated for 4 h with either vehicle or 1 EU/ml LPS following 2 h pretreatment with either vehicle or 10 μM PP2. LPS induced significant activation of pp60src, Lyn, Yes, but not Fyn. PP2 significantly suppressed the phosphorylation of Lyn (Fig. 4B), Yes (Fig. 4C) and Fyn (Fig. 4D) tyrosine kinases and exerted its greater inhibitory effect on pp60src activation (Fig. 4A).
Fig. 4.

The effect of the PP2 inhibitor is mainly exerted by the suppression of the pp60src-non-receptor tyrosine kinase. A: Western blot analysis of pY416 pp60src levels in HLMVEC treated with LPS or vehicle and pretreated with PP2 or vehicle. The band densities were analyzed by densitometry and normalized to pp60src expression. B: Western blot analysis of pLyn levels in HLMVEC treated with LPS or vehicle and pretreated with PP2 or vehicle. The band densities were analyzed by densitometry and normalized to Lyn expression. C: Western blot analysis of pYes levels in HLMVEC treated with LPS or vehicle and pretreated with PP2 or vehicle. The band densities were analyzed by densitometry and normalized to Yes expression. D: Western blot analysis of pFyn levels in HLMVEC treated with LPS or vehicle and pretreated with PP2 or vehicle. The band densities were analyzed by densitometry and normalized to Fyn expression. *P < 0.05, ***P < 0.001 vs. vehicle, #P < 0.01, ##P < 0.001 vs. LPS. Each blot shown represents 1 of 5 independent experiments. Bars represent SE.
Dominant negative pp60src adenovirus blocks the LPS-induced tyrosine phosphorylation of Hsp90 in HLMVEC.
HLMVEC were transduced with dominant-negative pp60src or GFP adenovirus. Cells infected with ad DN-pp60src exhibited dramatically lowered pp60src activity than those infected with adGFP (Fig. 5A, right). LPS (10 EU/ml)-treated and GFP-transduced cells showed elevated levels of Hsp90 phosphorylation compared with untreated cells. In contrast, LPS-treated, DN-pp60src transduced cells did not exhibit an increase in phosphorylated Hsp90 compared with untreated cells (Fig. 5A).
Fig. 5.
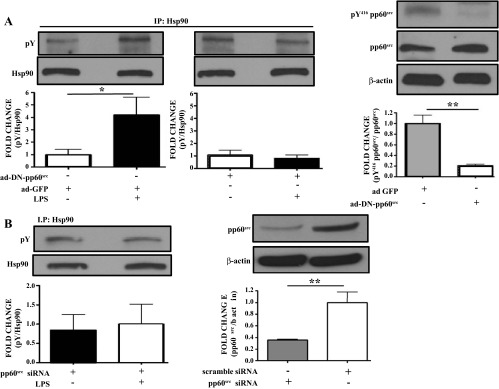
pp60src inhibition suppresses the LPS-induced Y phosphorylation of Hsp90. A: Western blot analysis of pY Hsp90 levels in HLMVEC transduced with ad-DN-pp60src or ad-GFP and exposed to LPS or vehicle. Western blotting was performed on samples immunoprecipitated with antibodies against Hsp90. Band density of pY and Hsp90 was analyzed by densitometry and normalized to Hsp90. *P < 0.05 vs. ad-GFP (left). Western blot analysis of pY416 pp60src and pp60src expression in HLMVEC transduced with ad-DN-pp60src or ad-GFP. Band density of pY416 pp60src was analyzed by densitometry and normalized to pp60src expression. β-Actin was used as an additional loading control. **P < 0.01 vs. ad-GFP (right). In all cases, each blot shown represents 1 of 3 independent experiments. Bars represent SE. B: Western blot analysis of pY Hsp90 levels in HLMVEC transduced with pp60src siRNA and exposed to LPS or vehicle. Band density of pY and Hsp90 was analyzed by densitometry and normalized to Hsp90 (left). Western blot analysis of pp60src expression in HLMVEC transfected with either pp60src siRNA or scrambled siRNA. Band density of pp60src was analyzed by densitometry and normalized to β-actin (right). **P < 0.01. In each case, the blot shown represents 1 of 3 independent experiments. Bars represent SE.
LPS does not induce phosphotyrosine levels of Hsp90 in pp60src silenced HLMVEC.
HLMVEC were transfected with siRNA especially designed for the knocking down of pp60src in human cells. Scrambled siRNA was used as control (Fig. 5B, right). Transfected cells were exposed for 4 h to either vehicle or 10 EU/ml LPS. LPS did not alter phosphotyrosine levels of Hsp90 in pp60src-silenced cells (Fig. 5B, left).
LPS phosphorylates the Y309 residue of Hsp90α in bovine pulmonary arterial endothelial cells.
BPAEC were treated for 4 h with either vehicle or 1 EU/ml LPS; following immunoprecipitation against Hsp90α antibody, the visible Hsp90α band from the Coomassie-stained gel was removed and subjected to mass spectrometry. LPS-treated cells exhibited phosphorylation at the Y309 site of the Hsp90a residue (Fig. 6).
Fig. 6.

LPS induces phosphorylation of Hsp90α at the Y309 residue. BPAEC were treated with 1 EU/ml LPS or vehicle for 4 h, immunoprecipitated with antibody against Hsp90α and subjected to mass spectrometry, as described in materials and methods. There is distinct phosphorylation of the Hsp90α Y309 residue (representative of 3 similar experiments).
Y300F mutated Hsp90β exhibits reduced binding to Hsp90 client proteins in HLMVEC.
HLMVEC were transfected with myc-Y300F-Hsp90β or myc-wt-Hsp90β plasmids. Cells transfected with the myc-Y300F-Hsp90β construct showed reduced association with the Hsp90 client proteins eNOS and pp60src compared with those transfected with the wild-type plasmid (Fig. 7A).
Fig. 7.
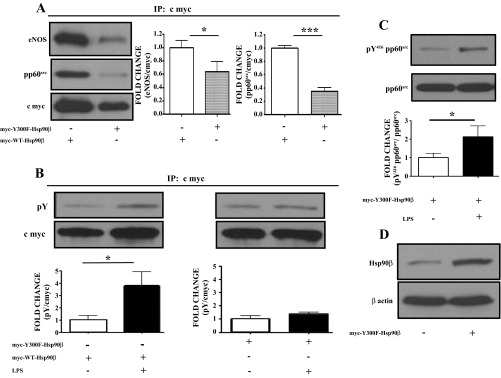
Y300F mutated Hsp90β exhibits reduced ability to bind the Hsp90 client proteins eNOS and pp60src and resists LPS-induced Y phosphorylation in HLMVEC. A: HLMVEC transfected with myc-Y300F-Hsp90β plasmid show a reduced ability to bind the Hsp90 client proteins eNOS and pp60src. Myc-WT-Hsp90β transfected cells were used as control. Band density of eNOS and pp60src was analyzed by densitometry and normalized to the c-myc tag. *P < 0.05, ***P < 0.001. Blots shown represent 1 of 3 independent experiments for eNOS or 4 independent experiments for pp60src. Bars represent SE. B: Western blot analysis of the expression levels of pY in HLMVEC transfected with myc-Y300F-Hsp90β or myc-WT-Hsp90β plasmids and exposed to LPS or vehicle. Band density of pY and c-myc was analyzed by densitometry and normalized to the c-myc tag. *P < 0.05. The blot shown represents 1 of 3 independent experiments. Bars represent SE. C: Western blot analysis of the expression levels of pY416pp60src in HLMVEC transfected with myc-Y300F-Hsp90β and exposed to LPS. Band density of pY416 pp60src was analyzed by densitometry and normalized to pp60src. The blot shown represents 1 of 6 independent experiments. Bars represent SE. D: Western blot analysis of the expression levels of Hsp90β in HLMVEC transfected with myc-Y300F-Hsp90β. The β actin band was used as a loading control.
Y300F mutated Hsp90β resists LPS-induced tyrosine phosphorylation in HLMVEC.
HLMVEC were transfected with myc-Y300F-Hsp90β or myc-wt-Hsp90β plasmids and were treated for 4 h with either vehicle or 10 EU/ml LPS. Cells transfected with the wild-type Hsp90β plasmid showed increased levels of Hsp90β phosphorylation compared with controls. In contrast, cells transfected with the myc-Y300F-Hsp90β construct resisted the LPS-induced tyrosine Hsp90β phosphorylation (Fig. 7B).
Transfection of HLMVEC with Y300F-mutated Hsp90β does not block LPS-induced pp60src phosphorylation.
HLMVEC were transfected with myc-Y300F-Hsp90β plasmid and were treated for 4 h with either vehicle or 10 EU/ml LPS. Myc-Y300F-Hsp90β-transfected cells exhibited significant LPS-induced pp60src phosphorylation (Fig. 7C). Figure 7D demonstrates higher expression levels of Hsp90β in transfected cells compared with untransfected cells.
Y300F mutated Hsp90β-transfected HLMVEC exhibit attenuated LPS-induced endothelial barrier dysfunction.
HLMVEC were transfected with myc-Y300F-Hsp90β or myc-wt-Hsp90β plasmids and 24 h later were seeded on gold electrode arrays. Twenty hours later, cells were exposed to PBS (vehicle) or LPS. PBS-treated cells maintained constant transendothelial resistance (TER) values (Fig. 8A), suggesting a confluent endothelial monolayer and minimal variability during the experiment. LPS (0.5 EU/ml) profoundly reduced TER in myc-wt-Hsp90β transfected cells. However, myc-Y300F-Hsp90β-transfected cells exhibited moderately reduced decrease in TER. Figure 8B demonstrates the increased expression levels of Hsp90β in the transfected cells, but also the high expression levels of constitutive Hsp90 in all HLMVEC.
Fig. 8.

Y300F mutated Hsp90β expression resists LPS-induced endothelial barrier dysfunction: PBS (vehicle) or LPS was added to the media of confluent transfected HLMVEC monolayers at 0 h. A gradual increase in endothelial permeability (reduced TER) was observed in the LPS-treated cells. The myc-Y300F-Hsp90β-transfected cells resisted to the LPS-induced endothelial barrier dysfunction: *,#P < 0.001 vs. corresponding untreated group of transfected cells; n = 4 per group. Error bars represent SE (A). Western blot analysis of the expression levels of Hsp90β and Hsp90 in HLMVEC transfected with myc-Y300F-Hsp90β and myc-wt-Hsp90β. The β-actin band was used as a loading control (B).
DISCUSSION
Hsp90 is a major regulator of important physiological processes conducted through “client” proteins, mostly enzymes and transcriptions factors, that it stabilizes and/or activates (48). Hsp90 inhibitors were initially developed to fight cancer, but ongoing research suggests that these compounds may also have a beneficial role in other diseases, since they exhibit strong anti-inflammatory properties (3, 7).
Hsp90 consists of COOH-, middle-, and NH2-terminal domains, the latter arranged in a two-layer α/β sandwich structure in which the helices form a pocket that is the binding site of adenine nucleotides and most Hsp90 inhibitors. The biological function of Hsp90 depends on its ability to hydrolyze and bind ATP (33). The middle segment of Hsp90 consists of an αβα domain at the NH2 terminus of the construct, connecting to a small αβα domain at the COOH terminus via a series of α-helices. This is the major site for client protein interaction. The COOH-terminal domain of Hsp90 is crucial for Hsp90 dimerization (33).
There are two major cytoplasmic isoforms of Hsp90: Hsp90α and Hsp90β. Hsp90α expression is inducible and lower compared with Hsp90β, which is constitutively expressed (12). Differences in the sequence between the α and β isoforms suggest that they may bind client proteins with different affinity (43). Other members of the Hsp90 family include Hsp90N, which is mainly associated with cellular transformation (17); GPR 94, found in the endoplasmic reticulum; and TRAP1, located in the mitochondria (11).
Hsp90 is regulated at several levels, including the ATPase cycle (16), association with conformation-specific cochaperones (36), and posttranslational modifications (35, 37), the latter including phosphorylation, acetylation, S-nitrosylation, oxidation, and ubiquitination (28). Hyperacetylation induced by HDAC inhibition can abrogate client protein, p23, and ATP binding while it enhances the binding of inhibitory drugs to the N domain (21). Nitric oxide-induced nitrosylation of the Hsp90 C domain inhibits chaperone and ATPase activity in endothelial cells (25, 31, 35). Changes in phosphorylation impact Hsp90 function (22). Phosphorylation at sites in the M and the C domains, specifically, and by different molecular mechanisms, can modulate conformational rearrangements during the ATPase cycle of Hsp90 (42). LPS induced tyrosine phosphorylation of Hsp90, in HLMVEC, in a time-dependent manner. Similar Hsp90 posttranslational modifications are induced when in complex with the P2X (7) receptor and by the cell cycle protein Swe1, a Hsp90 client protein that phosphorylates the conserved Tyr24 in yeast Hsp90. This phosphorylation is crucial for the association of Swe1 with Hsp90 and established the importance of Hsp90 in cell cycle regulation (29, 30). To our knowledge this is the first report of tyrosine phosphorylation of Hsp90 by a proinflammatory agent.
Hsp90 activation is involved in the induction of inflammation and Hsp90 inhibitors are able to counteract these effects (4, 9, 24). 17-Dimethylaminoethylamino-17-demethoxygeldanamycin reduces inflammation in lupus (39), blocks the NF-κB inflammatory cascade in immune stimulated macrophages (40) and substantially reduces necrosis-induced cytokine production and permeability increases in a model of endotoxin-induced uveitis (34). Moreover, the synthetic Hsp90 inhibitor EC144 appears to be efficacious in mouse models of endotoxin shock and arthritis (51). Other studies have reported a therapeutic benefit of Hsp90 inhibitors in experimental autoimmune encephalitis and arthritis (22, 24, 32).
Here we report for the first time that the Hsp90 inhibitor 17-AAG can block the LPS-induced tyrosine phosphorylation of Hsp90 in both human and bovine endothelial cells, suggesting that this is not a species-specific effect. Hsp90 inhibitors have been shown to block phosphorylation of major intracellular pathways. In particular, SNX-2122 has been reported to degrade both full-length HER2 and p95-HER2 and to downregulate HER2, AKT, and ERK signaling in breast cancer cells in vitro and in vivo (6). In breast cancer cells that express high levels of HER2, 17-AAG also causes rapid inhibition of Akt kinase activity (4). Tyrosine phosphorylation-dependent NF-κB activation is sensitive to pharmacological inhibition of Hsp90 by geldanamycin, most likely through c-Src. Geldanamycin inhibits tyrosine phosphorylation and degradation of IkBa in HeLa and Jurkat cells, resulting in NF-κB nuclear translocation and DNA binding, suggesting that Hsp90 plays a key role in NF-κB activation by oxidative stress (10).
The LPS-induced activation of pp60src in endothelial cells was first reported in 2008 by Chatterjee et al. (7) and was in line with other studies that introduced LPS-induced pp60Src activation in alveolar epithelial cells and macrophages (13, 23, 44). Src family tyrosine kinases (SFK) also play an important role in LPS-induced superoxide production and are responsible for the phosphorylation of the TLR4 in mouse RAW264 macrophages and in Chinese hamster ovary cells (8). In endothelial cells, pp60src is activated in response to a variety of stimuli, including H2O2, estrogen, and shear stress and leads to activation of eNOS (15). Furthermore, activated pp60src increases the nuclear translocation of c-Jun and p65 (46) via PI3K/Akt and MAPKs (19), regulates LPS-induced actin cytoskeleton rearrangement in macrophages (9), and controls NADPH oxidase activation and ROS production (45). Indeed, Severgnini et al. (38) have shown both in vivo and in vitro that inhibition of the Src kinases protects against LPS-induced acute lung injury. However, this is the first report of inhibition of the LPS-induced phosphorylation of Hsp90 by pp60src silencing or by antagonists.
The role of Hsp90 phosphorylation in inflammation remains largely unknown. In an effort to investigate the role of Hsp90 tyrosine phosphorylation in endothelial barrier function, we demonstrated overexpression of a nonphosphorylatable Hsp90 mutant to HLMVEC, increased resting barrier strength, and moderately but significantly attenuated LPS-induced hyperpermeability. The small magnitude of the protective effect is likely due to the small transfection efficacy of the mutant and the continuous presence of the phosphorylatable, constitutive Hsp90. This is likely the reason why the mutant was also unable to reduce LPS-induced pp60src activation. Indeed, when examined directly, the Y300F Hsp90β mutant exhibits profoundly reduced binding to two distinct Hsp90 client proteins. This agrees with previous studies demonstrating that the Y300 of Hsp90β is a major phosphorylation site by c-Src in VEGFR-2-stimulated COS-7 cells and that the mutant Y300F Hsp90 reduces the association with eNOS in response to VEGFR-2 (14). The hypothesis that tyrosine phosphorylation activates Hsp90 and improves its client binding efficacy is further supported by the observation that tyrosine-phosphorylated Hsp90 preferentially associates with the P2X7 receptor complex and that the levels of tyrosine phosphorylation are associated with the function of the receptor. Treatment of HEK293 cells with geldanamycin decreased the Hsp90 phosphotyrosine levels without a detectable changes in Hsp90 protein levels (1). Recently, Hsp90 tyrosine phosphorylation was shown to promote recruitment of AHA1, stimulate Hsp90 ATPase activity, and regulate the Hsp90 chaperone cycle (50). Phosphorylation of two serine residues by casein kinase 2 is also crucial for Hsp90 activity, whereas hyperphosphorylation of these sites decreases its activity (27). It has been proposed that a cycle of phosphorylation and dephosphorylation of Hsp90 is necessary for client binding and release (52).
Endothelial cells are both a target and a participant of inflammation. They contain a substantial amount of Hsp90, which is critically involved in the hyperpermeability caused by inflammation and, in the case of pulmonary microvascular endothelium, in the edema and subsequent lung dysfunction associated with ALI/ARDS. Our present data agree with aforementioned findings in other tissues and strongly suggest that endothelial Hsp90 phosphorylation mediates, at least in part, Hsp90 complex formation with certain client proteins that are involved in normal endothelial function as well as in mediating inflammatory responses. Furthermore, the present data indicate that in endothelial cells, since pp60src and Hsp90 phosphorylation/activation are abrogated by either Hsp90 or pp60src inhibition, they are tightly coordinated and constitute a powerful proinflammatory unit (Fig. 9).
Fig. 9.

Proposed model of Hsp90-pp60src interactions in LPS-activated endothelial cells.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants HL 06693, HL 093460 and HL 101902.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
N.B., V.H., C.D., R.R., C.M.S., S.K., A.J., G.T., D.J.F., and V.P. performed experiments; N.B., V.H., R.R., A.J., G.T., D.J.F., S.M.B., and J.D.C. analyzed data; N.B., R.R., D.J.F., S.M.B., and J.D.C. interpreted results of experiments; N.B. and V.H. prepared figures; N.B. drafted manuscript; D.J.F., S.M.B., V.P., and J.D.C. edited and revised manuscript; J.D.C. conception and design of research; J.D.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Bristol Myers Squibb and the National Cancer Institute for providing us with 17-AAG and Yevgeniy Kovalenkov, Medical College of Georgia, Vascular Biology Center, Georgia Regents University, Augusta, GA, for outstanding technical assistance.
REFERENCES
- 1.Adinolfi E, Kim M, Young MT, Di Virgilio F, Surprenant A. Tyrosine phosphorylation of HSP90 within the P2X7 receptor complex negatively regulates P2X7 receptors. J Biol Chem 278: 37344–37351, 2003 [DOI] [PubMed] [Google Scholar]
- 2.Antonov A, Snead C, Gorshkov B, Antonova GN, Verin AD, Catravas JD. Heat shock protein 90 inhibitors protect and restore pulmonary endothelial barrier function. Am J Respir Cell Mol Biol 39: 551–559, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barabutis N, Catravas JD. Anti-inflammatory activity of Hsp90 inhibitors in the human vasculature. Med Surg Urol 2: e104, 2013 [Google Scholar]
- 4.Basso AD, Solit DB, Munster PN, Rosen N. Ansamycin antibiotics inhibit Akt activation and cyclin D expression in breast cancer cells that overexpress HER2. Oncogene 21: 1159–1166, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Catravas JD, Snead C, Dimitropoulou C, Chang AS, Lucas R, Verin AD, Black SM. Harvesting, identification and barrier function of human lung microvascular endothelial cells. Vascul Pharmacol 52: 175–181, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chandarlapaty S, Scaltriti M, Angelini P, Ye Q, Guzman M, Hudis CA, Norton L, Solit DB, Arribas J, Baselga J, Rosen N. Inhibitors of HSP90 block p95-HER2 signaling in Trastuzumab-resistant tumors and suppress their growth. Oncogene 29: 325–334, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chatterjee A, Snead C, Yetik-Anacak G, Antonova G, Zeng J, Catravas JD. Heat shock protein 90 inhibitors attenuate LPS-induced endothelial hyperpermeability. Am J Physiol Lung Cell Mol Physiol 294: L755–L763, 2008 [DOI] [PubMed] [Google Scholar]
- 8.Check J, Byrd CL, Menio J, Rippe RA, Hines IN, Wheeler MD. Src kinase participates in LPS-induced activation of NADPH oxidase. Mol Immunol 47: 756–762, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen YJ, Hsieh MY, Chang MY, Chen HC, Jan MS, Maa MC, Leu TH. Eps8 protein facilitates phagocytosis by increasing TLR4-MyD88 protein interaction in lipopolysaccharide-stimulated macrophages. J Biol Chem 287: 18806–18819, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crevecoeur J, Merville MP, Piette J, Gloire G. Geldanamycin inhibits tyrosine phosphorylation-dependent NF-κB activation. Biochem Pharmacol 75: 2183–2191, 2008 [DOI] [PubMed] [Google Scholar]
- 11.Csermely P, Schnaider T, Soti C, Prohaszka Z, Nardai G. The 90-kDa molecular chaperone family: structure, function, and clinical applications. A comprehensive review. Pharmacol Ther 79: 129–168, 1998 [DOI] [PubMed] [Google Scholar]
- 12.Dezwaan DC, Freeman BC. HSP90: the Rosetta stone for cellular protein dynamics? Cell Cycle 7: 1006–1012, 2008 [DOI] [PubMed] [Google Scholar]
- 13.dos Santos CC, Han B, Andrade CF, Bai X, Uhlig S, Hubmayr R, Tsang M, Lodyga M, Keshavjee S, Slutsky AS, Liu M. DNA microarray analysis of gene expression in alveolar epithelial cells in response to TNFα, LPS, and cyclic stretch. Physiol Genomics 19: 331–342, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Duval M, Le Boeuf F, Huot J, Gratton JP. Src-mediated phosphorylation of Hsp90 in response to vascular endothelial growth factor (VEGF) is required for VEGF receptor-2 signaling to endothelial NO synthase. Mol Biol Cell 18: 4659–4668, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fulton D, Church JE, Ruan L, Li C, Sood SG, Kemp BE, Jennings IG, Venema RC. Src kinase activates endothelial nitric-oxide synthase by phosphorylating Tyr-83. J Biol Chem 280: 35943–35952, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Graf C, Stankiewicz M, Kramer G, Mayer MP. Spatially and kinetically resolved changes in the conformational dynamics of the Hsp90 chaperone machine. EMBO J 28: 602–613, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grammatikakis N, Vultur A, Ramana CV, Siganou A, Schweinfest CW, Watson DK, Raptis L. The role of Hsp90N, a new member of the Hsp90 family, in signal transduction and neoplastic transformation. J Biol Chem 277: 8312–8320, 2002 [DOI] [PubMed] [Google Scholar]
- 18.Grandel U, Grimminger F. Endothelial responses to bacterial toxins in sepsis. Crit Rev Immunol 23: 267–299, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Hsieh HL, Wang HH, Wu CY, Tung WH, Yang CM. Lipoteichoic acid induces matrix metalloproteinase-9 expression via transactivation of PDGF receptors and NF-κB activation in rat brain astrocytes. Neurotox Res 17: 344–359, 2010 [DOI] [PubMed] [Google Scholar]
- 20.Kaplan KB, Li R. A prescription for ‘stress’—the role of Hsp90 in genome stability and cellular adaptation. Trends Cell Biol 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell 18: 601–607, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Lees-Miller SP, Anderson CW. Two human 90-kDa heat shock proteins are phosphorylated in vivo at conserved serines that are phosphorylated in vitro by casein kinase II. J Biol Chem 264: 2431–2437, 1989 [PubMed] [Google Scholar]
- 23.Leu TH, Charoenfuprasert S, Yen CK, Fan CW, Maa MC. Lipopolysaccharide-induced c-Src expression plays a role in nitric oxide and TNFα secretion in macrophages. Mol Immunol 43: 308–316, 2006 [DOI] [PubMed] [Google Scholar]
- 24.Lucas R, Verin AD, Black SM, Catravas JD. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol 77: 1763–1772, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez-Ruiz A, Villanueva L, Gonzalez de Orduna C, Lopez-Ferrer D, Higueras MA, Tarin C, Rodriguez-Crespo I, Vazquez J, Lamas S. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc Natl Acad Sci USA 102: 8525–8530, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mimnaugh EG, Worland PJ, Whitesell L, Neckers LM. Possible role for serine/threonine phosphorylation in the regulation of the heteroprotein complex between the hsp90 stress protein and the pp60v-src tyrosine kinase. J Biol Chem 270: 28654–28659, 1995 [DOI] [PubMed] [Google Scholar]
- 27.Miyata Y. Protein kinase CK2 in health and disease: CK2: the kinase controlling the Hsp90 chaperone machinery. Cell Mol Life Sci 66: 1840–1849, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mollapour M, Neckers L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim Biophys Acta 1823: 648–655, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mollapour M, Tsutsumi S, Neckers L. Hsp90 phosphorylation, Wee1 and the cell cycle. Cell Cycle 9: 2310–2316, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mollapour M, Tsutsumi S, Truman AW, Xu W, Vaughan CK, Beebe K, Konstantinova A, Vourganti S, Panaretou B, Piper PW, Trepel JB, Prodromou C, Pearl LH, Neckers L. Threonine 22 phosphorylation attenuates Hsp90 interaction with cochaperones and affects its chaperone activity. Mol Cell 41: 672–681, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morra G, Verkhivker G, Colombo G. Modeling signal propagation mechanisms and ligand-based conformational dynamics of the Hsp90 molecular chaperone full-length dimer. PLoS Comput Biol 5: e1000323, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murata M, Miura Y, Hashiramoto A, Kitamura H, Kawasaki H, Shiozawa K, Yoshiya S, Baba H, Chihara K, Shiozawa S. Heat shock protein 90 is required for increased DNA binding activity of activator protein-1, a heterodimer of Fos/JunD, in rheumatoid synovial cells under inflammatory stimuli. Int J Mol Med 15: 649–653, 2005 [PubMed] [Google Scholar]
- 33.Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem 75: 271–294, 2006 [DOI] [PubMed] [Google Scholar]
- 34.Qin S, Ni M, Wang X, Maurier-Mahe F, Shurland DL, Rodrigues GA. Inhibition of RPE cell sterile inflammatory responses and endotoxin-induced uveitis by a cell-impermeable HSP90 inhibitor. Exp Eye Res 93: 889–897, 2011 [DOI] [PubMed] [Google Scholar]
- 35.Retzlaff M, Stahl M, Eberl HC, Lagleder S, Beck J, Kessler H, Buchner J. Hsp90 is regulated by a switch point in the C-terminal domain. EMBO Rep 10: 1147–1153, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samant RS, Clarke PA, Workman P. The expanding proteome of the molecular chaperone HSP90. Cell Cycle 11: 1301–1308, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scroggins BT, Robzyk K, Wang D, Marcu MG, Tsutsumi S, Beebe K, Cotter RJ, Felts S, Toft D, Karnitz L, Rosen N, Neckers L. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol Cell 25: 151–159, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Severgnini M, Takahashi S, Tu P, Perides G, Homer RJ, Jhung JW, Bhavsar D, Cochran BH, Simon AR. Inhibition of the Src and Jak kinases protects against lipopolysaccharide-induced acute lung injury. Am J Respir Crit Care Med 171: 858–867, 2005 [DOI] [PubMed] [Google Scholar]
- 39.Shimp SK, 3rd, Chafin CB, Regna NL, Hammond SE, Read MA, Caudell DL, Rylander M, Reilly CM. Heat shock protein 90 inhibition by 17-DMAG lessens disease in the MRL/lpr mouse model of systemic lupus erythematosus. Cell Mol Immunol 9: 255–266, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shimp SK, 3rd, Parson CD, Regna NL, Thomas AN, Chafin CB, Reilly CM, Rylander MN. HSP90 inhibition by 17-DMAG reduces inflammation in J774 macrophages through suppression of Akt and nuclear factor-κB pathways. Inflamm Res 61: 521–533, 2012 [DOI] [PubMed] [Google Scholar]
- 41.Soroka J, Buchner J. Mechanistic aspects of the Hsp90 phosphoregulation. Cell Cycle 11: 1870–1871, 2012 [DOI] [PubMed] [Google Scholar]
- 42.Soroka J, Wandinger SK, Mausbacher N, Schreiber T, Richter K, Daub H, Buchner J. Conformational switching of the molecular chaperone Hsp90 via regulated phosphorylation. Mol Cell 45: 517–528, 2012 [DOI] [PubMed] [Google Scholar]
- 43.Sreedhar AS, Kalmar E, Csermely P, Shen YF. Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett 562: 11–15, 2004 [DOI] [PubMed] [Google Scholar]
- 44.Su SC, Hua KF, Lee H, Chao LK, Tan SK, Yang SF, Hsu HY. LTA and LPS mediated activation of protein kinases in the regulation of inflammatory cytokines expression in macrophages. Clin Chim Acta 374: 106–115, 2006 [DOI] [PubMed] [Google Scholar]
- 45.Sumimoto H, Hata K, Mizuki K, Ito T, Kage Y, Sakaki Y, Fukumaki Y, Nakamura M, Takeshige K. Assembly and activation of the phagocyte NADPH oxidase. Specific interaction of the N-terminal Src homology 3 domain of p47phox with p22phox is required for activation of the NADPH oxidase. J Biol Chem 271: 22152–22158, 1996 [DOI] [PubMed] [Google Scholar]
- 46.Tang CH, Hsu CJ, Yang WH, Fong YC. Lipoteichoic acid enhances IL-6 production in human synovial fibroblasts via TLR2 receptor, PKCδ and c-Src dependent pathways. Biochem Pharmacol 79: 1648–1657, 2010 [DOI] [PubMed] [Google Scholar]
- 47.Tiruppathi C, Malik AB, Del Vecchio PJ, Keese CR, Giaever I. Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. Proc Natl Acad Sci USA 89: 7919–7923, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 10: 537–549, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wandinger SK, Suhre MH, Wegele H, Buchner J. The phosphatase Ppt1 is a dedicated regulator of the molecular chaperone Hsp90. EMBO J 25: 367–376, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu W, Mollapour M, Prodromou C, Wang S, Scroggins BT, Palchick Z, Beebe K, Siderius M, Lee MJ, Couvillon A, Trepel JB, Miyata Y, Matts R, Neckers L. Dynamic tyrosine phosphorylation modulates cycling of the HSP90-P50 (CDC37)-AHA1 chaperone machine. Mol Cell 47: 434–443, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yun TJ, Harning EK, Giza K, Rabah D, Li P, Arndt JW, Luchetti D, Biamonte MA, Shi J, Lundgren K, Manning A, Kehry MR. EC144, a synthetic inhibitor of heat shock protein 90, blocks innate and adaptive immune responses in models of inflammation and autoimmunity. J Immunol 186: 563–575, 2011 [DOI] [PubMed] [Google Scholar]
- 52.Zhao YG, Gilmore R, Leone G, Coffey MC, Weber B, Lee PW. Hsp90 phosphorylation is linked to its chaperoning function. Assembly of the reovirus cell attachment protein. J Biol Chem 276: 32822–32827, 2001 [DOI] [PubMed] [Google Scholar]