

Abstract
Obesity is a risk factor for stroke, but the early effects of high-fat diet (HFD) on neurovascular function and ischemic stroke outcomes remain unclear. The goal of this study was to test the hypotheses that HFD beginning early in life 1) impairs neurovascular coupling, 2) causes cerebrovascular dysfunction, and 3) worsens short-term outcomes after cerebral ischemia. Functional hyperemia and parenchymal arteriole (PA) reactivity were measured in rats after 8 wk of HFD. The effect of HFD on basilar artery function after middle cerebral artery occlusion (MCAO) and associated O-GlcNAcylation were assessed. Neuronal cell death, infarct size, hemorrhagic transformation (HT) frequency/severity, and neurological deficit were evaluated after global ischemia and transient MCAO. HFD caused a 10% increase in body weight and doubled adiposity without a change in lipid profile, blood glucose, and blood pressure. Functional hyperemia and PA relaxation were decreased with HFD. Basilar arteries from stroked HFD rats were more sensitive to contractile factors, and acetylcholine-mediated relaxation was impaired. Vascular O-GlcNAcylated protein content was increased with HFD. This group also showed greater mortality rate, infarct volume, HT occurrence rate, and HT severity and poor functional outcome compared with the control diet group. These results indicate that HFD negatively affects neurovascular coupling and cerebrovascular function even in the absence of dyslipidemia. These early cerebrovascular changes may be the cause of greater cerebral injury and poor outcomes of stroke in these animals.
Keywords: cerebral ischemia, high-fat diet, hemorrhagic transformation, neurovascular coupling, vascular dysfunction
obesity is an independent risk factor for acute ischemic stroke (AIS) (19, 36). An alarming recent report showed that the prevalence of AIS dramatically increased in children and young adults, which positively correlated with increases in risk factors including obesity, lipid disorders, and diabetes (13). Clinical studies also suggest that obesity is an independent predictor of unfavorable functional outcome and mortality in AIS patients treated with tissue plasminogen activator (tPA), the only therapeutic option these patients have (39, 40). Given that stroke is the leading cause of disability and that the obesity epidemic is on the rise these clinical and social problems are expected to get worse, and therefore early interventions are necessary. While experimental studies in genetic or diet-induced obesity models have shown increased cerebral infarct size and poor outcomes of stroke (7, 25, 32, 33, 41), the early impact of a high-fat diet (HFD) before the development of obesity on AIS injury and functional outcomes is not known.
It is known that the brain relies heavily on constant blood flow for proper function. Two important mechanisms that contribute to the regulation of cerebral blood perfusion are autoregulatory behavior of cerebral vessels and functional hyperemia upon increased neuronal activity (11, 16, 20). HFD can negatively affect vascular function, as demonstrated by increased myogenic tone and endothelial dysfunction in diet-induced as well as genetic models of obesity (7, 8, 24, 32, 33). The effect of a HFD on neurovascular coupling and cerebrovascular reactivity after an ischemic insult especially in the absence of metabolic abnormalities is unknown. To address this key deficit in our knowledge, the present study tested the hypotheses that HFD 1) impairs neurovascular coupling, 2) causes cerebrovascular dysfunction, and 3) worsens outcomes after cerebral ischemia, even in the absence of obesity.
METHODS
Animals.
This study was conducted in accordance with the National Institutes of Health guidelines for the care and use of animals in research and was approved by the Division of Laboratory Animal Services at the Georgia Health Sciences University. Male Wistar rats (Harlan Laboratories, Indianapolis, IN; 4–5 wk old, n = 64) were fed either an isocaloric control diet (CD, 10% fat) or a HFD (45% fat; Research Diets, New Brunswick, NJ) for 8 wk ad libitum. Blood pressure was measured by tail cuff (Kent Scientific, Torrington, CT), and blood glucose levels were measured with a glucometer (FreeStyle, Abbott Diabetes Care, Alameda, CA).
Metabolic parameters.
At death, blood was collected and processed for plasma analyses. Adipose tissue from the subcutaneous, peritoneal, and epididymal depots was collected and weighed separately. Total adiposity (all depots combined) was normalized to body weight and expressed as percent body weight. Plasma insulin (ALPCO Diagnostics, Salem, NH), triglycerides, and cholesterol (Wako USA, Richmond, VA) were measured.
Measurement of functional hyperemia.
Functional hyperemia was assessed 2 days prior to ischemia injury by measuring the cerebral blood flow (CBF) change in the somatosensory cortex upon whisker stimulation (21, 22). Animals were anesthetized with ketamine-xylazine (100 and 10 mg/kg) injection, and trimmed contralateral whiskers were gently stroked at a frequency of 2.5 Hz with a cotton tip attached to a vortex. The PIM3 laser Doppler scanning system (LDS, Perimed, Ardmore, PA) was programmed to scan an area covering somatosensory cortex, which is supplied by the middle cerebral artery (MCA), without tissue contact. CBF changes were expressed as percent increase relative to resting levels.
Brain slice preparation.
Parenchymal arteriole (PA) function was assessed with a well-established brain slice preparation (4, 5, 15). After death, the brain was removed and 300-μm-thick coronal slices were cut in ice-cold artificial cerebrospinal fluid (aCSF) containing (mM) 3 KCl, 120 NaCl, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 10 glucose, and 0.4 l-ascorbic acid, equilibrated with 95% O2-5% CO2 (3). Ascorbic acid was added to reduce cell swelling associated with oxidative stress. An aCSF with identical composition was used for bath perfusion in all experiments, except for those assessing the effects of high external K+ concentration ([K+]), in which control aCSF contained 4.2 mM KCl and KCl replaced NaCl to increase [K+] to 10 mM. Osmolality of aCSF was ∼290 mosmol/kgH2O. After the slicing procedure, slices were kept at room temperature in aCSF equilibrated with 95% O2-5% CO2 (pH ∼7.45) until use.
Video microscopy.
Diameter changes in cortical arterioles (<30-μm internal diameter) were recorded with an upright Zeiss Axioscope 2FS microscope (Carl Zeiss USA, Thornwood, NY) equipped with infrared-differential interference contrast (IR-DIC) optics, a water-immersion objective, and an EMCCD camera (iXon+885, Andor Tech, South Windsor, CT). Images were acquired at 1 frame/s and visualized and stored with IQ software (Andor Tech). The slices were perfused with aCSF (35 ± 2°C) gassed with 95% O2-5% CO2 and were allowed to equilibrate for ≥10 min prior to the beginning of recording. Only one arteriole per slice was recorded. Slices were perfused with the thromboxane A2 receptor agonist U-46619 to induce vasoconstriction, and test solutions were applied in the constant presence of U-46619 after a stable preconstriction was attained. Vessels that did not respond to U-46619 were not included in the analysis. Data from arteriolar diameter (IR-DIC) experiments were analyzed with custom software created by Dr. Adrian D. Bonev (Univ. of Vermont). Changes in the internal (luminal) diameter of arterioles were determined from averaged measurements taken from multiple points across the arteriolar lumen. Baseline diameter (represented as 100%) was determined during the first ∼10 min of sampling, before any experimental stimulation. All arteriolar diameter values are expressed as percentage relative to baseline. Vascular tone is expressed as “degree of constriction” relative to baseline.
Models of ischemia.
Focal cerebral ischemia (FCI) was induced by transient MCA occlusion (MCAO) as previously described (10). Briefly, all animals were anesthetized with 2% isoflurane via inhalation. The right MCA was occluded for 3 h with a 19- to 21-mm 3-0 surgical nylon filament, which was introduced from the external carotid artery lumen into the internal carotid artery to block the origin of the MCA. The rectal temperature was maintained at 37°C with a heating pad (Fine Science Tools, Foster City, CA). The cerebral perfusion was monitored with LDS to confirm successful occlusion or reperfusion. In a subset of animals, global cerebral ischemia (GCI) was induced (10-min occlusion, 7-day reperfusion) as an alternative method of ischemia. For GCI, all animals (except sham control animals) underwent four-vessel occlusion performed as described previously (43). Briefly, 24 h after electrocautery of the vertebral arteries, the common carotid arteries (CCAs) were occluded with aneurysm clips to induce 10-min forebrain ischemia. Animals that lost their righting reflex within 30 s and whose pupils were dilated and unresponsive to light during occlusion were selected for the experiments. The clips were then removed, and the blood flow through the CCAs was confirmed before the wound was sutured. The animals of the sham group underwent identical procedures except that the CCAs were not occluded. Rectal temperature was maintained at 36.5–37.5°C throughout the experiment with a thermal blanket.
Isolated vessel studies.
At 24 h after FCI, 2-mm basilar artery segments were isolated and mounted for myograph for isometric tension recordings as described previously (27). Concentration-response curves to serotonin [5-hydroxytryptamine (5-HT), 1 nM to 100 μM], endothelin-1 (ET-1, 0.01 nM to 0.1 μM), and the stable analog of the endoperoxide prostaglandin H2 (U-46619, 0.1 nM to 10 μM) were performed to evaluate vascular contractility. Endothelium-dependent relaxation to acetylcholine (ACh, 1 nM to 1 μM) was assessed after vessels were constricted to 60% of the baseline tension with phenylephrine (PE). Sensitivity (EC50) and maximum response (Rmax) values were calculated from the respective concentration-response equations (27).
Evaluation of O-GlcNAcylation.
O-GlcNAcylation-modified protein levels in the basilar arteries were determined by immunoblotting as previously described (28) with anti-O-GlcNAc antibody CTD 110.6 (1:2,000; Pierce Biotechnology, Rockford, IL). All membranes were stripped and reblotted with anti-actin antibody to ensure equal protein loading.
Infarct, edema, and hemorrhagic transformation analysis.
Brains from animals that died overnight after MCAO before euthanasia at 24 h were not processed for evaluation of ischemic injury but were included in the mortality data. The infarct volume was measured after 2,3,5-triphenyltetrazolium chloride (TTC) staining as previously described (10). Edema is reported as percent increase in ischemic hemisphere size versus the contralateral hemisphere. After staining, the hemispheres were separated and deep frozen for tissue hemoglobin (Hb) quantification with a QuantiChrom kit (BioAssay Systems, Hayward, CA) (35). Hemorrhagic transformation (HT) occurrence rate (presence of macroscopic bleeding) and severity (excess Hb, μg/mg protein in the ischemic hemisphere) are reported.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end staining.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end (TUNEL) staining was performed on the free-floating coronal sections of GCI groups at 7 days after reperfusion with the In Situ Cell Death Detection Kit (Roche Diagnostics, Indianapolis, IN) as described previously (43). Samples were analyzed with a LSM510 Meta confocal microscope. For quantitative analyses, the number of TUNEL-positive cells per 250-μm length of medial CA1 pyramidal cell layer was counted bilaterally in four or five sections per animal to provide a single value for each animal. A mean ± SE was calculated from the data.
Behavioral measurements.
Neurobehavioral tests were assessed and scored in a blinded fashion by video recording before MCAO surgery and before death in each animal. The items tested in Bederson's score included 1) spontaneous ipsilateral circling, graded from 2 to 0; 2) hindlimb retraction and 3) forelimb flexion, graded from 1 to 0, respectively; and 4) resistance to push, graded from 1 to 0. Beam walking ability tested the stability of the animal traversing a 2.4-cm-wide, 80-cm-long beam and was graded on a 7-point scale. A composite score was given by combining all the above tests, with a greater score representing better neurological outcome. Grip strength was measured with a standard grip strength meter (Columbus Instrument, Columbus, OH). The rat was gently held with its forepaws grasping the pull-bar and then pulled back consistently. The digital recording obtained from three trials was averaged.
Statistics.
Data are expressed as means ± SE or as scatterplots with median where appropriate. Contractile responses were calculated as a percentage of KCl (120 mM)-induced contraction. Concentration-response curves were fitted with a nonlinear interactive fitting program (GraphPad Software, La Jolla, CA), and two pharmacological parameters were obtained: the maximal effect generated by the agonist (or Emax) and −log EC50 (or pD2). Statistical analyses of Emax and pD2 values as well as infarct size, Hb, grip strength, and percent change in CBF were performed with Student's t-test. HT occurrence was compared by χ2-test, and neurological deficit by composite score was analyzed with the Mann-Whitney test. For analysis of mortality, Fisher's exact test was used. Values of P < 0.05 were considered statistically significant.
RESULTS
Metabolic parameters.
Eight-week HFD significantly increased body weight and adiposity without affecting plasma lipids (Table 1). Adipose tissue in all depots (subcutaneous, peritoneal, and epididymal) was increased. There were no differences in blood glucose, blood pressure, or plasma insulin levels.
Table 1.
Metabolic parameters of CD and HFD groups
| CD (n = 18) | HFD (n = 18) | |
|---|---|---|
| Body wt, g | 452 ± 9 | 494 ± 9* |
| Adiposity, % body wt | 5.2 ± 0.5 | 9.3 ± 1.2* |
| Triglyceride, mg/dl | 57 ± 4 | 69 ± 9 |
| Cholesterol, mg/dl | 49 ± 3 | 61 ± 8 |
| Insulin, ng/ml | 1.4 ± 0.2 | 1.2 ± 0.2 |
| SBP, mmHg | 112 ± 2 | 115 ± 3 |
Values are means ± SE for n rats. CD, control diet; HFD, high-fat diet; SBP, systolic blood pressure.
P < 0.05.
Effect of HFD on cerebrovascular function.
The effect of HFD on cerebrovascular function was assessed by several methods looking at vessels of different caliber. First, functional hyperemia was measured to evaluate the response of smaller arterioles by using the relative change in CBF upon whisker stimulation. As shown in Fig. 1, A and B, HFD animals displayed blunted change in CBF, indicating impaired neurovascular coupling.
Fig. 1.
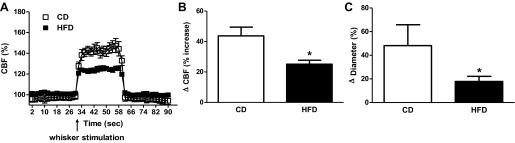
Functional hyperemia is compromised by high-fat diet (HFD). A: tracing of changes in cerebral blood flow (CBF) as measured by laser Doppler directed at 2 mm posterior and 5 mm lateral to bregma during whisker stimulation demonstrates that HFD blunts neurovascular coupling (n = 11/group). CD, control diet. B: quantitative analysis of total change (Δ) in CBF upon neuronal stimulation. C: K+-induced relaxations of parenchymal arterioles in brain slices are decreased in HFD-fed animals (n = 4/group). Values are means ± SE. *P < 0.05.
Next, the tone and relaxation properties of PAs were measured in slice preparations. We previously showed that the degree of tone in PAs dictates the polarity of the vascular response to vasoactive signals released by activated astrocytes, with decreased tone favoring constrictions and increased tone favoring dilations (3). To determine whether the HFD induced any change in vascular tone, cortical PAs were exposed to 150 nM U-46619 to induce arteriolar constriction. While no statistically significant differences were achieved, arterioles from the HFD group showed lower baseline tone (23.9 ± 4.8%, n = 11) compared with the control group (35.7 ± 9.9%, n = 6). The values of the control group were comparable to those previously reported by us in Sprague-Dawley rats fed chow diet (3). In agreement with our previous observations, reduced tone such as that observed in the HFD group resulted in a reduced vasodilatory response to 10 mM K+ (Fig. 1C).
Third, the contractile and dilatory responses of basilar arteries before and after focal ischemic injury were determined. There was no effect of HFD on these functions if the animals were not subjected to stroke (data not shown). However, when basilar arteries were tested at 24 h after MCAO, the concentration-response curves to several vasoconstrictors including 5-HT, ET-1, and U-46619 were left-shifted, indicating enhanced sensitivity, as well as greater maximum responses (Fig. 2, A–C). Endothelium-dependent relaxation was also significantly impaired in the HFD group (Fig. 2D). O-GlcNAc levels in the basilar arteries of HFD-fed animals were significantly greater, suggesting that this posttranslational modification can be the underlying mechanism of increased contractility in basilar arteries (Fig. 3).
Fig. 2.
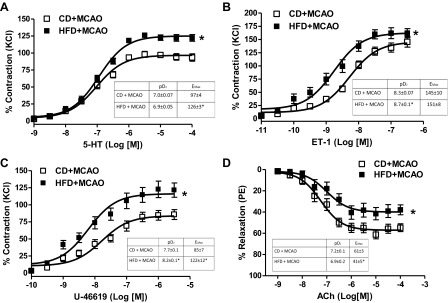
Effect of HFD on basilar artery function after focal cerebral ischemia [middle cerebral artery occlusion (MCAO)]-reperfusion. HFD increased the contractile response to multiple agonists (A–C) and also reduced endothelium-dependent relaxation (D). Experimental values of contraction were calculated relative to the contractile response produced by 120 mM KCl, which was taken as 100% (n = 8/group). Values are means ± SE. *P < 0.05.
Fig. 3.
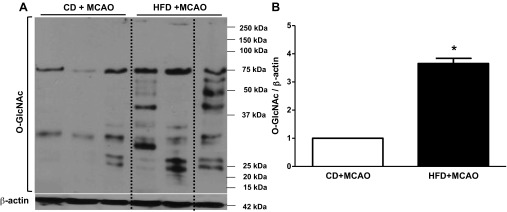
O-GlcNAcylation in basilar artery after focal cerebral ischemia-reperfusion. An increase in total O-GlcNAc-protein content was seen in the HFD-fed group after MCAO (n = 6/group). A representative Western blot image of O-GlcNAc-modified proteins and actin control is given in A, and cumulative data are summarized in B. Representative images were selected from the same membrane, and splices are indicated by dashed lines. Values are means ± SE. *P < 0.05, n = 4.
Effect of HFD on neurovascular injury after ischemia-reperfusion.
When focal ischemic injury was induced by 3-h MCAO and 21-h reperfusion, the percent drop in CBF after occlusion (40 ± 5% in CD and 38 ± 4% in HFD compared with baseline) or recovery after reperfusion (17 ± 3% in CD and 20 ± 11% in HFD compared with occlusion) was similar in both groups, but infarct size was higher in the HFD group than in the control group (Fig. 4A). Mortality rate was 11% (2 of 18) and 33% (6 of 18) in CD and HFD groups, respectively (P = 0.09). When ischemic injury was induced by 10-min GCI followed by 7-day reperfusion, mortality was 50% in the HFD group. Hippocampal CA1 sections were collected from animals that survived the surgery, and TUNEL staining was performed to access apoptotic cell death. There was no difference in apoptotic cell death between sections from CD and HFD rats (Fig. 4B). In the focal ischemia model, there was no difference in edema between the groups but the incidence of macroscopic HT as well as tissue Hb levels were increased in the HFD group (Fig. 5).
Fig. 4.
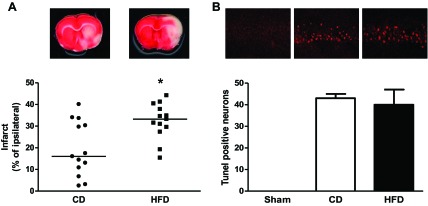
Effect of HFD on neuronal injury in different models of cerebral ischemia. Focal ischemia (A) induced by 3-h MCAO and 21-h reperfusion increased infarct size in the HFD group, but 10-min global ischemia (B) did not impact neuronal death in the hippocampus (n = 10–18/group). Values are medians in A and means ± SE in B. *P < 0.05.
Fig. 5.
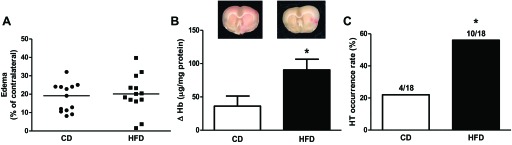
Effect of HFD on vascular function after focal cerebral ischemia-reperfusion. The balanced edema percentage (A) was not significantly higher, but the occurrence rate (C) and the severity (B) of hemorrhagic transformation (HT) determined by excess Hb in the brain were greater in the HFD group (n = 18/group). Values are medians in A and means ± SE in B. *P < 0.05.
Effect of HFD on functional outcome after ischemia-reperfusion.
A composite score derived from Bederson's score and beam walking showed poorer HFD rat performance on the behavioral tests compared with the CD group (Fig. 6A; 7.8 ± 1.3 in CD vs. 4.0 ± 0.8 in HFD, P < 0.05). However, there was no difference on grip strength (Fig. 6B; 1.11 ± 0.07 kgF in CD vs. 0.90 ± 0.08 kgF in HFD, P = 0.07).
Fig. 6.

Effect of HFD on functional outcomes after focal cerebral ischemia-reperfusion. A composite neurological deficit score including Bederson's score (including 1) spontaneous ipsilateral circling, graded from 2 to 0, 2) hindlimb retraction, 3) forelimb flexion, and 4) resistance to push, scored either 1 or 0, respectively) and beam walk graded from 1 to 7 was measured. A: composite score was lower in HFD-fed rats, indicating poor outcome. B: grip strength was not affected. (n = 18/group). Values are medians in A and means ± SE in B. *P < 0.05.
DISCUSSION
This study provides novel information about the early impact of HFD on cerebrovascular function and stroke outcomes in the absence of overt metabolic changes. First, HFD impairs communication between neurons and penetrating arterioles even in the absence of an ischemic insult. Second, ischemic injury serves as a second hit and causes large-artery dysfunction in stroked HFD rats that is not otherwise detectable in HFD-alone animals. Third, stroke in HFD-fed animals that do not have obesity or metabolic derangement worsens neurovascular injury and functional outcomes. Collectively, these data suggest that detrimental effects of HFD start early in the disease process and preventive measures should be implemented as early as possible.
HFD or obesity is a major risk factor for vascular dysfunction. It was realized several decades ago that high intake of saturated fat in the diet significantly enhanced the development of the atherosclerotic and autoimmune lesions in aorta of the autoimmune-prone B/W mice, which were known to develop severe glomerulonephritis and vasculitis (12). Numerous studies thereafter demonstrated that HFD impaired the structure and function and increased the lesion in different vascular beds (18, 31, 37, 42). Recent studies that focused on the cerebral vasculature have found that the dilator response to ACh was impaired in cerebral arterioles of HFD-fed apoE−/− mice (24) or in basilar artery of HFD-fed peroxisome proliferator-activated receptor (PPAR)-γ knockdown mice (2). In the present study, we found that relatively short-term administration of a nonatherogenic HFD impaired the ability of smaller arterioles to dilate and altered the contractile and dilatory properties of basilar arteries only after ischemic injury. Interestingly, these detrimental changes in cerebrovascular function were in the absence of overt obesity. There is no definition of obesity in animal models such as it is clearly defined in humans as body mass index (BMI) > 30. A person has traditionally been considered to be obese if he or she is >20% over ideal weight. In our animals total body weight increased by 10%, and this was mainly adipose mass. Thus the changes we observed in this model are mainly the effect of HFD and not obesity per se.
Cerebral vascular function is closely regulated by central nervous system activity, especially astrocytes whose processes are in direct contact with both synapses and blood vessels (20). Previous reports demonstrated the contribution of astrocytes in neurovascular coupling through K+ signaling (3, 9). In the present study, we evaluated whether K+-mediated vasodilation is disrupted after HFD treatment. We found that whisker stimulation-induced functional hyperemia (in vivo) and K+-induced vasodilation (in vitro) are reduced in the HFD group. The data suggest that PAs from the HFD group had impaired vascular function. Given the lack of increased blood flow response following whisker stimulation, future studies addressing the role of astrocytes in activity-dependent vascular responses are needed to better define whether HFD only affected vascular function or if it also altered the activity of upstream mechanisms such as that of K+ signaling by astrocytes.
In the present study we found no significant effect of HFD on surviving neuronal cell number in the GCI model even with different methods (NeuN staining, data not shown), which is consistent with another report that used 60-day Western HFD in Sprague-Dawley rats and also found no effect of HFD on neuronal cell death or survival after GCI (1). While we did not assess functional end points after GCI in our study, the study by Arvanitidis et al. (1) did assess functional outcome with the Morris water maze and found no significant effect of HFD, a finding consistent with the lack of effect of HFD on neuronal cell death or survival in both our and their studies. In a preliminary study, we utilized an even longer HFD period of 10 wk, with the thought that a longer duration may be needed to observe an effect in the GCI model. However, 10-wk HFD also had no significant effect on neuronal cell death/survival after GCI. It is not clear as to why HFD increased neuronal damage in the FCI but not GCI model of cerebral ischemia. It is known that the pathophysiological mechanisms differ between the two models (e.g., a more delayed neuronal cell death occurring in vulnerable brain regions after GCI), which might contribute to the difference. In addition, the duration of ischemia is also quite different between the two models (3 h in FCI vs. 10 min in GCI), which might contribute to differences in effects. While the mechanisms underlying the differential effect of HFD on neuronal cell death/survival in the two ischemia models requires further study, the significant effect of HFD observed in the FCI model is of potential translational importance. This is especially true considering that, of the two ischemia models, the FCI (MCAO) model is generally accepted as the most translationally relevant model of ischemic stroke, as >75% of strokes in humans involve occlusion of the MCA.
Obesity is an independent risk factor and may affect other risk factors for stroke such as hypertension, diabetes, and hyperlipidemia. Experimental studies have shown that either HFD or genetically induced obesity was accompanied by increased cerebrovascular remodeling, promoted hypertension, and increased infarct size in either transient or permanent focal ischemia models (7, 32). HFD-fed apoE−/− mice with hyperlipidemia also had increased infarct volume (23). However, another report showed that 1-mo HFD had no effect on the cerebral ischemia outcome (26). In the present study, 8-wk HFD resulted in significantly larger infarct volume after transient focal ischemia induced by suture occlusion of MCA, which is comparable to previous reports. When a global ischemia model was employed, there was no difference in hippocampal neuronal death between the groups, which was also reported by another group (1). These findings suggest that the duration of the diet and the method of ischemia are important for the extent and localization of neuronal injury. While we do not know the potential mechanisms contributing to greater neurovascular injury and poor outcomes in our model, it is possible that proper regulation of cerebral perfusion after stroke contributes to unfavorable outcomes. Since large arteries like the basilar artery contribute significantly to total cerebrovascular resistance and are major determinants of microvascular pressure, dysregulation of basilar artery function may worsen stroke injury by altering cerebral perfusion after stroke. In this context, it is highly possible that exacerbated release of vasoactive factors, such as ET-1, released into the circulation may be mediating this response. In a recent elegant study from Dr. Cipolla's group, investigators showed that plasma from stroked hyperglycemic animals can affect cerebrovascular function through peroxynitrite generation and ET-1 (34). In another study, we showed that stroke decreases the dilatory ability of basilar arteries in regular chow-fed animals compared with sham treatment (6), and administration of atrasentan, an ET receptor antagonist, at reperfusion prevented this response. While the experimental conditions of that particular study were different, maximum relaxation observed in sham-treated rats (∼50%) was reduced to ∼25% and this was normalized by ET receptor antagonism. In the present study, we do not have a sham treatment group, but it is possible that even CD-fed animals may be displaying some degree of dysfunction and this is exacerbated in HFD. We have previously shown that HFD increases plasma ET-1 (38). Given these findings, the ET system may play a role in exacerbated stroke injury in our model and will be further pursued.
Along the same lines, in light of our recent studies showing that augmented O-GlcNAcylation increases vascular reactivity to ET-1 (28), we next assessed whether this posttranslational modification is a potential downstream mechanism contributing to HFD-induced vascular dysfunction. As recently reviewed, there are multiple targets that are regulated by O-GlcNAcylation in the vasculature (30). A positive correlation between phosphorylation of the MAPK cascade (ERK1/2 and p38) and nuclear O-GlcNAcylation was observed in fetal human cardiac myocytes exposed to high glucose (14). Previous work from our group has shown that O-GlcNAcylation-induced increased reactivity of aorta to PE was prevented by a PKC inhibitor or a Rho kinase inhibitor, respectively (17, 29). Our present finding of increased O-GlcNAc levels in basilar arteries of HFD-fed animals after stroke merits further studies to determine the mechanisms linking HFD to increased O-GlcNAcylation as well as linking O-GlcNAcylation to cerebrovascular dysfunction. In a preliminary study, we found that HFD alone caused a small increase (1.5 fold) in O-GlcNAc levels compared with a fourfold increase observed in this study with HFD + MCAO. It is of great interest to determine whether blockade of increased O-GlcNAc levels prevents vascular dysfunction and improves stroke outcomes.
Perspectives and Significance
In the present study, the important findings of impaired neurovascular communication, large-artery dysfunction, and augmented neurovascular injury suggest that even short-term HFD without obesity or metabolic imbalance may be detrimental to the cerebrovasculature and exacerbate the response to cerebral ischemia. We recognize that there are limitations to this study such as evaluation of the outcome only at 24 h and inclusion of only male rats. Given that AIS has dramatically increased in children and young adults, which is positively correlated with increases in risk factors including obesity, lipid disorders, and diabetes (13), further studies are warranted to explore the underlying mechanisms by which HFD worsens short- and long-term stroke outcome in both female and male animal models.
GRANTS
A. Ergul is a research pharmacologist at the Charlie Norwood Department of Veterans Affairs (VA) Medical Center. This work was supported in part by VA Merit Award BX00347 (to A. Ergul), a Georgia Health Sciences University Diabetes and Obesity Discovery Institute Synergy Award (S. P. Didion, J. A. Filosa, D. W. Brann, R. C. Tostes, and A. Ergul), an American Heart Association Established Investigator Award (0740002N to A. Ergul), and National Institutes of Health Grants HL-089884 and HL-107632 to S. P. Didion, HL-089067 to J. A. Filosa, NS-050730-08 to D. W. Brann, and NS-054688 to A. Ergul.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: W.L., R.P., D.C., W.D., Q.Z., and V.V.L. performed experiments; W.L., R.P., D.C., W.D., Q.Z., and V.V.L. analyzed data; W.L., R.C.T., and A.E. interpreted results of experiments; W.L. and V.V.L. prepared figures; W.L., R.P., J.A.F., and D.W.B. drafted manuscript; W.L., R.C.T., and A.E. edited and revised manuscript; S.P.D., J.A.F., D.W.B., R.C.T., and A.E. conception and design of research; S.P.D., J.A.F., D.W.B., and A.E. approved final version of manuscript.
REFERENCES
- 1.Arvanitidis AP, Corbett D, Colbourne F. A high fat diet does not exacerbate CA1 injury and cognitive deficits following global ischemia in rats. Brain Res 1252: 192–200, 2009 [DOI] [PubMed] [Google Scholar]
- 2.Beyer AM, de Lange WJ, Halabi CM, Modrick ML, Keen HL, Faraci FM, Sigmund CD. Endothelium-specific interference with peroxisome proliferator activated receptor gamma causes cerebral vascular dysfunction in response to a high-fat diet. Circ Res 103: 654–661, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blanco VM, Stern JE, Filosa JA. Tone-dependent vascular responses to astrocyte-derived signals. Am J Physiol Heart Circ Physiol 294: H2855–H2863, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brahma B, Forman RE, Stewart EE, Nicholson C, Rice ME. Ascorbate inhibits edema in brain slices. J Neurochem 74: 1263–1270, 2000 [DOI] [PubMed] [Google Scholar]
- 5.Collingridge GL. The brain slice preparation: a tribute to the pioneer Henry McIlwain. J Neurosci Methods 59: 5–9, 1995 [DOI] [PubMed] [Google Scholar]
- 6.Coucha M, Li W, Ergul A. The effect of endothelin receptor A antagonism on basilar artery endothelium-dependent relaxation after ischemic stroke. Life Sci 91: 676–680, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deutsch C, Portik-Dobos V, Smith AD, Ergul A, Dorrance AM. Diet-induced obesity causes cerebral vessel remodeling and increases the damage caused by ischemic stroke. Microvasc Res 78: 100–106, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Didion SP, Lynch CM, Faraci FM. Cerebral vascular dysfunction in TallyHo mice: a new model of Type II diabetes. Am J Physiol Heart Circ Physiol 292: H1579–H1583, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Dunn KM, Nelson MT. Potassium channels and neurovascular coupling. Circ J 74: 608–616, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ergul A, Elgebaly MM, Middlemore ML, Li W, Elewa H, Switzer JA, Hall C, Kozak A, Fagan SC. Increased hemorrhagic transformation and altered infarct size and localization after experimental stroke in a rat model type 2 diabetes. BMC Neurol 7: 33, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faraci FM, Heistad DD. Regulation of large cerebral arteries and cerebral microvascular pressure. Circ Res 66: 8–17, 1990 [DOI] [PubMed] [Google Scholar]
- 12.Fernandes G, Alonso DR, Tanaka T, Thaler HT, Yunis EJ, Good RA. Influence of diet on vascular lesions in autoimmune-prone B/W mice. Proc Natl Acad Sci USA 80: 874–877, 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.George MG, Tong X, Kuklina EV, Labarthe DR. Trends in stroke hospitalizations and associated risk factors among children and young adults, 1995–2008. Ann Neurol 70: 713–721, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Gross BJ, Kraybill BC, Walker S. Discovery of O-GlcNAc transferase inhibitors. J Am Chem Soc 127: 14588–14589, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Hajos N, Mody I. Establishing a physiological environment for visualized in vitro brain slice recordings by increasing oxygen supply and modifying aCSF content. J Neurosci Methods 183: 107–113, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol 100: 1059–1064, 2006 [DOI] [PubMed] [Google Scholar]
- 17.Hardy DM, Tostes RC, Webb RC. O-GlcNAcylation augments protein kinase C mediated vasoconstriction (Abstract). FASEB J 24, Suppl: 603.8comment>, 2010 [Google Scholar]
- 18.Helfenstein T, Fonseca FA, Ihara SS, Bottos JM, Moreira FT, Pott H, Jr, Farah ME, Martins MC, Izar MC. Impaired glucose tolerance plus hyperlipidaemia induced by diet promotes retina microaneurysms in New Zealand rabbits. Int J Exp Pathol 92: 40–49, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubert HB, Feinleib M, McNamara PM, Castelli WP. Obesity as an independent risk factor for cardiovascular disease: a 26-year follow-up of participants in the Framingham Heart Study. Circulation 67: 968–977, 1983 [DOI] [PubMed] [Google Scholar]
- 20.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci 10: 1369–1376, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Kaarisalo MM, Raiha I, Sivenius J, Immonen-Raiha P, Lehtonen A, Sarti C, Mahonen M, Torppa J, Tuomilehto J, Salomaa V. Diabetes worsens the outcome of acute ischemic stroke. Diabetes Res Clin Pract 69: 293–298, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Kelly-Cobbs AI, Prakash R, Coucha M, Knight RA, Li W, Ogbi SN, Johnson M, Ergul A. Cerebral myogenic reactivity and blood flow in Type 2 diabetic rats: role of peroxynitrite in hypoxia-mediated loss of myogenic tone. J Pharmacol Exp Ther 342: 407–415, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim JH, Park SH, Bae SS, Hong KW, Kim YD, Park KP, Choi BT, Shin HK. Combinatorial effect of probucol and cilostazol in focal ischemic mice with hypercholesterolemia. J Pharmacol Exp Ther 338: 451–457, 2011 [DOI] [PubMed] [Google Scholar]
- 24.Kitayama J, Faraci FM, Lentz SR, Heistad DD. Cerebral vascular dysfunction during hypercholesterolemia. Stroke 38: 2136–2141, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Kumari R, Willing LB, Patel SD, Krady JK, Zavadoski WJ, Gibbs EM, Vannucci SJ, Simpson IA. The PPAR-gamma agonist, darglitazone, restores acute inflammatory responses to cerebral hypoxia-ischemia in the diabetic ob/ob mouse. J Cereb Blood Flow Metab 30: 352–360, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langdon KD, Clarke J, Corbett D. Long-term exposure to high fat diet is bad for your brain: exacerbation of focal ischemic brain injury. Neuroscience 182: 82–87, 2011 [DOI] [PubMed] [Google Scholar]
- 27.Li W, Sachidanandam K, Ergul A. Comparison of selective versus dual endothelin receptor antagonism on cerebrovascular dysfunction in diabetes. Neurol Res 33: 185–191, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lima VV, Giachini FR, Carneiro FS, Carneiro ZN, Saleh MA, Pollock DM, Fortes ZB, Carvalho MH, Ergul A, Webb RC, Tostes RC. O-GlcNAcylation contributes to augmented vascular reactivity induced by endothelin 1. Hypertension 55: 180–188, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lima VV, Giachini FR, Carneiro FS, Carvalho MH, Fortes ZB, Webb RC, Tostes RC. O-GlcNAcylation contributes to the vascular effects of ET-1 via activation of the RhoA/Rho-kinase pathway. Cardiovasc Res 89: 614–622, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lima VV, Giachini FR, Hardy DM, Webb RC, Tostes RC. O-GlcNAcylation: a novel pathway contributing to the effects of endothelin in the vasculature. Am J Physiol Regul Integr Comp Physiol 300: R236–R250, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naderali EK, Williams G. Prolonged endothelial-dependent and -independent arterial dysfunction induced in the rat by short-term feeding with a high-fat, high-sucrose diet. Atherosclerosis 166: 253–259, 2003 [DOI] [PubMed] [Google Scholar]
- 32.Osmond JM, Mintz JD, Dalton B, Stepp DW. Obesity increases blood pressure, cerebral vascular remodeling, and severity of stroke in the Zucker rat. Hypertension 53: 381–386, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osmond JM, Mintz JD, Stepp DW. Preventing increased blood pressure in the obese Zucker rat improves severity of stroke. Am J Physiol Heart Circ Physiol 299: H55–H61, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palomares SM, Gardner-Morse I, Sweet JG, Cipolla MJ. Peroxynitrite decomposition with FeTMPyP improves plasma-induced vascular dysfunction and infarction during mild but not severe hyperglycemic stroke. J Cereb Blood Flow Metab 32: 1035–1045, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qin Z, Karabiyikoglu M, Hua Y, Silbergleit R, He Y, Keep RF, Xi G. Hyperbaric oxygen-induced attenuation of hemorrhagic transformation after experimental focal transient cerebral ischemia. Stroke 38: 1362–1367, 2007 [DOI] [PubMed] [Google Scholar]
- 36.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart Disease and Stroke Statistics—2012 Update: a report from the American Heart Association. Circulation 125: e2–e220, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rudel LL, Parks JS, Sawyer JK. Compared with dietary monounsaturated and saturated fat, polyunsaturated fat protects African green monkeys from coronary artery atherosclerosis. Arterioscler Thromb Vasc Biol 15: 2101–2110, 1995 [DOI] [PubMed] [Google Scholar]
- 38.Sachidanandam K, Hutchinson JR, Elgebaly MM, Mezzetti EM, Wang MH, Ergul A. Differential effects of diet-induced dyslipidemia and hyperglycemia on mesenteric resistance artery structure and function in type 2 diabetes. J Pharmacol Exp Ther 328: 123–130, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarikaya H, Arnold M, Engelter ST, Lyrer PA, Mattle HP, Michel P, Odier C, Weder B, Siebel P, Mueller F, Ballinari P, Georgiadis D, Baumgartner RW. Outcome of intravenous thrombolysis in stroke patients weighing over 100 kg. Cerebrovasc Dis 32: 201–206, 2011 [DOI] [PubMed] [Google Scholar]
- 40.Sarikaya H, Elmas F, Arnold M, Georgiadis D, Baumgartner RW. Impact of obesity on stroke outcome after intravenous thrombolysis. Stroke 42: 2330–2332, 2011 [DOI] [PubMed] [Google Scholar]
- 41.Vannucci SJ, Willing LB, Goto S, Alkayed NJ, Brucklacher RM, Wood TL, Towfighi J, Hurn PD, Simpson IA. Experimental stroke in the female diabetic, db/db, mouse. J Cereb Blood Flow Metab 21: 52–60, 2001 [DOI] [PubMed] [Google Scholar]
- 42.Wilde DW, Massey KD, Walker GK, Vollmer A, Grekin RJ. High-fat diet elevates blood pressure and cerebrovascular muscle Ca2+ current. Hypertension 35: 832–837, 2000 [DOI] [PubMed] [Google Scholar]
- 43.Zhang QG, Raz L, Wang R, Han D, De Sevilla L, Yang F, Vadlamudi RK, Brann DW. Estrogen attenuates ischemic oxidative damage via an estrogen receptor alpha-mediated inhibition of NADPH oxidase activation. J Neurosci 29: 13823–13836, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]