

Abstract
Myofibroblast differentiation induced by transforming growth factor-β (TGF-β) is characterized by the expression of smooth muscle α-actin (SMA) and extracellular matrix proteins. We and others have previously shown that these changes are regulated by protein kinase A (PKA). Adrenomedullin (ADM) is a vasodilator peptide that activates cAMP/PKA signaling through the calcitonin-receptor-like receptor (CRLR) and receptor-activity-modifying proteins (RAMP). In this study, we found that recombinant ADM had little effect on cAMP/PKA in quiescent human pulmonary fibroblasts, whereas it induced a profound activation of cAMP/PKA signaling in differentiated (by TGF-β) myofibroblasts. In contrast, the prostacyclin agonist iloprost was equally effective at activating PKA in both quiescent fibroblasts and differentiated myofibroblasts. TGF-β stimulated a profound expression of CRLR with a time course that mirrored the increased PKA responses to ADM. The TGF-β receptor kinase inhibitor SB431542 abolished expression of CRLR and attenuated the PKA responses of cells to ADM but not to iloprost. CRLR expression was also dramatically increased in lungs from bleomycin-treated mice. Functionally, ADM did not affect initial differentiation of quiescent fibroblasts in response to TGF-β but significantly attenuated the expression of SMA, collagen-1, and fibronectin in pre-differentiated myofibroblasts, which was accompanied by decreased contractility of myofibroblasts. Finally, sensitization of ADM signaling by transgenic overexpression of RAMP2 in myofibroblasts resulted in enhanced survival and reduced pulmonary fibrosis in the bleomycin model of the disease. In conclusion, differentiated pulmonary myofibroblasts gain responsiveness to ADM via increased CRLR expression, suggesting the possibility of using ADM for targeting pathological myofibroblasts without affecting normal fibroblasts.
Keywords: myofibroblast, TGF-β, adrenomedullin, PKA, fibrosis
idiopathic pulmonary fibrosis (IPF) is a progressive, fatal disease characterized by parenchymal fibrosis and structural distortion of the lungs. Age-adjusted mortality due to pulmonary fibrosis is increasing (45), and it poses a vexing clinical challenge given the lack of proven efficacious therapy. IPF is thought to be a disorder of abnormal wound healing (18, 64), in which the initial trigger to the fibrotic response is injury to the alveolar epithelial cell, followed by an exuberant, non-resolving, wound-healing response (43, 51, 58). Injury of alveolar epithelial cells is thought to result in the elaboration of a fibrinous provisional matrix and the activation of several pro-inflammatory, pro-coagulant, and pro-fibrotic mediators, of which transforming growth factor-β1 (TGF-β1) is the most established (1, 25, 53). Fibroblasts, under stimulation of TGF-β1, respond by altering their gene expression profile with de novo expression of cytoskeletal and contractile proteins normally found within smooth muscle cells, modified focal adhesion complexes (20), and components of the extracellular matrix (30, 46, 60). These smooth muscle (SM)-like fibroblasts are called myofibroblasts and display a phenotype that is in an intermediate state between fibroblasts and smooth muscle cells (7, 21, 48). Several cytoskeletal and SM proteins are expressed in myofibroblasts, including SM α-actin (SMA), the most established marker for myofibroblast differentiation (12, 21, 47, 60). Functionally, this phenotypic switch is thought to increase the ability of the myofibroblasts to attach to the remodeling matrix and facilitate wound contraction, an important role of the myofibroblast. Additionally, induction of myofibroblast phenotype is also associated with secretion of extracellular matrix proteins (collagen isoforms, cellular fibronectin, etc.) and of pro-fibrotic factors [connective tissue growth factor (CTGF), insulin-like growth factor (IGF-1) etc. (30, 31)], thus perpetuating the ongoing tissue remodeling and fibrosis. Myofibroblasts are invariably found in histological sections of human lung specimens from patients with pulmonary fibrosis and are thought to be a critical pathogenic mechanism responsible for the progressive nature of IPF. Therefore, disrupting cellular mechanisms that induce and maintain the myofibroblast phenotype may be a potential strategy to attenuate the ongoing fibrotic response in pulmonary fibrosis.
In vitro studies have demonstrated that differentiation, proliferation, and extracellular matrix deposition by pulmonary myofibroblasts can be diminished by activators of cAMP and protein kinase A (PKA) (13, 22, 23, 27, 28, 33, 50, 59), suggesting that cAMP/PKA signaling may be a potential therapeutic target for the treatment of pulmonary fibrosis. In vivo studies have demonstrated a protective effect of prostaglandin E2 or prostacyclin analog iloprost in the bleomycin model of pulmonary fibrosis (5, 68). However, identification of suitable agonists acting through cAMP/PKA signaling that could be effective in humans remains unaccomplished.
Adrenomedullin (ADM) is a vasodilator peptide known to act through an atypical G-protein-coupled receptor complex consisting of the seven-transmembrane calcitonin receptor-like receptor (CRLR) and co-receptors, receptor-activity-modifying proteins (RAMP), which are required for the transport of CRLR to the plasma membrane and provide a high affinity binding of ADM to CRLR (35). Most of the known cellular effects of ADM are associated with cAMP/PKA signaling (11). Previous studies have demonstrated that ADM administration or gene delivery attenuates hypertension, cardiac remodeling, and renal injury in various animal models (3, 9, 41, 42, 44, 54, 62, 63). In the lung, adrenomedullin attenuates acute lung injury in animal models (24, 37, 57), attenuates pulmonary hypertension in animal models and in humans (34, 38, 39), and regulates allergen-induced airway hyperresponsiveness in mice (67). However, regulation of myofibroblast function and pulmonary fibrosis by ADM signaling has been poorly investigated, and this is the focus of this study.
METHODS
Isolation and primary culture of human lung fibroblasts.
Human lung fibroblasts were isolated as described previously (50). Briefly, tissue samples from explanted lungs from patients undergoing lung transplantation were obtained and placed in DMEM with 100 U/ml streptomycin, 250 ng/ml amphotericin B, and 100 U/ml penicillin. Alveolated lung tissue was minced, washed in PBS, and plated on 10-cm plates in growth media containing DMEM supplemented with 10% FBS, 2 mM L-glutamine, 100 U/ml streptomycin, 250 ng/ml amphotericin B, and 100 U/ml penicillin. Expanded populations of fibroblasts were subsequently subcultured after 4–5 days, resulting in the development of a homogenous fibroblast population. All primary cultures were used from passage 3 to 10.
DNA transduction.
Adenovirus-mediated gene transduction was performed by incubating cells with desired adenoviruses (500 plaque-forming units/cell) together with GeneJammer reagent (Stratagene, La Jolla, CA) to facilitate the efficiency of transduction.
Reagents.
Recombination-deficient adenovirus expressing adrenomedullin (Ad-ADM) and control adenovirus (Ad-LacZ) was provided by the University of Iowa Gene Transfer Vector Core Facility with permission from Dr. Timo Hautala (49). TGF-β1 was from EMD Biosciences (Gibbstown, NJ). Antibodies against SM α-actin were from Sigma, collagen-1 antibodies were from Cedarlane, fibronectin antibodies were from BD Transduction, and VASP antibodies were from Calbiochem. Pharmaceutical-grade bleomycin (Bleocip) was from Cipla. The cAMP ELISA kit was purchased from Enzo Lifesciences.
Cell lysis and Western blotting.
After stimulation of cells with desired agonists, cells were lysed in the radioimmunoprecipitation (RIPA) buffer containing 25 mM HEPES (pH 7.5), 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 2 mM EDTA, 2 mM EGTA, 10% glycerol, 1 mM NaF, 200 μM sodium orthovanadate, and protease inhibitor cocktail (Sigma). Cells were scraped, sonicated, a sample was taken for the measurement of protein concetration, and the remainder was boiled in Laemmli buffer for 5 min. The samples were normalized to the protein content, subjected to polyacrylamide gel electrophoresis, analyzed by Western blotting with desired primary antibodies and corresponding horseradish peroxidase-conjugated secondary antibodies, and developed by an enhanced chemilumeniscence reaction (Pierce). The digital chemilumeniscent pictures were imaged by a Luminescent Image Analyzer LAS-4000 (Fujifilm).
cAMP assay.
Cells were treated with 200 μM IBMX (Cayman Chemicals) for 30 min followed by stimulation with 100 nM ADM for indicated times. The cAMP assay was performed on acetylated samples following Enzo Lifescience's protocol. Conditions were performed in triplicate.
Reverse-transcription quantitative real-time PCR.
RNA STAT-60 (TEL-Test) was used to isolate total RNA following the manufacturer's protocol. RNA was random primed and reverse-transcribed with iScript cDNA synthesis kit (Bio-Rad), according to the manufacturer's protocols. Real-time PCR analysis was performed using iTaq SYBR Green supermix with ROX (Bio-Rad) in a MyIQ single-color real-time PCR detection system (Bio-Rad).
Contractile force measurements.
Fibroblast contractility was quantified using monolayer traction force microscopy (2, 29, 56, 61). Briefly, following the desired treatments, cells were allowed to spread overnight on collagen-coated polyacrylamide gel substrates (Young's modulus = 4kPa) embedded with fluorescent beads. By tracking displacements of beads that were directly underneath the cells and solving for the inverse problem of stresses that induce those displacements, the contractility of the cell cluster called the traction was quantified. The tractions were averaged over nine fields (∼1,000 cells per field) per condition.
Animal studies.
C57/B6 mice with a RAMP2 transgene under the control of the smooth muscle actin promoter (TG-RAMP2) were kindly provided by Dr. Walter Born and were used previously (32, 55). Eight- to ten-week-old WT or Tg-RAMP2 mice weighing 25 g were intratracheally instilled with 1 U/kg bleomycin (Bleocip, Cipla). Lungs were removed 15 days or 21 days after bleomycin administration as indicated. Left lungs were formalin fixed and paraffin embedded, and the lung sections were stained with H&E or SM-α-actin antibody by immunohistochemistry. The images of the stained sections were obtained by the CRi Pannoramic whole slide scanner. Right lungs were snap frozen and processed for mRNA or hydroxyproline content measurements.
Hydroxyproline assay.
The hydroxyproline assay was performed as described previously (10). Briefly, right lungs were homogenized in 6 N hydrochloric acid and hydrolyzed for 12 h at 110°C. An aliquot was evaporated, resuspended in citrate-acetate buffer with chloramine T, and left at room temperature for 20 min. Ehrlich's solution was then added, and samples were heated at 65°C for 15 min. After cooling to room temperature, absorbance was measured at 550 nm. Hydroxyproline content was determined against a standard curve generated from pure hydroxyproline.
Statistical analysis.
All of the data represent the results of three independent experiments. Quantitative data were analyzed by the Student's t-test. Values of P < 0.05 were considered as statistically significant.
RESULTS
To examine the effect of ADM on PKA activation, we used PKA-substrate antibodies recognizing proteins phosphorylated at the PKA consensus site (RRXpS/pT, Cell Signaling). As shown in Fig. 1A, quiescent human pulmonary fibroblasts responded to ADM very modestly, whereas in TGF-β1 pre-differentiated myofibroblasts, a large number of proteins were detected by Western blotting with PKA-substrate antibodies. We also assessed phosphorylation of a well established PKA substrate, vasodilator-stimulated phosphoprotein (VASP), by electrophoretic mobility shift assay, whose specificity as a reporter for PKA activity was demonstrated by us previously (6). As shown in Fig. 1A, only half of VASP was phosphorylated in response to ADM in quiescent fibroblasts, whereas all detectable VASP molecules were phosphorylated in differentiated myofibroblasts, as confirmed by SM-α-actin staining. Furthermore, ADM-induced modest phosphorylation of VASP was transient in quiescent fibroblasts, whereas in pre-differentiated myofibroblasts, VASP phosphorylation lasted for >2 h (Fig. 1B). Consistent with the VASP shift assay, ADM produced a far more profound and sustained increase in cAMP levels in pre-differentiated myofibroblasts compared with quiescent fibroblasts (Fig. 1C). The enhanced responsiveness of differentiated myofibroblasts to ADM was not due to a general increase in sensitization toward cAMP/PKA signaling, since the PKA responses of quiescent fibroblasts and differentiated myofibroblasts to the prostacyclin agonist iloprost were similar (Fig. 1D).
Fig. 1.

Increased protein kinase A (PKA) responses to adrenomedullin (ADM) but not to iloprost in differentiated myofibroblasts compared with quiescent fibroblasts. Human lung fibroblasts were kept quiescent or treated with 1 ng/ml TGF-β1 for 3 days to induce myofibroblast differentiation. Cells were then stimulated for indicated times with 100 nM ADM (A–C) or 1 μM iloprost (D). A, B, and D: equal amounts of cell lysates were analyzed by Western blotting with indicated antibodies. The percent of phosphorylated (shifted) over total VASP (% P-VASP) is indicated below VASP Western blots. C: cells were pretreated with 200 μM 3-Isobutyl-1-methylxanthine (IBMX) 30 min before stimulation with 100 nM ADM for indicated times. Equal amounts of cell extracts were examined for cAMP levels by ELISA.
To elucidate a potential mechanism of increased PKA activation by ADM in differentiated myofibroblasts, we examined the expression levels of CRLR. As shown in Fig. 2A, treatment of fibroblasts with TGF-β1 resulted in a fivefold increase in CRLR mRNA expression with a time course mirroring that of increased differentiation and PKA responsiveness to ADM (Fig. 2B). Treatment of cells with the TGF-β receptor kinase inhibitor SB431542 abolished TGF-β1-induced CRLR expression (Fig. 2C) and blocked the PKA responses of cells to ADM (Fig. 2D), suggesting the causative relation between these processes. Importantly, SB431542 did not affect iloprost-induced VASP phosphorylation (Fig. 2D). Finally, CRLR mRNA was also increased in mouse lungs following intratracheal administration of bleomycin as a model of pulmonary fibrosis (Fig. 2, E and F).
Fig. 2.
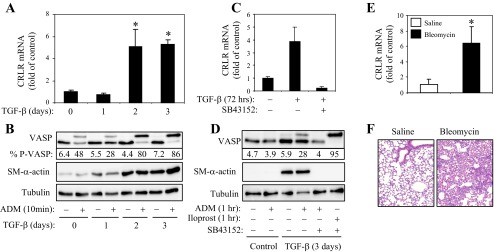
Upregulation of calcitonin-receptor-like receptor (CRLR) in differentiated myofibroblasts and in the lungs from bleomycin treated mice. A and B: time-dependent effect of TGF-β1 (1 ng/ml, 72 h) on the expression of CRLR mRNA (A) and on VASP phosphporylation (B) in human lung fibroblasts. C and D: effect of SB431542 (10 μM) on TGF-β1 (1 ng/ml, 3 days)-induced CRLR mRNA expression (C) or on VASP phosphorylation induced by 100 nM ADM or 1 μM iloprost (D). The % P-VASP is indicated below the VASP Western blots (B and D). E and F: bleomycin-induced upregulation of CRLR in mouse lungs. Pulmonary fibrosis was induced in 8- to 10-wk-old male C57Bl/6 mice by a single intratracheal administration of 1 U/kg bleomycin for 21 days. Lungs were then collected and analyzed for CRLR mRNA by real-time qPCR (E), or paraffin-embedded sections were stained for H&E (F). Shown are means ± SD from five lungs per condition (E).
We have previously shown that PKA activators such as forskolin or iloprost are potent inhibitors of myofibroblast differentiation (50). Therefore, we next examined whether the differential PKA responses of quiescent fibroblasts and myofibroblasts to ADM translate functionally to the regulation of expression of myofibroblast marker proteins. Pretreatment of quiescent fibroblasts with ADM shortly before stimulation with TGF-β1 had no effect on the increased expression of SM-α-actin, collagen-1, or fibronectin, either at mRNA or protein levels (Fig. 3, A and B). In contrast, when myofibroblast differentiation by TGF-β was induced for 48 h and then treated with ADM for an additional 24 h, there was a significant decrease in the expression of SM-α-actin and collagen-1α1 mRNA, and there was a trend toward a decrease in fibronectin mRNA levels (Fig. 4A). However, we did not observe significant changes in protein levels of these differentiation markers under these conditions (Fig. 4B). Given the transient nature of PKA responses of myofibroblasts to a single application of recombinant ADM (∼2 h; Fig. 1B), we examined whether sustained ADM application would attenuate the expression of these proteins in myofibroblasts, utilizing adenovirus-mediated transduction of secreted ADM (Ad-ADM). Quiescent fibroblasts were treated with TGF-β1 for 24 h to induce differentiation and then transduced with Ad-ADM or control virus (Ad-LacZ) for an additional 48 h. As shown in Fig. 5A, Ad-ADM transduction resulted in PKA activation as determined by the electrophoretic mobility VASP shift assay. Importantly, transduction with Ad-ADM, but not with the control Ad-LacZ, resulted in a dramatic decrease in the expression of SM-α-actin, collagen-1, and fibronectin proteins (Fig. 5A) and in contraction of HLFs (Fig. 5B).
Fig. 3.

Recombinant ADM does not inhibit initial differentiation of quiescent fibroblasts. Quiescent human lung fibroblasts were pretreated with 100 nM ADM for 5 min followed by stimulation with 1 ng/ml TGF-β1 for 24 h. Cell extracts were then analyzed for the expression of desired mRNAs by real-time qPCR (A) or for proteins by Western blotting (B). The representative densitometry expressed as percent of TGF-β-treated condition is indicated below the corresponding Western blots.
Fig. 4.

Effect of recombinant ADM on the expression of differentiation markers in pre-differentiated myofibroblasts. Quiescent human fibroblasts were treated with or without 1 ng/ml TGF-β1 for 48 h, followed by 100 nM ADM for an additional 24 h. Cell extracts were then analyzed for the expression of desired mRNAs by real-time qPCR (A) or for proteins by Western blotting (B). The representative densitometry expressed as percent of TGF-β-treated condition is indicated below the corresponding Western blots.
Fig. 5.

Sustained application of ADM attenuates the expression of differentiation markers in myofibroblasts and reduces their contractile force. Quiescent fibroblasts were treated with TGF-β1 for 24 h and then transduced with Ad-ADM or control Ad-LacZ adenoviruses for an additional 48 h. A: cell extracts were analyzed for the expression of desired proteins by Western blotting. The representative densitometry expressed as percent of TGF-β-treated condition is indicated below the corresponding Western blots. *Significant differences between Ad-LacZ and Ad-ADM conditions (P < 0.05; n = 3). B: equal numbers of cells were reseeded on collagen-coated polyacrylamide gel substrates (Young's modulus = 4 kPa) and allowed to spread overnight. The contractility of the cell cluster was quantified using monolayer traction microscopy as described in methods. Plotted is the average ± SD contraction over nine fields per condition.
Since ADM also has been reported to stimulate nitric oxide (NO) production (19, 52) and to affect cell proliferation (26, 65), we sought to examine whether these processes contributed to the effects of ADM on myofibroblast differentiation by using a general inhibitor of NO synthesis, l-NG-nitroarginine methyl ester (l-NAME). However, TGF-β, ADM, l-NAME, or their combination had no significant effect on proliferation of fibroblasts (data not shown). Furthermore, l-NAME (1 mM) did not reverse the inhibition of TGF-β-induced myofibroblast differentiation by ADM (data not shown), suggesting that NO production does not mediate these effects of ADM.
Finally, to examine whether activation of ADM signaling in myofibroblasts in vivo would affect the development of pulmonary fibrosis in bleomycin model of the disease, we utilized mice with transgenic overexpression of RAMP2 under the control of SM-α-actin promoter, which previously has been shown to increase the density of functional adrenomedullin receptors and sensitize vascular smooth muscle responses to ADM (55). Because differentiating myofibroblasts express SM-α-actin, they also express the RAMP2 transgene, which was confirmed in cultured lung myofibroblasts from these mice (data not shown). As shown in Fig. 6, intratracheal administration of bleomycin (1 U/kg) into WT mice resulted in ∼50% survival and a significant decrease in pulmonary fibrosis (as assessed by both the Ashcroft scoring system and by a hydroxyproline assay for collagen content), and was accompanied by accumulation of SM-α-actin-positive cells in the lungs from surviving mice. In contrast, all RAMP2-transgenic mice survived 15 days after bleomycin administration, had a significantly decreased pulmonary fibrosis, and had a decreased amount of SM-α-actin-positive cells in the lung compared with WT mice (Fig. 6).
Fig. 6.

Bleomycin-induced pulmonary fibrosis is decreased in mice with transgenic overexpression of RAMP2. Pulmonary fibrosis was induced in 8- to 10-wk-old male wild-type (WT) C57Bl/6 mice or mice with transgenic overexpression of RAMP2 under the control of SM-α-actin promoter (RAMP2-tg) by a single intratracheal administration of 1 U/kg bleomycin for 15 days. A: survival curves for WT and RAMP2-tg mice. B: lung sections were stained for H&E, and scored blindly (using the Ashcroft scoring system) over 20–25 fields (×10 magnification) covering the whole lung section, and the average score and standard deviation were calculated and plotted. C: collagen content in mouse lungs as measured by hydroxyproline assay. D: representative images (×20 magnification) of lung sections from bleomycin-treated WT or RAMP2-tg mice, stained with H&E or SM-α-actin antibodies.
DISCUSSION
This study describes the following novel findings. First, we show that, upon TGF-β treatment, differentiated human pulmonary myofibroblasts acquire responsiveness to ADM in terms of cAMP/PKA signaling, whereas quiescent fibroblasts poorly respond to ADM. Functionally, this translates to the inhibition of sustained myofibroblast differentiation and cell contractility by ADM. Second, we show for the first time that TGF-β is a potent inducer of CRLR expression, which may explain the increased responsiveness of differentiated myofibroblasts to ADM. Furthermore, we found that CRLR expression is greatly increased in fibrotic lungs from bleomycin-treated mice, suggesting that signaling through this receptor could be exploited for treatment of pulmonary fibrosis. Third, we demonstrate that sensitization of CRLR signaling to endogenous ADM via transgenic overexpression of RAMP2 in myofibroblasts significantly improves the outcomes of bleomycin-induced pulmonary fibrosis.
The increased responsiveness of human pulmonary myofibroblasts to ADM (Fig. 1) is of potential practical significance, since it suggests approaches for targeting pathological myofibroblasts by ADM without affecting normal quiescent fibroblasts. Importantly, PKA responses of myofibroblasts to another cAMP-linked agonist, iloprost, were not different from quiescent fibroblasts in our studies. However, a recent paper has reported that ADM inhibited collagen synthesis by presumably quiescent human fetal lung fibroblasts in response to TGF-β (17). This data contrasts to our results in that addition of ADM to human adult lung fibroblasts shortly before TGF-β treatment (Fig. 3A) or even 8 h post-TGF-β treatment (data not shown) had no effect on TGF-β-induced production of collagen, fibronectin, or SM-α-actin; only when myofibroblasts start to become differentiated do they begin responding to ADM in terms of decreased expression of these differentiation markers (Figs. 4 and 5). Moreover, we have reproduced our results using separate primary cultures from three human lungs (data not shown). It is possible that this discrepancy could be due to differences in CRLR expression levels between the fetal lung fibroblasts in their study (17) and the adult lung fibroblasts in our experiments. One could wonder whether TGF-β treatment would further increase the responsiveness of fetal lung fibroblasts to ADM. Of importance is the data published by others showing that ADM had no effect on collagen and SM-α-actin production by quiescent cardiac fibroblasts unless adenylyl cyclase 6 (AC6) is overexpressed in these cells (54), which is consistent with our results.
We also report for the first time that CRLR expression is dramatically increased by TGF-β in human pulmonary fibroblasts and in mouse lungs during bleomycin-induced pulmonary fibrosis (Fig. 2). Importantly, the time course of CRLR expression paralleled that of increased PKA responsiveness to ADM (Fig. 2, A and B), and attenuation of CRLR expression by the TGF-β receptor kinase inhibitor SB431542 resulted in decreased PKA responses of cells to ADM but not to iloprost (Fig. 2, C and D), suggesting a causative relationship between these processes. Previous studies have shown that CRLR expression is induced by hypoxia in a manner dependent on hypoxia-inducible factor 1α (HIF-1α), driving the CRLR promoter through the HIF-response element (HRE) (40). Given that TGF-β can stabilize HIF-1α protein under normoxic conditions in vascular smooth muscle cells (16) and in HepG2 or HT1080 cells (36), it is possible that HIF-1α mediates TGF-β-induced CRLR expression in human pulmonary fibroblasts, which will be investigated in the future.
While our studies were in progress, another group reported that intraperitoneal administration of ADM attenuated the development of bleomycin-induced pulmonary fibrosis in mice (8). In that study, ADM administration was associated with a dramatic reduction of cytokines, such as tumor necrosis factor-α, interleukin-1β, and TGF-β1 in bleomycin-treated mice. Since ADM is known to modulate the activity of the immune system (14, 15, 66), this is a likely mechanism for the protective effect of ADM in their study (8). Our in vivo experiments utilized transgenic overexpression of the CRLR co-activator RAMP2 in SM α-actin-expressing cells, which has been shown to increase the density of functional adrenomedullin receptors and sensitize vascular smooth muscle responses to ADM (55). We found that these mice showed an increased survival and a significantly attenuated fibrosis induced by bleomycin administration (Fig. 6). Although our results are consistent with the reported protective role for ADM (8), our model has key differences and is likely to involve a distinct mechanism for protection against fibrosis. Because the RAMP2 transgene is under the SM-α-actin promoter, it is expected to express in smooth muscle cells and in myofibroblasts but not in immune cells. Therefore, it is unlikely that the cytokine component is affected in our model as much as it was attenuated after systemic intraperitoneal administration of ADM (8). In accordance with this notion, the initial weight loss of mice after bleomycin administration was not different between the WT and RAMP2 transgenic mice (data not shown), indirectly suggesting that the response to injury was unaffected. Regarding a potential contribution of the cardiovascular component, it has been shown that both basal and angiotensin II-induced increase in blood pressure, as well as the overall cardiovascular profile, are similar between the WT and RAMP2-transgenic mice, unless challenged with ADM (55). Given that, in our experiments, no treatment with ectopic ADM was performed and the levels of endogenous ADM mRNA in mouse lungs were unchanged upon bleomycin treatment (data not shown), we cautiously postulate that the protective effect of the RAMP2 transgene can be largely attributed to alterations in myofibroblast function, at least in the bleomycin model of pulmonary fibrosis.
Previous studies have demonstrated the increased sensitivity of vascular smooth muscle cells from RAMP2 transgenic mice to ADM but not to CRLR/RAMP1-specific ligand, calcitonin gene-related peptide (CGRP) (55), suggesting a selective role for endogenous ADM in the protective effect of RAMP2 transgene on the development of pulmonary fibrosis in our mouse model. However, RAMP2 can also interact with vasoactive intestinal polypeptide/pituitary adenylate cyclase activating peptide receptor (VPAC1), parathyroid hormone receptor (PTH1), or glucagon receptor (4). In the case of VPAC1, RAMP2 had no effect on ligand binding affinity but modulated the specificity of G-protein signaling by promoting phospho-inositide generation without affecting cAMP production in response to various VPAC1 ligands (4). Therefore, the precise role of ADM or other potential ligands for RAMP2-modified receptors in regulation of pulmonary fibrosis remains to be determined.
GRANTS
This study was supported by National Institutes of Health Awards R01 GM-85058 (N. O. Dulin), K08 HL-093367-01A1 (N. Sandbo), T32 HL-007237 (J. Kach), and T32 HD-009007 (N. Sandbo), and American Heart Association Fellowships 10PRE4190120 (J. Kach) and 10PRE2630163 (N. Sandbo).
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.K., S.D.B., A.I.S., K.G.B., and N.O.D. conception and design of research; J.K., N. Sandbo, N. Sethakorn, J.W., E.B.R., J.L., X.T., K.R., and N.O.D. performed experiments; J.K., N. Sandbo, N. Sethakorn, J.W., E.B.R., J.L., X.T., K.R., R.K., A.I.S., K.G.B., and N.O.D. analyzed data; J.K., N. Sandbo, J.W., E.B.R., J.L., X.T., K.R., R.K., A.I.S., K.G.B., and N.O.D. interpreted results of experiments; J.K., E.B.R., J.L., X.T., K.R., R.K., A.I.S., K.G.B., and N.O.D. prepared figures; J.K. drafted manuscript; J.K., A.I.S., K.G.B., and N.O.D. edited and revised manuscript; J.K., N. Sandbo, N. Sethakorn, J.W., E.B.R., J.L., X.T., S.D.B., K.R., R.K., A.I.S., K.G.B., and N.O.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Timo Hautala for providing adenovirus encoding adrenomedullin and Dr. Walter Born for providing RAMP2 transgenic mice.
REFERENCES
- 1. Broekelmann TJ, Limper AH, Colby TV, McDonald JA. Transforming growth factor β 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc Natl Acad Sci USA 88: 6642–6646, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Butler JP, Tolic-Norrelykke IM, Fabry B, Fredberg JJ. Traction fields, moments, and strain energy that cells exert on their surroundings. Am J Physiol Cell Physiol 282: C595–C605, 2002 [DOI] [PubMed] [Google Scholar]
- 3. Chao J, Kato K, Zhang JJ, Dobrzynski E, Wang C, Agata J, Chao L. Human adrenomedullin gene delivery protects against cardiovascular remodeling and renal injury. Peptides 22: 1731–1737, 2001 [DOI] [PubMed] [Google Scholar]
- 4. Christopoulos A, Christopoulos G, Morfis M, Udawela M, Laburthe M, Couvineau A, Kuwasako K, Tilakaratne N, Sexton PM. Novel receptor partners and function of receptor activity-modifying proteins. J Biol Chem 278: 3293–3297, 2003 [DOI] [PubMed] [Google Scholar]
- 5. Dackor RT, Cheng J, Voltz JW, Card JW, Ferguson CD, Garrett RC, Bradbury JA, DeGraff LM, Lih FB, Tomer KB, Flake GP, Travlos GS, Ramsey RW, Jr, Edin ML, Morgan DL, Zeldin DC. Prostaglandin E(2) protects murine lungs from bleomycin-induced pulmonary fibrosis and lung dysfunction. Am J Physiol Lung Cell Mol Physiol 301: L645–L655, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davis A, Hogarth K, Fernandes D, Solway J, Niu J, Kolenko V, Browning D, Miano JM, Orlov SN, Dulin NO. Functional significance of protein kinase A activation by endothelin-1 and ATP: negative regulation of SRF-dependent gene expression by PKA. Cell Signal 15: 597–604, 2003 [DOI] [PubMed] [Google Scholar]
- 7. Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen 13: 7–12, 2005 [DOI] [PubMed] [Google Scholar]
- 8. Di Paola R, Talero E, Galuppo M, Mazzon E, Bramanti P, Motilva V, Cuzzocrea S. Adrenomedullin in inflammatory process associated with experimental pulmonary fibrosis. Respir Res 12: 41, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dobrzynski E, Wang C, Chao J, Chao L. Adrenomedullin gene delivery attenuates hypertension, cardiac remodeling, and renal injury in deoxycorticosterone acetate-salt hypertensive rats. Hypertension 36: 995–1001, 2000 [DOI] [PubMed] [Google Scholar]
- 10. Eitzman DT, McCoy RD, Zheng X, Fay WP, Shen T, Ginsburg D, Simon RH. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J Clin Invest 97: 232–237, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eto T. A review of the biological properties and clinical implications of adrenomedullin and proadrenomedullin N-terminal 20 peptide (PAMP), hypotensive and vasodilating peptides. Peptides 22: 1693–1711, 2001 [DOI] [PubMed] [Google Scholar]
- 12. Evans JN, Kelley J, Krill J, Low RB, Adler KB. The myofibroblast in pulmonary fibrosis. Chest 83: 97S–98S, 1983 [DOI] [PubMed] [Google Scholar]
- 13. Fine A, Goldstein RH. The effect of PGE2 on the activation of quiescent lung fibroblasts. Prostaglandins 33: 903–913, 1987 [DOI] [PubMed] [Google Scholar]
- 14. Gonzalez-Rey E, Chorny A, O'Valle F, Delgado M. Adrenomedullin protects from experimental arthritis by down-regulating inflammation and Th1 response and inducing regulatory T cells. Am J Pathol 170: 263–271, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gonzalez-Rey E, Chorny A, Varela N, Robledo G, Delgado M. Urocortin and adrenomedullin prevent lethal endotoxemia by down-regulating the inflammatory response. Am J Pathol 168: 1921–1930, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gorlach A, Diebold I, Schini-Kerth VB, Berchner-Pfannschmidt U, Roth U, Brandes RP, Kietzmann T, Busse R. Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: role of the p22(phox)-containing NADPH oxidase. Circ Res 89: 47–54, 2001 [DOI] [PubMed] [Google Scholar]
- 17. Hao SL, Yu ZH, Qi BS, Luo JZ, Wang WP. The antifibrosis effect of adrenomedullin in human lung fibroblasts. Exp Lung Res 37: 615–626 [DOI] [PubMed] [Google Scholar]
- 18. Hardie WD, Glasser SW, Hagood JS. Emerging concepts in the pathogenesis of lung fibrosis. Am J Pathol 175: 3–16, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hayakawa H, Hirata Y, Kakoki M, Suzuki Y, Nishimatsu H, Nagata D, Suzuki E, Kikuchi K, Nagano T, Kangawa K, Matsuo H, Sugimoto T, Omata M. Role of nitric oxide-cGMP pathway in adrenomedullin-induced vasodilation in the rat. Hypertension 33: 689–693, 1999 [DOI] [PubMed] [Google Scholar]
- 20. Hinz B. Masters and servants of the force: the role of matrix adhesions in myofibroblast force perception and transmission. Eur J Cell Biol 85: 175–181, 2006 [DOI] [PubMed] [Google Scholar]
- 21. Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function multiple origins. Am J Pathol 170: 1807–1816, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang S, Wettlaufer SH, Hogaboam C, Aronoff DM, Peters-Golden M. Prostaglandin E(2) inhibits collagen expression and proliferation in patient-derived normal lung fibroblasts via E prostanoid 2 receptor and cAMP signaling. Am J Physiol Lung Cell Mol Physiol 292: L405–L413, 2007 [DOI] [PubMed] [Google Scholar]
- 23. Huang SK, Wettlaufer SH, Chung J, Peters-Golden M. Prostaglandin E2 inhibits specific lung fibroblast functions via selective actions of PKA and Epac-1. Am J Respir Cell Mol Biol 39: 482–489, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Itoh T, Obata H, Murakami S, Hamada K, Kangawa K, Kimura H, Nagaya N. Adrenomedullin ameliorates lipopolysaccharide-induced acute lung injury in rats. Am J Physiol Lung Cell Mol Physiol 293: L446–L452, 2007 [DOI] [PubMed] [Google Scholar]
- 25. Kaminski N, Allard JD, Pittet JF, Zuo F, Griffiths MJ, Morris D, Huang X, Sheppard D, Heller RA. Global analysis of gene expression in pulmonary fibrosis reveals distinct programs regulating lung inflammation and fibrosis. Proc Natl Acad Sci USA 97: 1778–1783, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kano H, Kohno M, Yasunari K, Yokokawa K, Horio T, Ikeda M, Minami M, Hanehira T, Takeda T, Yoshikawa J. Adrenomedullin as a novel antiproliferative factor of vascular smooth muscle cells. J Hypertens 14: 209–213, 1996 [DOI] [PubMed] [Google Scholar]
- 27. Kohyama T, Ertl RF, Valenti V, Spurzem J, Kawamoto M, Nakamura Y, Veys T, Allegra L, Romberger D, Rennard SI. Prostaglandin E(2) inhibits fibroblast chemotaxis. Am J Physiol Lung Cell Mol Physiol 281: L1257–L1263, 2001 [DOI] [PubMed] [Google Scholar]
- 28. Kolodsick JE, Peters-Golden M, Larios J, Toews GB, Thannickal VJ, Moore BB. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol 29: 537–544, 2003 [DOI] [PubMed] [Google Scholar]
- 29. Krishnan R, Klumpers DD, Park CY, Rajendran K, Trepat X, van Bezu J, van Hinsbergh VW, Carman CV, Brain JD, Fredberg JJ, Butler JP, van Nieuw Amerongen GP. Substrate stiffening promotes endothelial monolayer disruption through enhanced physical forces. Am J Physiol Cell Physiol 300: C146–C154, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leask A, Abraham DJ. TGF-β signaling and the fibrotic response. FASEB J 18: 816–827, 2004 [DOI] [PubMed] [Google Scholar]
- 31. Leask A, Holmes A, Abraham DJ. Connective tissue growth factor: a new and important player in the pathogenesis of fibrosis. Curr Rheumatol Rep 4: 136–142, 2002 [DOI] [PubMed] [Google Scholar]
- 32. Liang L, Tam CW, Pozsgai G, Siow R, Clark N, Keeble J, Husmann K, Born W, Fischer JA, Poston R, Shah A, Brain SD. Protection of angiotensin II-induced vascular hypertrophy in vascular smooth muscle-targeted receptor activity-modifying protein 2 transgenic mice. Hypertension 54: 1254–1261, 2009 [DOI] [PubMed] [Google Scholar]
- 33. Liu X, Ostrom RS, Insel PA. cAMP-elevating agents and adenylyl cyclase overexpression promote an antifibrotic phenotype in pulmonary fibroblasts. Am J Physiol Cell Physiol 286: C1089–C1099, 2004 [DOI] [PubMed] [Google Scholar]
- 34. Matsui H, Shimosawa T, Itakura K, Guanqun X, Ando K, Fujita T. Adrenomedullin can protect against pulmonary vascular remodeling induced by hypoxia. Circulation 109: 2246–2251, 2004 [DOI] [PubMed] [Google Scholar]
- 35. McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, Solari R, Lee MG, Foord SM. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature 393: 333–339, 1998 [DOI] [PubMed] [Google Scholar]
- 36. McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM. Transforming growth factor β1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem 281: 24171–24181, 2006 [DOI] [PubMed] [Google Scholar]
- 37. Muller HC, Witzenrath M, Tschernig T, Gutbier B, Hippenstiel S, Santel A, Suttorp N, Rosseau S. Adrenomedullin attenuates ventilator-induced lung injury in mice. Thorax 65: 1077–1084 [DOI] [PubMed] [Google Scholar]
- 38. Nagaya N, Kyotani S, Uematsu M, Ueno K, Oya H, Nakanishi N, Shirai M, Mori H, Miyatake K, Kangawa K. Effects of adrenomedullin inhalation on hemodynamics and exercise capacity in patients with idiopathic pulmonary arterial hypertension. Circulation 109: 351–356, 2004 [DOI] [PubMed] [Google Scholar]
- 39. Nagaya N, Okumura H, Uematsu M, Shimizu W, Ono F, Shirai M, Mori H, Miyatake K, Kangawa K. Repeated inhalation of adrenomedullin ameliorates pulmonary hypertension and survival in monocrotaline rats. Am J Physiol Heart Circ Physiol 285: H2125–H2131, 2003 [DOI] [PubMed] [Google Scholar]
- 40. Nikitenko LL, Smith DM, Bicknell R, Rees MC. Transcriptional regulation of the CRLR gene in human microvascular endothelial cells by hypoxia. FASEB J 17: 1499–1501, 2003 [DOI] [PubMed] [Google Scholar]
- 41. Nishikimi T, Yoshihara F, Mori Y, Kangawa K, Matsuoka H. Cardioprotective effect of adrenomedullin in heart failure. Hypertens Res 26, Suppl: S121–127, 2003 [DOI] [PubMed] [Google Scholar]
- 42. Niu P, Shindo T, Iwata H, Iimuro S, Takeda N, Zhang Y, Ebihara A, Suematsu Y, Kangawa K, Hirata Y, Nagai R. Protective effects of endogenous adrenomedullin on cardiac hypertrophy, fibrosis, and renal damage. Circulation 109: 1789–1794, 2004 [DOI] [PubMed] [Google Scholar]
- 43. Noble PW, Homer RJ. Back to the future: historical perspective on the pathogenesis of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 33: 113–120, 2005 [DOI] [PubMed] [Google Scholar]
- 44. Okumura H, Nagaya N, Kangawa K. Adrenomedullin infusion during ischemia/reperfusion attenuates left ventricular remodeling and myocardial fibrosis in rats. Hypertens Res 26, Suppl: S99–S104, 2003 [DOI] [PubMed] [Google Scholar]
- 45. Olson AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, Brown KK. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med 176: 277–284, 2007 [DOI] [PubMed] [Google Scholar]
- 46. Phan SH. Fibroblast phenotypes in pulmonary fibrosis. Am J Respir Cell Mol Biol 29: S87–S92, 2003 [PubMed] [Google Scholar]
- 47. Phan SH. Role of the myofibroblast in pulmonary fibrosis. Kidney Int Suppl 54: S46–S48, 1996 [PubMed] [Google Scholar]
- 48. Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West Myofibroblasts AB., I Paracrine cells important in health and disease. Am J Physiol Cell Physiol 277: C1–C9, 1999 [DOI] [PubMed] [Google Scholar]
- 49. Rauma-Pinola T, Paakko P, Ilves M, Serpi R, Romppanen H, Vuolteenaho O, Ruskoaho H, Hautala T. Adrenomedullin gene transfer induces neointimal apoptosis and inhibits neointimal hyperplasia in injured rat artery. J Gene Med 8: 452–458, 2006 [DOI] [PubMed] [Google Scholar]
- 50. Sandbo N, Kregel S, Taurin S, Bhorade S, Dulin NO. Critical role of serum response factor in pulmonary myofibroblast differentiation induced by TGF-β. Am J Respir Cell Mol Biol 41: 332–338, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Selman M, Pardo A. Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-talk disorder. Respir Res 3: 3, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shimekake Y, Nagata K, Ohta S, Kambayashi Y, Teraoka H, Kitamura K, Eto T, Kangawa K, Matsuo H. Adrenomedullin stimulates two signal transduction pathways, cAMP accumulation and Ca2+ mobilization, in bovine aortic endothelial cells. J Biol Chem 270: 4412–4417, 1995 [DOI] [PubMed] [Google Scholar]
- 53. Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-β1 induces prolonged severe fibrosis in rat lung. J Clin Invest 100: 768–776, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc Natl Acad Sci USA 102: 437–442, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tam CW, Husmann K, Clark NC, Clark JE, Lazar Z, Ittner LM, Gotz J, Douglas G, Grant AD, Sugden D, Poston L, Poston R, McFadzean I, Marber MS, Fischer JA, Born W, Brain SD. Enhanced vascular responses to adrenomedullin in mice overexpressing receptor-activity-modifying protein 2. Circ Res 98: 262–270, 2006 [DOI] [PubMed] [Google Scholar]
- 56. Tambe DT, Hardin CC, Angelini TE, Rajendran K, Park CY, Serra-Picamal X, Zhou EH, Zaman MH, Butler JP, Weitz DA, Fredberg JJ, Trepat X. Collective cell guidance by cooperative intercellular forces. Nature Materials 10: 469–475, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tao W, Shu YS, Miao QB, Zhu YB. Attenuation of hyperoxia-induced lung injury in rats by adrenomedullin. Inflammation 35: 150–157, 2012 [DOI] [PubMed] [Google Scholar]
- 58. Thannickal VJ, Toews GB, White ES, Lynch JP, 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med 55: 395–417, 2004 [DOI] [PubMed] [Google Scholar]
- 59. Thomas PE, Peters-Golden M, White ES, Thannickal VJ, Moore BB. PGE(2) inhibition of TGF-β1-induced myofibroblast differentiation is Smad-independent but involves cell shape and adhesion-dependent signaling. Am J Physiol Lung Cell Mol Physiol 293: L417–L428, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3: 349–363, 2002 [DOI] [PubMed] [Google Scholar]
- 61. Trepat X, Wasserman MR, Angelini TE, Millet E, Weitz DA, Butler JP, Fredberg JJ. Physical forces during collective cell migration. Nature Physics 5: 426–430, 2009 [Google Scholar]
- 62. Wang C, Dobrzynski E, Chao J, Chao L. Adrenomedullin gene delivery attenuates renal damage and cardiac hypertrophy in Goldblatt hypertensive rats. Am J Physiol Renal Physiol 280: F964–F971, 2001 [DOI] [PubMed] [Google Scholar]
- 63. Wei X, Zhao C, Jiang J, Li J, Xiao X, Wang DW. Adrenomedullin gene delivery alleviates hypertension and its secondary injuries of cardiovascular system. Hum Gene Ther 16: 372–380, 2005 [DOI] [PubMed] [Google Scholar]
- 64. White ES, Lazar MH, Thannickal VJ. Pathogenetic mechanisms in usual interstitial pneumonia/idiopathic pulmonary fibrosis. J Pathol 201: 343–354, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Withers DJ, Coppock HA, Seufferlein T, Smith DM, Bloom SR, Rozengurt E. Adrenomedullin stimulates DNA synthesis and cell proliferation via elevation of cAMP in Swiss 3T3 cells. FEBS Lett 378: 83–87, 1996 [DOI] [PubMed] [Google Scholar]
- 66. Wong LY, Cheung BM, Li YY, Tang F. Adrenomedullin is both proinflammatory and antiinflammatory: its effects on gene expression and secretion of cytokines and macrophage migration inhibitory factor in NR8383 macrophage cell line. Endocrinology 146: 1321–1327, 2005 [DOI] [PubMed] [Google Scholar]
- 67. Yamamoto H, Nagase T, Shindo T, Teramoto S, Aoki-Nagase T, Yamaguchi Y, Hanaoka Y, Kurihara H, Ouchi Y. Adrenomedullin insufficiency increases allergen-induced airway hyperresponsiveness in mice. J Appl Physiol 102: 2361–2368, 2007 [DOI] [PubMed] [Google Scholar]
- 68. Zhu Y, Liu Y, Zhou W, Xiang R, Jiang L, Huang K, Xiao Y, Guo Z, Gao J. A prostacyclin analogue, iloprost, protects from bleomycin-induced pulmonary fibrosis in mice. Respir Res 11: 34, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]