

Abstract
Lung inflammation can result from exposure to multiple types of inflammatory stimuli. Fibroblasts, key structural cells in the lung that are integral to inflammation and wound healing, produce inflammatory mediators after exposure to stimuli such as IL-1β. We and others have shown that the NF-κB member RelB has anti-inflammatory properties in mice. Little is known, however, about the anti-inflammatory role of RelB in human cells and how it functions. MicroRNAs (miRNAs), a novel class of small, noncoding RNAs, can mediate inflammatory signaling pathways, including NF-κB, through regulation of target gene expression. Our goal was to analyze the anti-inflammatory properties of RelB in human lung fibroblasts. We hypothesized that RelB regulates inflammatory mediator production in lung fibroblasts in part through a mechanism involving miRNAs. To accomplish this, we transfected human lung fibroblasts with a plasmid encoding RelB and small interfering (si)RNA targeting RelB mRNA to overexpress and downregulate RelB, respectively. IL-1β, a powerful proinflammatory stimulus, was used to induce NF-κB-driven inflammatory responses. RelB overexpression reduced IL-1β-induced cyclooxygenase (Cox)-2, PGE2, and cytokine production, and RelB downregulation increased Cox-2 expression and PGE2 production. Furthermore, RelB overexpression increased IL-1β-induced expression of miRNA-146a, an NF-κB-dependent miRNA with anti-inflammatory properties, whereas RelB downregulation reduced miRNA-146a. miR-146a overexpression ablated the effects of RelB downregulation on IL-1β-induced Cox-2, PGE2, and IL-6 production, suggesting that RelB mediates IL-1β-induced inflammatory mediator production in lung fibroblasts through miRNA-146a. RelB and miRNA-146a may therefore be new therapeutic targets in the treatment of lung inflammation caused by various agents and conditions.
Keywords: inflammation, fibroblasts, nuclear factor-κB, RelB, microRNA-146a
inflammation, the normal physiological response to tissue damage and disruptions in homeostasis, may arise in the lung after exposure to various inhaled insults (14, 34). Stimulation of the inflammatory response promotes the accumulation of leukocytes, particularly neutrophils, macrophages, and lymphocytes, at the site of insult (14, 34). Inflammatory stimuli also induce the production of proinflammatory cytokines and other mediators such as cyclooxygenase (Cox)-2 (43, 44), which increases the production of proinflammatory eicosanoids such as PGE2 (3, 4, 28). Importantly, prolonged synthesis of these mediators can promote chronic inflammation and the pathogenesis of inflammatory diseases (50).
Lung fibroblasts, previously thought to be passive structural cells, are now recognized as crucial cells that incite inflammation and promote wound healing (1, 3, 4, 39). In addition to producing and depositing collagen, fibroblasts produce inflammatory mediators, including PGE2 and various cytokines and chemokines, such as IL-6, IL-8, and monocyte chemoattractant protein (MCP)-1, that regulate other cell types and promote inflammatory cell infiltration and activation (4, 28, 39). Stimulation of fibroblasts therefore heightens the sensitivity of the tissues in which they reside and can promote disease pathogenesis (4). Numerous signaling pathways are involved in these processes, but the NF-κB family is intimately linked to inflammation and the manifestation of diseases (7, 8).
The NF-κB signaling pathway is integral to numerous cellular processes, including inflammation, cell growth, and differentiation (8, 16). Activation of the canonical NF-κB pathway typically induces inflammation, but the noncanonical NF-κB member RelB has been reported to possess potent anti-inflammatory properties (3, 29, 43). RelB-deficient mice exhibit severe multiorgan inflammation, particularly in the lung (46), and kidney fibroblasts from RelB-deficient mice exhibit a heightened sensitivity to inflammatory stimuli (49). Furthermore, we have previously shown that RelB dampens cigarette smoke extract-induced PGE2 production in mouse lung fibroblasts (3) and that targeted overexpression of RelB ablates cigarette smoke-induced lung inflammation in mice in vivo (29). Little is known, however, about the anti-inflammatory properties of RelB in human cells, and the mechanisms underlying these properties remain poorly understood.
MicroRNAs (miRNAs) have emerged as a novel class of RNA molecules involved in numerous cellular functions, including cell growth, proliferation, and apoptosis (31, 36). Synthesized as large precursor RNAs and enzymatically converted into small, ∼22-nt fragments, mature miRNAs regulate gene expression by blocking mRNA translation and promoting mRNA degradation (31, 33). miRNA expression is upregulated during inflammation, and key miRNAs regulate inflammatory mediator production (31). Several miRNAs are also targets of NF-κB, and others regulate the translation of NF-κB proteins (25, 26, 41, 51). miRNAs may therefore be key players in RelB-mediated regulation of inflammation.
The present study was aimed at testing the anti-inflammatory effects of RelB in a more potentially clinically relevant human model of inflammatory mediator production and identifying the mechanism(s) responsible for these effects. We hypothesized that RelB regulates inflammatory mediator production in human lung fibroblasts through a mechanism involving miRNAs. Establishing a link between RelB and miRNAs in the context of inflammatory mediator production would provide necessary insights into the development of novel therapeutic strategies for inflammation and, potentially, diseases of the lung.
METHODS
Cell culture.
Normal adult human lung fibroblasts were obtained and characterized as previously described (19) and used between passages 4 and 10. Cultures were maintained at 37°C in humidified 5% CO2-95% air and cultured in minimum essential medium (MEM; Invitrogen, Carlsbad, CA) supplemented with 10% FBS (HyClone Laboratories, Logan, UT) and 0.1% gentamicin (15750-060, Invitrogen) until confluent. To induce inflammatory mediator production, fibroblasts were incubated in serum-free MEM for 24 h and treated with IL-1β (201-LB, R&D Systems, Minneapolis, MN) at either 1 ng/ml or 50 pg/ml in serum-free MEM for 8 h.
Plasmid transfections.
To transiently overexpress RelB, a plasmid containing the human RelB gene fused to green fluorescent protein (GFP; EX-G0029-M03, GeneCopoeia, Germantown, MD) was used. A GFP-only plasmid was used as a control (EX-EGFP-M03, GeneCopoeia). Fibroblasts (1 × 106 cells) were trypsinized, suspended in Ingenio Electroporation solution (MIR 50115, Mirus Bio, Madison, WI) with 14 μg DNA, transfected using a Nucleofector II Device (Lonza, Basel, Switzerland), and incubated for 24 h in MEM containing 10% FBS under normal culture conditions. Cells were then incubated in serum-free MEM for 24 h before IL-1β treatment as described above.
Small interfering RNA and miRNA mimic transfections.
RelB small interfering (si)RNA 1 and 2 were purchased from Ambion's Silencer Select predesigned siRNA library (s11917 and s11918, respectively, Ambion, Grand Island, NY). A Silencer Select Negative Control siRNA was used as a control (4390846, Ambion). miRNA-146a (miR-146a) and control mimics were also obtained from Ambion (4464066-MC10722 and AM17110, respectively). Cells were grown to 70% confluence in six-well plates as described above and treated with siRNAs and/or miRNA mimics mixed with Lipofectamine 2000 (11688-019, Invitrogen) in OptiMEM I (Invitrogen) at 100 μM for 24 h according to the manufacturers' instructions. Cells were incubated in serum-free MEM for a further 24 h before IL-1β treatment as described above.
Western blot analysis.
Cells were homogenized with lysis buffer containing SDS and a protease inhibitor cocktail (P8340, Sigma-Aldrich, St. Louis, MO). Total protein concentration was determined with a bicinchoninic acid detection assay (Pierce, Rockford, IL). Protein (1 μg) was separated via 8% SDS-PAGE, transferred onto Immobilon-P (Millipore), and blocked with 10% nonfat dry milk in 0.1% Tween 20 in PBS. Antibodies targeting Cox-2 (160106, Cayman Chemical, Ann Arbor, MI), RelB (sc-226, Santa Cruz Biotechnology, Santa Cruz, CA), β-tubulin (2146, Cell Signaling Biotechnology, Beverly, MA), and total actin (CP01, Calbiochem, San Diego, CA) were diluted according to the manufacturers' instructions. Horseradish peroxidase-conjugated secondary antibodies were obtained from Jackson Immunoresearch (West Grove, PA). Protein was visualized using Immobilon Western chemiluminescent horseradish peroxidase substrate (Millipore, Billerica, MA) and developed on Classic blue-sensitive X-ray film (Laboratory Product Sales, Rochester, NY) with an X-OMAT 1000A film developer (Eastman Kodak, Rochester, NY). Densitometric analysis was performed with Discovery Series Quantity One 1-D analysis software (Bio-Rad, Hercules, CA).
Cytokine and PGE2 detection.
Cell culture supernatants were collected at the conclusion of each experiment. IL-6, IL-8, and MCP-1 were detected in the supernatants by ELISA as previously described (48). IL-6 antibodies were purchased from BD Pharmingen (554543 and 554546, San Diego, CA). IL-8 antibodies were purchased from Pierce (M-801 and M-802-B). MCP-1 antibodies were purchased from R&D Systems (MAB679 and BAF279). IL-6 and MCP-1 standards were purchased from R&D Systems (206-IL and 279-MC, respectively). The IL-8 standard was purchased from GIBCO (PHC0884, Carlsbad, CA). PGE2 was detected by competitive enzyme immunoassay as previously described (20).
RNA extraction and quantitative RT-PCR.
Total cell RNA was extracted using TRIzol lysis reagent (79306, Qiagen, Valencia, CA) and isolated with a miRNeasy Mini kit according to the manufacturer's instructions (217004, Qiagen). Total RNA concentrations were determined with a NanoDrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE). cDNA was generated using a TaqMan microRNA reverse transcription kit (4366596, Applied Biosystems) and quantified via real-time PCR with a TaqMan Universal PCR Master Mix (4324018, Applied Biosystems) and an iCycler iQ5 PCR thermal cycler (Bio-Rad). miRNA primers were also obtained from Applied Biosystems (miR-21: 4427975-000397, miR-101: 4427975-002253, miR-125a: 4427975-002198, miR-146a: 4427975-000468, and U6: 4395470-001973).
Statistical analysis.
Statistical analyses were performed with GraphPad Prism (version 4.0, GraphPad Software, San Diego, CA). All results are presented as means ± SE. Statistically significant differences were assessed with two-way ANOVA and a Bonferroni post hoc test.
RESULTS
RelB overexpression and downregulation were achieved in human lung fibroblasts.
We first established an in vitro model in which we could test the potential anti-inflammatory role of RelB in human lung fibroblasts. To accomplish this, we determined that modulation of RelB expression could be accomplished in human lung fibroblasts in vitro. To overexpress RelB, a plasmid containing the human RelB gene fused to GFP was introduced via transfection, and whole cell protein lysates were collected after a 24-h incubation under normal culture conditions. Control groups received a GFP-only plasmid. No noticeable differences were seen between untransfected and GFP-transfected cells. As shown in Fig. 1A, RelB overexpression was detected by Western blot analysis after RelB-GFP gene delivery. GFP expression was observed by immunofluorescent microscopy 24 h posttransfection (data not shown).
Fig. 1.

RelB overexpression and downregulation was achieved in human lung fibroblasts. A: normal adult human lung fibroblasts were transfected with either a green fluorescent protein (GFP) control plasmid or a RelB-GFP expression plasmid, and total protein lysates were collected 48 h posttreatment for Western blot analysis. RelB overexpression was confirmed by the presence of a RelB-GFP band ∼27 kDa larger than RelB. All RelB and RelB-GFP bands were detected with the same antibody on the same gel and are shown separated by white space. B: normal adult human lung fibroblasts were transfected with either a control small interfering (si)RNA or a RelB siRNA, and total protein lysates were collected 48 h posttreatment. RelB downregulation was confirmed by Western blot analysis.
To decrease RelB expression, two siRNAs targeting different locations in human RelB mRNA were introduced into fibroblasts as described above. Control groups received a nonspecific, scrambled siRNA. After treatment, cells were incubated for 24 h under normal conditions, and RelB expression was detected by Western blot analysis. The results shown in Fig. 1B demonstrate that RelB expression could be dramatically reduced with both siRNAs. All subsequent siRNA experiments were conducted using both RelB siRNAs separately, and both siRNAs produced comparable results, thereby excluding the possibility of off-target effects. Collectively, these data show that both RelB overexpression and downregulation can be achieved in human lung fibroblasts in vitro using these techniques. No noticeable changes in cell growth or viability were observed in our experiments.
RelB overexpression decreased IL-1β-induced proinflammatory mediator production in human lung fibroblasts.
To determine whether or not RelB had anti-inflammatory effects in human lung fibroblasts, we first tested the ability of RelB overexpression to dampen inflammatory mediator production in primary human lung fibroblasts in vitro. Cells were transfected with either a RelB-GFP plasmid or a GFP-only control plasmid as described above and were incubated for 24 h to allow for gene expression. Cells were serum starved for an additional 24 h and subsequently treated with IL-1β at 1 ng/ml for 8 h to induce inflammatory mediator production. After treatment, whole cell protein extracts and cell supernatants were collected for analysis, and Cox-2 expression was analyzed by Western blot analysis (Fig. 2A). IL-1β significantly increased Cox-2 protein expression, but RelB overexpression significantly ablated this increase by approximately threefold (Fig. 2B). Furthermore, IL-1β-induced PGE2 was significantly decreased after RelB overexpression (Fig. 2C). Proinflammatory cytokine and chemokine production was detected in cell supernatants by ELISA. As shown in Fig. 3A, IL-1β-induced secretion of IL-6 was significantly reduced after RelB overexpression. IL-1β-induced production of neutrophil (IL-8) and macrophage (MCP-1) chemokines was also significantly reduced (Fig. 3, B and C, respectively).
Fig. 2.

RelB overexpression decreased IL-1β induced cyclooxygenase (Cox)-2 expression and PGE2 production in human lung fibroblasts. Normal adult human lung fibroblasts were transfected with either a GFP control plasmid or a RelB-GFP expression plasmid, serum starved for 24 h, and treated with IL-1β at 1 ng/ml for 8 h. A: Cox-2 and RelB expression was detected in whole cell protein extracts by Western blot analysis. IL-1β-induced Cox-2 expression was reduced in cells in which RelB expression was increased. The Western blots shown are representative of three separate experiments. Samples from each blot were transposed from different locations on the same gel and are shown separated by white space. RelB and RelB-GFP bands were also transposed from different locations on the same blot. B: densitometric analysis of all three blots confirmed a significant 8-fold increase in Cox-2 expression upon IL-1β treatment and a significant, ∼3-fold reduction in Cox-2 expression after RelB overexpression (n = 3/group). C: PGE2 production (detected in the supernatants of these cells) was significantly increased upon IL-1β treatment and significantly reduced upon RelB overexpression (n = 3/group). **P < 0.01 and ***P < 0.001 by two-way ANOVA with a Bonferroni post hoc test.
Fig. 3.
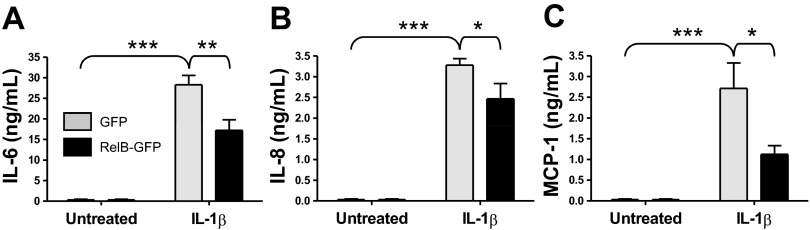
RelB overexpression decreased IL-1β induced proinflammatory cytokine and chemokine production in human lung fibroblasts. Normal adult human lung fibroblasts were transfected with either a GFP control plasmid or a RelB-GFP expression plasmid, serum starved for 24 h, and treated with IL-1β at 1 ng/ml for 8 h. Proinflammatory cytokine and chemokine production was detected in supernatants from these cells by ELISA. IL-1β treatment significantly increased the secretion of IL-6 (A), IL-8 (B), and monocyte chemoattractant protein (MCP)-1 (C), whereas RelB overexpression significantly reduced the release of each cytokine. *P < 0.05, **P < 0.01, and ***P < 0.001, two-way ANOVA with a Bonferroni post hoc test (n = 3/group).
RelB downregulation increased IL-1β-induced Cox-2 expression and PGE2 production in human lung fibroblasts.
Increasing RelB expression decreased IL-1β-induced inflammatory mediator production, so we next hypothesized that decreasing RelB expression would heighten inflammatory mediator production. Fibroblasts were treated with either a control siRNA or a RelB siRNA for 24 h as described above to decrease RelB expression, serum starved for an additional 24 h, and treated with IL-1β at 50 pg/ml for 8 h, after which whole cell protein extracts and cell supernatants were collected for analysis. Cox-2 and RelB expression were detected by Western blot analysis (Fig. 4A). Cells in which RelB expression was targeted by RelB siRNA exhibited heightened IL-1β-induced Cox-2 expression relative to IL-1β-treated control groups. Statistical significance was determined via densitometric analysis (Fig. 4B). IL-1β-induced PGE2 production was also significantly increased in RelB-depleted groups (Fig. 4C). Proinflammatory cytokine and chemokine production was detected in cell supernatants as described above. RelB downregulation, however, did not further enhance IL-1β-induced production of IL-8 or MCP-1 (Fig. 5, B and C). IL-6 production, while not significantly affected, trended upward in RelB siRNA-treated groups (Fig. 5A).
Fig. 4.

RelB downregulation increased IL-1β induced Cox-2 expression and PGE2 production in human lung fibroblasts. Normal adult human lung fibroblasts were transfected with either a control siRNA or RelB siRNA 1, serum starved for 24 h, and treated with IL-1β at 50 pg/ml for 8 h. A: Cox-2 and RelB expression was detected in whole cell protein extracts by Western blot analysis. IL-1β-induced Cox-2 expression was increased in cells in which RelB expression was decreased. The Western blots shown are representative of three separate experiments. Samples from each blot were transposed from different locations on the same gel and are shown separated by white space. B: densitometric analysis of all three blots confirmed a significant increase in Cox-2 expression after RelB downregulation. C: IL-1β-induced PGE2 production (detected in supernatants of these cells) was significantly increased upon RelB downregulation. Comparable results were obtained with RelB siRNA 2, which was used in separate experiments (not shown). **P < 0.01 and ***P < 0.001 by two-way ANOVA with a Bonferroni post hoc test (n = 3/group).
Fig. 5.
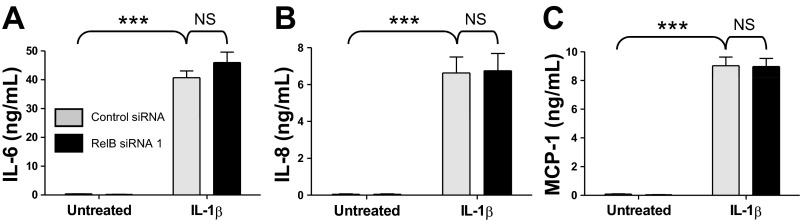
RelB downregulation did not significantly affect IL-1β-induced proinflammatory cytokine production in human lung fibroblasts. Normal adult human lung fibroblasts were transfected with either a control siRNA or RelB siRNA 1, serum starved for 24 h, and treated with IL-1β at 50 pg/ml for 8 h. Proinflammatory cytokine and chemokine production was detected in supernatants from these cells by ELISA. IL-1β-induced production of IL-6 (A), IL-8 (B), and MCP-1 (C) was not significantly affected upon RelB downregulation. Comparable results were obtained with RelB siRNA 2, which was used in separate experiments. ***P < 0.001 and not significant (NS) by two-way ANOVA with a Bonferroni post hoc test (n = 6/group).
RelB overexpression and downregulation affect IL-1β-induced miR-146a production in human lung fibroblasts.
We were next interested in determining if RelB expression regulated IL-1β-induced inflammatory mediator production through a mechanism involving miRNAs. Five miRNAs linked to inflammation, lung disease, and NF-κB (miR-21, miR-101, miR-125a, miR-146a, and miR-155) were initially selected for analysis in our RelB overexpression and downregulation models (17, 30, 36, 40). We hypothesized that overexpression of RelB would increase IL-1β-induced miRNA production in lung fibroblasts. Fibroblasts were transfected with either a RelB-GFP plasmid or a GFP-only plasmid and incubated for 24 h as described above, serum starved for 24 h, and treated with IL-1β at 1 ng/ml for 8 h. Expression of miR-21, miR-101, miR-125a, and miR-155 was not significantly affected by either IL-1β or RelB overexpression (Fig. 6). miR-146a expression, however, was significantly increased upon IL-1β treatment, and IL-1β-induced miR-146a expression was significantly heightened upon RelB overexpression.
Fig. 6.
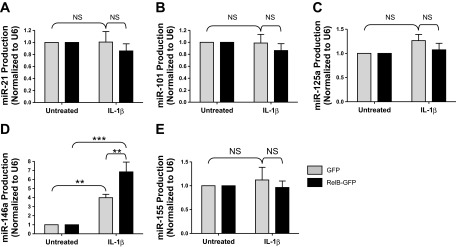
RelB overexpression increases IL-1β-induced miRNA-146a (miR-146a) production in human lung fibroblasts. Normal adult human lung fibroblasts were transfected with either a GFP control plasmid or a RelB-GFP expression plasmid, serum starved for 24 h, and treated with IL-1β at 1 ng/ml for 8 h. Total RNA was extracted, and miRNA production was detected by quantitative RT-PCR. Production of miR-146a was significantly increased upon IL-1β treatment, and IL-1β-induced miRNA-146a production was significantly increased upon RelB downregulation (D). miR-21, miR-101, miR-125a, and miR-155 were not significantly affected by either IL-1β or RelB overexpression in our model (A–C and E). *P < 0.05 and ***P < 0.001 by two-way ANOVA with a Bonferroni post hoc test (n = 3/group).
Since RelB overexpression increases IL-1β-induced miR-146a production, we next hypothesized that RelB downregulation would decrease IL-1β-induced miRNA production. Fibroblasts were treated with a RelB siRNA for 24 h to reduce RelB expression, serum starved for 24 h, and treated with IL-1β at 50 pg/ml for 8 h. Total RNA was extracted for analysis by quantitative RT-PCR. As shown in Fig. 7, RelB downregulation significantly reduced IL-1β-induced miR-146a production. The production of miR-21, miR-101, and miR-125a, however, was not significantly affected by either IL-1β or RelB downregulation in our model.
Fig. 7.

RelB downregulation decreased IL-1β induced miR-146a production in human lung fibroblasts. Normal adult human lung fibroblasts were transfected with either a control siRNA or RelB siRNA, serum starved for 24 h, and treated with IL-1β at 50 pg/ml for 8 h. Total RNA was extracted, and miRNA production was detected by quantitative RT-PCR. Production of miR-146a was significantly increased upon IL-1β treatment, whereas IL-1β-induced miRNA-146a production was significantly reduced upon RelB downregulation. miR-21, miR-101, and miR-125a were not significantly affected by either IL-1β or RelB downregulation in our model. *P < 0.05 and ***P < 0.001 by two-way ANOVA with a Bonferroni post hoc test (n = 3/group).
miR-146a overexpression dampened the effects of RelB downregulation on IL-1β-induced inflammatory mediator production in human lung fibroblasts.
To determine if the concurrent RelB-mediated changes in IL-1β-induced proinflammatory mediator production and miR-146a expression are linked, we overexpressed miR-146a in our RelB downregulation model. Fibroblasts were treated with a RelB siRNA and a miR-146a mimic simultaneously for 24 h, serum starved for 24 h, and treated with IL-1β at 50 pg/ml for 8 h. Total protein was extracted for Western blot analysis. As shown in Fig. 8A, RelB downregulation noticeably increased IL-1β-induced Cox-2 expression. Overexpression of miR-146a, however, dramatically reduced IL-1β-induced Cox-2 expression in both control fibroblasts and those in which RelB was downregulated. miR-146a also significantly reduced the effect of RelB downregulation on IL-1β-induced PGE2 production (Fig. 8B). IL-1β-induced IL-6 production was also significantly reduced in RelB-depleted fibroblasts after miR-146a overexpression (Fig. 8C), although IL-1β-induced IL-8 production was not significantly affected (Fig. 8D). miR-146a overexpression therefore dampened the effects of RelB downregulation on IL-1β-induced Cox-2 expression and PGE2 production, suggesting that RelB regulates IL-1β-induced production of these mediators in part through a mechanism involving miR-146a.
Fig. 8.

miR-146a overexpression dampened the effects of RelB downregulation on IL-1β-induced inflammatory mediator production in normal human lung fibroblasts. Normal adult human lung fibroblasts were transfected with either a control siRNA or RelB siRNA and simultaneously either a control mimic or a mimic for miR-146a. Cells were serum starved for 24 h and treated with IL-1β at 50 pg/ml for 8 h. Total protein was extracted and analyzed by Western blot analysis. A: IL-1β-induced Cox-2 expression was dramatically increased with RelB downregulation. Overexpression of miR-146a, however, dampened IL-1β-induced Cox-2 expression both under otherwise normal conditions and in the context of RelB downregulation. Samples from each blot were transposed from different locations on the same gel and are shown separated by white space. Untreated and IL-1β-treated groups, however, were analyzed on separate blots. B: IL-1β-induced PGE2 production (also increased upon RelB downregulation) was significantly reduced with miR-146a overexpression. C: IL-1β-induced IL-6 production was significantly reduced in RelB-depleted fibroblasts with miR-146a overexpression. D: IL-1β-induced IL-8 production was not significantly affected by either RelB downregulation or miR-146a overexpression. **P < 0.01 and NS by two-way ANOVA with a Bonferroni post hoc test (n = 3/group).
DISCUSSION
The present study demonstrates that RelB regulates IL-1β-induced proinflammatory mediator production in human lung fibroblasts in part through a mechanism involving miR-146a. RelB overexpression reduced IL-1β-induced Cox-2 expression, PGE2 production, and proinflammatory cytokine production, whereas RelB downregulation heightened Cox-2 expression and PGE2 production. IL-1β-induced secretion of IL-6, IL-8, and MCP-1, however, was not significantly affected by RelB downregulation. RelB downregulation also significantly decreased IL-1β-induced miR-146a production, and miR-146a overexpression ablated the stimulatory effects of RelB downregulation on IL-1β-induced Cox-2, PGE2, and IL-6 production.
The NF-κB pathway is responsible for numerous cellular processes, particularly inflammation (7, 9, 16). Activation of canonical NF-κB by cytokines such as IL-1β promotes inflammatory mediator production (8), but relatively little research has focused on the anti-inflammatory properties of the noncanonical NF-κB member RelB as well as the mechanisms underlying these properties. The present study was designed to test the anti-inflammatory effects of RelB against IL-1β, a robust and widely used inflammatory stimulus linked to multiple lung diseases (13, 18, 21, 47), in a human in vitro model using lung fibroblasts, a class of structural cells that are integral during inflammation, wound healing, and disease pathogenesis (1, 3, 4, 39). We also sought to elucidate the mechanism(s) linking RelB and inflammatory mediator production.
miRNAs have emerged as a novel class of RNA molecules involved in numerous cellular functions, including inflammation and lung disease (31, 33, 36). We hypothesized that RelB regulates IL-1β-induced inflammatory mediator production in lung fibroblasts through miRNAs, although we were unsure which miRNAs may be involved. The findings shown in Fig. 7 demonstrate that RelB downregulation significantly attenuated IL-1β-induced production of miR-146a, an NF-κB-dependent miRNA with anti-inflammatory properties (15, 36, 42), in human lung fibroblasts. Similarly, the results shown in Fig. 6 demonstrate that RelB overexpression significantly increased IL-1β-induced miR-146a production in human lung fibroblasts. miR-146a binds to the 3′-untranslated region of Cox-2 mRNA, and expression of miR-146a inhibits Cox-2 expression and PGE2 production in human chronic obstructive pulmonary disease (COPD) lung fibroblasts (36). miR-146a also regulates the translation of other proteins involved in inflammation (15, 26, 31). We therefore predicted that RelB increases miR-146a production and that RelB regulates inflammatory mediator production through a mechanism involving miR-146a. Our data clearly suggest that the production of miR-146a induced by IL-1β involves RelB, which is consistent with a previous report (15).
miR-146a contains three NF-κB-binding sites in its promoter sequence (42). Analysis of multiple NF-κB binding sites has shown that RelB dimers typically bind to sites containing the sequence GGGRNNTTYY, which differs slightly from the classical NF-κB consensus sequence (GGGRNNYYCC) (12). Comparison of two of the binding sites in the miR-146a promoter, GGGATTTCCC and GGGACTTTCC (42), suggests that RelB is capable of binding to these sites. RelB may therefore alter the expression of miR-146a by targeting these sequences. Interestingly, miR-146a has also been shown to downregulate RelB expression in monocytes (15), but RelB expression was not affected after miR-146a overexpression in our model (Fig. 8A). Variations in RelB/miR-146a signaling may therefore be cell type or immune function dependent.
Recent research has indicated that cells contain feedback control mechanisms designed to reduce and reverse the prolonged effects of exposure to inflammatory stimuli (37, 38). Interestingly, RelB expression was increased after IL-1β treatment (Figs. 2A and 4A), an effect that has been seen elsewhere (45). RelB is a target of canonical NF-κB activation (11), which is intriguing considering its previously documented anti-inflammatory properties (3, 29, 48). IL-1β also increased miR-146a expression (Fig. 7), so both RelB and miR-146a may be part of a negative feedback loop designed to control inflammation. Our findings here suggest that RelB reduces inflammatory mediator production in part through a miR-146a-mediated mechanism and that sustained inflammation may result after dysregulation of this mechanism. Further exploration of this pathway may yield new insights into the pathogenesis of lung inflammation and possibly new anti-inflammatory treatment strategies.
Interestingly, whereas RelB overexpression and downregulation significantly affected IL-1β-induced Cox-2 expression and PGE2 production, IL-1β-induced cytokine and chemokine production was not affected as dramatically. As shown in Fig. 5, RelB downregulation did not significantly affect IL-1β-induced cytokine and chemokine production. Analysis via TargetScan confirmed the presence of miR-146a seed matches in the 3′-untranslated region of Cox-2 mRNA but not in the mRNA of IL-6, IL-8, and MCP-1 (22), suggesting that miR-146a does not directly regulate the production of these cytokines. miR-146a does, however, suppress IL-6 and IL-8 production in multiple cell types, including fibroblasts (10, 23, 32), and regulates the translation of other proteins that affect IL-6, IL-8, and MCP-1 production, including IL-1 receptor-associated kinase 1 and TNF-α receptor-associated factor 6 (42). miR-146a may therefore be directly blocking translation of Cox-2 in our model and simultaneously may be indirectly regulating production of other inflammatory mediators, including IL-6, IL-8, and MCP-1, by regulating the translation of proteins upstream of these cytokines. Additionally, RelB may also be reducing IL-1β-induced cytokine and chemokine production independently of miR-146a. RelB dimers have been shown to be inhibitory (27), so overexpression of RelB may displace other transcription factors bound to the promoter regions of these mediators, particularly other NF-κB members, thereby inhibiting the transcription of these genes. Downregulation of RelB would therefore not affect the transcription of these genes as their promoter regions would remain unaffected. However, since miR-146a overexpression significantly reduced IL-1β-induced IL-6 secretion in RelB-depleted fibroblasts (Fig. 8C), it is likely that miR-146a is somehow involved in IL-1β-induced IL-6 production and that a combination of these mechanisms is present in our model.
Further research is needed to fully elucidate the implications of our study. Our findings suggest that miR-146a overexpression reduces the effects of RelB downregulation on IL-1β-induced Cox-2 expression and PGE2 production, but it is possible that miR-146a dampens the production of these mediators independent of RelB. Additional experiments are needed to illuminate the interaction between miR-146a and RelB in our model. Luciferase or chromatin immunoprecipitation assays, for example, may be used to determine that RelB binds to the NF-κB sites in the miR-146a promoter region. The role of p65 in the regulation of miR-146a may also be addressed in future studies. Experiments detailing the effects of miR-146a inhibition after RelB overexpression should also be performed. Moreover, it is unclear how significant this mechanism is in the greater context of lung inflammation, which is a highly complex process involving numerous cell types and signaling pathways. It is probable that other miRNAs and other downstream targets are also responsible for the anti-inflammatory properties of RelB and that these may differ among cell types. Furthermore, as previously mentioned, miR-146a has numerous potential targets that regulate numerous other cellular functions (24, 35, 36), so excessive sustained manipulation of miR-146a may dysregulate other functions and increase susceptibility to other diseases.
New and efficacious strategies for the treatment of lung inflammation and disease are needed (5, 6). As an example, current therapies for COPD, a debilitating lung disease that affects over 10 million people in the United States and is the fourth leading cause of death worldwide (29a), cannot prevent or reverse the associated pathological manifestations (5). Corticosteroid administration, the most common treatment for COPD exacerbations, primarily targets symptoms and is often ineffective after prolonged treatment periods (6). Our findings demonstrate, for the first time, that RelB regulates inflammatory mediator production in part through a mechanism involving miR-146a. Exploitation of the RelB/miR-146a pathway may promote the development of new therapeutic insights and previously unexplored avenues of research. Manipulation of RelB and/or miR-146a may also increase corticosteroid efficacy in treating COPD exacerbations. Next-generation methods of gene therapy, such as electroporation and small molecule-based strategies using siRNAs, miRNA mimics, and antagomirs, may be used to manipulate these targets. Upregulation of RelB and of miR-146a may be novel and powerful therapeutic strategies for treatment of human lung inflammation and diseases.
GRANTS
This work was supported by National Institutes of Health Grants HL-088325, HL-088325-02S1, ES-01247, T32-ES-07026, and T32-HL-066988.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: D.H.M., C.F.W., T.H.T., S.L.S., S.B.M., P.J.S., and R.P.P. conception and design of research; D.H.M. performed experiments; D.H.M., C.F.W., and T.H.T. analyzed data; D.H.M., C.F.W., T.H.T., S.L.S., S.B.M., P.J.S., and R.P.P. interpreted results of experiments; D.H.M. prepared figures; D.H.M. drafted manuscript; D.H.M., C.F.W., T.H.T., S.L.S., S.B.M., P.J.S., and R.P.P. edited and revised manuscript; D.H.M., C.F.W., T.H.T., S.L.S., S.B.M., P.J.S., and R.P.P. approved final version of manuscript.
REFERENCES
- 2. Baglole CJ, Bushinsky SM, Garcia TM, Kode A, Rahman I, Sime PJ, Phipps RP. Differential induction of apoptosis by cigarette smoke extract in primary human lung fibroblast strains: implications for emphysema. Am J Physiol Lung Cell Mol Physiol 291: L19–L29, 2006 [DOI] [PubMed] [Google Scholar]
- 3. Baglole CJ, Maggirwar SB, Gasiewicz TA, Thatcher TH, Phipps RP, Sime PJ. The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-κB family member RelB. J Biol Chem 283: 28944–28957, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baglole CJ, Ray DM, Bernstein SH, Feldon SE, Smith TJ, Sime PJ, Phipps RP. More than structural cells, fibroblasts create and orchestrate the tumor microenvironment. Immunol Invest 35: 297–325, 2006 [DOI] [PubMed] [Google Scholar]
- 5. Barnes PJ. Future advances in COPD therapy. Respiration 68: 441–448, 2001 [DOI] [PubMed] [Google Scholar]
- 6. Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet 373: 1905–1917, 2009 [DOI] [PubMed] [Google Scholar]
- 7. Barnes PJ, Karin M. Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med 336: 1066–1071, 1997 [DOI] [PubMed] [Google Scholar]
- 8. Beinke S, Ley SC. Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem J 382: 393–409, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beinke S, Robinson MJ, Hugunin M, Ley SC. Lipopolysaccharide activation of the TPL-2/MEK/extracellular signal-regulated kinase mitogen-activated protein kinase cascade is regulated by IκB kinase-induced proteolysis of NF-κB1 p105. Mol Cell Biol 24: 9658–9667, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bhaumik D, Scott GK, Schokrpur S, Patil CK, Orjalo AV, Rodier F, Lithgow GJ, Campisi J. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging 1: 402–411, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bren GD, Solan NJ, Miyoshi H, Pennington KN, Pobst LJ, Paya CV. Transcription of the RelB gene is regulated by NF-κB. Oncogene 20: 7722–7733, 2001 [DOI] [PubMed] [Google Scholar]
- 12. Britanova LV, Makeev VJ, Kuprash DV. In vitro selection of optimal RelB/p52 DNA-binding motifs. Biochem Biophys Res Commun 365: 583–588, 2008 [DOI] [PubMed] [Google Scholar]
- 13. Catley MC, Chivers JE, Cambridge LM, Holden N, Slater DM, Staples KJ, Bergmann MW, Loser P, Barnes PJ, Newton R. IL-1β-dependent activation of NF-κB mediates PGE2 release via the expression of cyclooxygenase-2 and microsomal prostaglandin E synthase. FEBS Lett 547: 75–79, 2003 [DOI] [PubMed] [Google Scholar]
- 14. Choi EY, Santoso S, Chavakis T. Mechanisms of neutrophil transendothelial migration. Front Biosci 14: 1596–1605, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Etzrodt M, Cortez-Retamozo V, Newton A, Zhao J, Ng A, Wildgruber M, Romero P, Wurdinger T, Xavier R, Geissmann F, Meylan E, Nahrendorf M, Swirski FK, Baltimore D, Weissleder R, Pittet MJ. Regulation of monocyte functional heterogeneity by miR-146a and Relb. Cell Rep 1: 317–324, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grossmann M, Nakamura Y, Grumont R, Gerondakis S. New insights into the roles of ReL/NF-κB transcription factors in immune function, hemopoiesis and human disease. Int J Biochem Cell Biol 31: 1209–1219, 1999 [DOI] [PubMed] [Google Scholar]
- 17. Kim SW, Ramasamy K, Bouamar H, Lin AP, Jiang D, Aguiar RC. MicroRNAs miR-125a and miR-125b constitutively activate the NF-κB pathway by targeting the tumor necrosis factor α-induced protein 3 (TNFAIP3, A20). Proc Natl Acad Sci USA 109: 7865–7870, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1β induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest 107: 1529–1536, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R, Honnons S, Jones C, Isern NG, Hu JZ, Nathan SD, Grant G, Phipps RP, Sime PJ. Lactic acid is elevated in IPF and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am J Respir Crit Care Med 186: 740–751, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koumas L, Phipps RP. Differential COX localization and PG release in Thy-1+ and Thy-1− human female reproductive tract fibroblasts. Am J Physiol Cell Physiol 283: C599–C608, 2002 [DOI] [PubMed] [Google Scholar]
- 21. Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1β causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol 32: 311–318, 2005 [DOI] [PubMed] [Google Scholar]
- 22. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20, 2005 [DOI] [PubMed] [Google Scholar]
- 23. Li G, Luna C, Qiu J, Epstein DL, Gonzalez P. Modulation of inflammatory markers by miR-146a during replicative senescence in trabecular meshwork cells. Invest Ophthalmol Vis Sci 51: 2976–2985, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li J, Huang J, Dai L, Yu D, Chen Q, Zhang X, Dai K. miR-146a, an IL-1β responsive miRNA, induces vascular endothelial growth factor and chondrocyte apoptosis by targeting Smad4. Arthritis Res Ther 14: R75, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li T, Morgan MJ, Choksi S, Zhang Y, Kim YS, Liu ZG. MicroRNAs modulate the noncanonical transcription factor NF-κB pathway by regulating expression of the kinase IKKα during macrophage differentiation. Nat Immunol 11: 799–805, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma X, Becker Buscaglia LE, Barker JR, Li Y. MicroRNAs in NF-κB signaling. J Mol Cell Biol 3: 159–166, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marienfeld R, May MJ, Berberich I, Serfling E, Ghosh S, Neumann M. RelB forms transcriptionally inactive complexes with RelA/p65. J Biol Chem 278: 19852–19860, 2003 [DOI] [PubMed] [Google Scholar]
- 28. Martey CA, Pollock SJ, Turner CK, O'Reilly KM, Baglole CJ, Phipps RP, Sime PJ. Cigarette smoke induces cyclooxygenase-2 and microsomal prostaglandin E2 synthase in human lung fibroblasts: implications for lung inflammation and cancer. Am J Physiol Lung Cell Mol Physiol 287: L981–L991, 2004 [DOI] [PubMed] [Google Scholar]
- 29. McMillan DH, Baglole CJ, Thatcher TH, Maggirwar S, Sime PJ, Phipps RP. Lung-targeted overexpression of the NF-κB member RelB inhibits cigarette smoke-induced inflammation. Am J Pathol 179: 125–133, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29a. National Health Interview Survey National Health Interview Survey: research for the 1995–2004 redesign. Vital Health Stat 2: 1–119, 1999 [PubMed] [Google Scholar]
- 30. Niu J, Shi Y, Tan G, Yang CH, Fan M, Pfeffer LM, Wu ZH. DNA damage induces NF-κB-dependent microRNA-21 up-regulation and promotes breast cancer cell invasion. J Biol Chem 287: 21783–21795, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31. Oglesby IK, McElvaney NG, Greene CM. MicroRNAs in inflammatory lung disease–master regulators or target practice? Respir Res 11: 148, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Perry MM, Williams AE, Tsitsiou E, Larner-Svensson HM, Lindsay MA. Divergent intracellular pathways regulate interleukin-1β-induced miR-146a and miR-146b expression and chemokine release in human alveolar epithelial cells. FEBS Lett 583: 3349–3355, 2009 [DOI] [PubMed] [Google Scholar]
- 33. Pottelberge GR, Mestdagh P, Bracke KR, Thas O, Durme YM, Joos GF, Vandesompele J, Brusselle GG. MicroRNA expression in induced sputum of smokers and patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 183: 898–906, 2011 [DOI] [PubMed] [Google Scholar]
- 34. Profita M, Sala A, Bonanno A, Riccobono L, Ferraro M, La Grutta S, Albano GD, Montalbano AM, Gjomarkaj M. Chronic obstructive pulmonary disease and neutrophil infiltration: role of cigarette smoke and cyclooxygenase products. Am J Physiol Lung Cell Mol Physiol 298: L261–L269, 2010 [DOI] [PubMed] [Google Scholar]
- 35. Rusca N, Monticelli S. MiR-146a in immunity and disease. Mol Biol Int 2011: 437301, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sato T, Liu X, Nelson A, Nakanishi M, Kanaji N, Wang X, Kim M, Li Y, Sun J, Michalski J, Patil A, Basma H, Holz O, Magnussen H, Rennard SI. Reduced miR-146a increases prostaglandin E2 in chronic obstructive pulmonary disease fibroblasts. Am J Respir Crit Care Med 182: 1020–1029, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol 25: 101–137, 2007 [DOI] [PubMed] [Google Scholar]
- 38. Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O'Neill LA, Perretti M, Rossi AG, Wallace JL. Resolution of inflammation: state of the art, definitions and terms. FASEB J 21: 325–332, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smith RS, Smith TJ, Blieden TM, Phipps RP. Fibroblasts as sentinel cells. Synthesis of chemokines and regulation of inflammation. Am J Pathol 151: 317–322, 1997 [PMC free article] [PubMed] [Google Scholar]
- 40. Strillacci A, Griffoni C, Sansone P, Paterini P, Piazzi G, Lazzarini G, Spisni E, Pantaleo MA, Biasco G, Tomasi V. MiR-101 downregulation is involved in cyclooxygenase-2 overexpression in human colon cancer cells. Exp Cell Res 315: 1439–1447, 2009 [DOI] [PubMed] [Google Scholar]
- 41. Sun X, Icli B, Wara AK, Belkin N, He S, Kobzik L, Hunninghake GM, Vera MP, Registry M, Blackwell TS, Baron RM, Feinberg MW. MicroRNA-181b regulates NF-κB-mediated vascular inflammation. J Clin Invest 122: 1973–1990, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA 103: 12481–12486, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thatcher TH, Maggirwar SB, Baglole CJ, Lakatos HF, Gasiewicz TA, Phipps RP, Sime PJ. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-κB component RelB. Am J Pathol 170: 855–864, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thatcher TH, McHugh NA, Egan RW, Chapman RW, Hey JA, Turner CK, Redonnet MR, Seweryniak KE, Sime PJ, Phipps RP. Role of CXCR2 in cigarette smoke-induced lung inflammation. Am J Physiol Lung Cell Mol Physiol 289: L322–L328, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vardar-Sengul S, Arora S, Baylas H, Mercola D. Expression profile of human gingival fibroblasts induced by interleukin-1β reveals central role of nuclear factor-κB in stabilizing human gingival fibroblasts during inflammation. J Periodontol 80: 833–849, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, Lira SA, Bravo R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/Rel family. Cell 80: 331–340, 1995 [DOI] [PubMed] [Google Scholar]
- 47. Wewers MD, Dare HA, Winnard AV, Parker JM, Miller DK. IL-1β-converting enzyme (ICE) is present and functional in human alveolar macrophages: macrophage IL-1β release limitation is ICE independent. J Immunol 159: 5964–5972, 1997 [PubMed] [Google Scholar]
- 48. Xi X, McMillan DH, Lehmann GM, Sime PJ, Libby RT, Huxlin KR, Feldon SE, Phipps RP. Ocular fibroblast diversity: implications for inflammation and ocular wound healing. Invest Ophthalmol Vis Sci 52: 4859–4865, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xia Y, Pauza ME, Feng L, Lo D. RelB regulation of chemokine expression modulates local inflammation. Am J Pathol 151: 375–387, 1997 [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang H, Cai B. The impact of tobacco on lung health in China. Respirology 8: 17–21, 2003 [DOI] [PubMed] [Google Scholar]
- 51. Zhou R, Hu G, Liu J, Gong AY, Drescher KM, Chen XM. NF-κB p65-dependent transactivation of miRNA genes following Cryptosporidium parvum infection stimulates epithelial cell immune responses. PLoS Pathog 5: e1000681, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]