

Abstract
Human heart failure has been associated with a low level of thin-filament protein phosphorylation and an increase in calcium sensitivity of contraction relative to both “control” human heart tissue and tissue from small animal models. However, diverse strategies of human tissue procurement and the reliance on tissue obtained from subjects with end-stage heart failure suggest this may be an incomplete characterization. Therefore, we evaluated cardiac left ventricular (LV) biopsy samples from patients with aortic stenosis undergoing valve replacement who presented either with LV hypertrophy and preserved systolic function (Hyp) or with LV dilation and reduced ejection fraction (Dil). In Hyp, total troponin I (TnI) phosphorylation was markedly increased and myosin light chain 2 (MLC2) phosphorylation was unchanged relative to a control group of patients with normal LV function. Conversely, in Dil, total TnI phosphorylation was significantly reduced compared with control subjects and MLC2 phosphorylation was increased. Site-specific analysis of TnI phosphorylation revealed phenotype-specific differences such that Hyp samples demonstrated significant increases in phosphorylation at serine 22/23 and Dil samples had significant decreases at serine 43. The ratio of phosphorylation at the two sites was biased toward serine 22/23 in Hyp and toward serine 43/45 in Dil. Western blot analysis showed that protein phosphatase-1 was reduced in Hyp and protein phosphatase-2 was reduced in Dil. These data suggest that posttranslational modifications of sarcomeric proteins, both singly and in combination, are stage specific. Defining these changes in progressive heart disease may provide important diagnostic and treatment information.
Keywords: heart failure, hypertrophy, signal transduction, stenosis, phosphorylation
the cardiac muscle accommodation to a chronic pressure load is complex and multifactorial (12). Data derived mostly from small animal models have shown that these cardiac responses include myocellular hypertrophy accompanied by changes in gene expression (largely a recapitulation of the fetal gene program), an increase in interstitial fibrosis, cell drop-out, and transcriptional and posttranslational changes in the contractile elements themselves (9, 10, 14, 44). The accumulated response has been referred to as “pathological hypertrophy.” Despite the fact that the broad characteristics of this phenotype have been evident for some time, there remain a number of unanswered questions. Two in particular are vexing: First, since the geometric, and by extension the mechanical, response to chronic pressure loading can manifest differently, as either hypertrophy with preserved contractile function or dilation with depressed contractile function, does this represent a continuum or a divergent set of responses? Second, since large animals and humans do not evidence marked changes in myosin heavy chain (MHC) gene expression in response to chronic pressure loading (19, 28, 39), are there other biochemical alterations in the contractile machinery of hearts of large mammals and humans that might presage these distinct phenotypes and that might be more functionally relevant in larger hearts?
To approach these questions we have examined the biochemical characteristics of the contractile elements isolated from human hearts subjected to chronic aortic stenosis. The progressive pressure overload of aortic stenosis leads initially to concentric left ventricular (LV) hypertrophy and ultimately to cardiac failure (15), making this an ideal context within which to address these issues. We examined cardiac muscle from two groups of aortic stenosis patients, one group with LV hypertrophy and preserved contractility (Hyp) and a second group with ventricular dilation and reduced contractility (Dil). Samples were collected in the operating room (OR) in a rapid and highly controlled fashion that allowed analysis of endogenous contractile protein phosphorylation, and, strikingly, the extent of phosphorylation both of troponin I (TnI) and of the regulatory myosin light chain 2 (MLC2) differed according to clinical status. A significant increase in TnI phosphorylation was associated with hypertrophy, whereas a decrease in TnI phosphorylation and an increase in MLC2 phosphorylation were seen with LV dilation. Moreover, the TnI changes reflected a shift in site-specific phosphorylation, with preservation of the putative PKA-dependent sites at serine 22/23 in the hypertrophic hearts and a significant reduction in the dilated hearts. Similarly, phosphorylation of the PKC site at serine 43 was preserved in the hypertrophic hearts and was reduced in the dilated samples, although to a lesser degree than serine 22/23. This pattern of phosphorylation is similar to that seen by our lab and others in other models of hypertrophy and dilation and suggests a reversible biochemical context that contributes to the contractile alterations that characterize these pathological circumstances.
METHODS
Tissue procurement.
Tissue samples were obtained from a total of 15 human hearts: 6 with aortic stenosis, hypertrophy, and preserved contractile function; 4 with aortic stenosis and ventricular dilation; and 5 control hearts. LV biopsies (∼10–40 μg) were obtained under direct observation from the anterior midwall of the heart just after the administration of cold cardioplegia, rinsed in cold saline, and immediately placed in liquid N2 (41). Less than 30 s elapsed between tissue excision and fast freezing. Samples were then transported in liquid nitrogen to the lab and stored in liquid nitrogen until the time of analysis. All experimental samples were obtained from patients undergoing aortic valve replacement. Control tissues were obtained from individuals undergoing coronary artery bypass graft (CABG) surgery in whom echocardiographically determined LV ejection fraction was in the normal range (50–65%), there were no regional wall motion abnormalities, and no LV hypertrophy was seen. Furthermore, there were no Doppler flow abnormalities across the aortic valve or any other evidence of intrinsic aortic valve disease in any of the control subjects. The Colorado Multicenter Institutional Review Board approved the protocol (no. 06-1100) for the collection, storage, and analysis of human tissue, and all patients gave informed consent prior to collection.
Tissue extraction and gel electrophoresis.
Small samples (∼10 μg wet wt) of the left ventricle were homogenized in 8 M urea, 2.5 M thiourea, 4% CHAPS, 10 mM EDTA, and a cocktail of protease and phosphatase inhibitors (42). For quantification of phosphorylation, samples were separated by 12.5% SDS-PAGE and fixed and stained with ProQ Diamond phosphoprotein gel stain (PQD; Invitrogen). After destaining, gels were imaged with a Typhoon Gel Imager (GE Life Sciences). Gels were rinsed with water and stained with BioSafe Coomassie brilliant blue (CBB; Bio-Rad) for detection of total proteins. Phosphorylation was calculated by dividing the PQD signal by the CBB signal (see Statistical analysis).
For separation of α- and β-myosin, samples were run by modified 6% SDS-PAGE (separating acrylamide-bis ratio 1:100; resolving gel buffer pH 9.0; running gel buffer pH 8.2; β-mercaptoethanol 600 μl/l inner gel buffer). Gels were run overnight at 4°C and stained with BioSafe CBB protein stain (40).
Western blotting.
For determination of phosphorylation site changes in TnI, we used phosphospecific antibodies that were generated by PhosphoSolutions (Aurora, CO) and have been carefully validated (unpublished observations). In our hands, the specificity of these antibodies for site-specific TnI phosphorylation is superior to that of other commercially available antibodies (data not shown). Gels were run as above, and proteins were transferred to polyvinylidene difluoride (PVDF). Membranes were blocked in 5% BSA for 1 h, rinsed with Tris-buffered saline-Tween 20 (TBST), and incubated in the appropriate antibody overnight at 4°C. Membranes were washed and incubated with secondary antibody for 1 h at room temperature. After washing, the proteins were visualized with enhanced chemiluminescence.
For determination of phosphatase expression, gels were run as above and proteins were transferred to PVDF. Membranes were blocked with 5% BSA for 1 h, rinsed with TBST, and incubated in primary antibody [anti-protein phosphatase (PP)1A or anti-PP2A] overnight at 4°C. Membranes were washed and incubated with secondary antibody for 1 h at room temperature. After washing, the proteins were visualized with enhanced chemiluminescence.
Antibodies used were anti-TnI (total) (Fitzgerald; 1:2,500); anti-pS22/23 TnI, anti-pS43 TnI, and anti-pS150 TnI (PhosphoSolutions; 1:1,000); anti-PP1A and anti-PP2A (Santa Cruz Biotechnology, Santa Cruz, CA; 1:1,000); and anti-sarcomeric actinin (Sigma, 1:1,000). Secondary anti-mouse and anti-rabbit were from Sigma (1:50,000).
Statistical analysis.
Differences in total phosphorylation (SDS-PAGE) were determined by dividing the PQD signal for each protein either by the CBB signal for itself [myosin binding protein C (MyBPC), TnI, and MLC2] or by the CBB signal for the actin/TnT (for TnT normalization, as we were unable to effectively measure TnT alone). Similarly, the ratio of α- to β-MHC was determined by densitometry of the CBB-stained gel, and percent α-MHC was expressed as the α signal divided by the total (α + β). Site-specific phosphorylation was quantified by densitometric analysis as the ratio of the phosphospecific signal to the total protein signal. All data are expressed as means ± SE. Comparisons between groups (control, Hyp, Dil) were made by one-way ANOVA followed by Fisher's least significant difference test. A value of P < 0.05 was considered significant.
RESULTS
Table 1 describes the clinical characteristics of the patients studied. The three groups of patients were of slightly varying ages (control 55 ± 1 yr, Hyp 64 ± 6 yr, Dil 61 ± 3 yr; P = 0.184), but this was not statistically different. Aortic valve area as determined by echocardiography was slightly greater in the patients with hypertrophy, and ventricular volumes were significantly larger and ejection fraction was lower in the patients with LV dilation (ejection fraction in control 66 ± 2%; Hyp 66 ± 3%; Dil 43 ± 5%; P < 0.001). The control patients were slightly younger and had slightly larger ventricular cavitary dimensions relative to those with hypertrophy, and wall thickness was greater in those with LV hypertrophy (not shown). Two of the control patients were taking beta-blocker therapy, whereas one of the aortic stenosis patients with hypertrophy and two of those with dilation were on beta-blocker therapy. All of the aortic stenosis patients in both groups had fibrocalcific disease of the aortic root, and none had a congenitally bicuspid valve.
Table 1.
Clinical characteristics of patients studied
| n (M/F) | Age, yr | LV EDD, cm | LV ESD, cm | AVA, cm2 | EF, % | |
|---|---|---|---|---|---|---|
| Control | 5 (4/1) | 55 ± 1 | 5.5 ± 0.2 | 3.5 ± 0.2 | NL | 66 ± 2 |
| Hyp | 6 (4/2) | 64 ± 6 | 4.6 ± 0.2* | 3.0 ± 0.2 | 0.71 ± 0.1 | 66 ± 3 |
| Dil | 4 (3/1) | 61 ± 3 | 6.0 ± 0.3 | 4.9 ± 0.4* | 0.56 ± 0.1 | 43 ± 5* |
Values are means ± SE for n patients. Control, normal left ventricular (LV) function; Hyp, LV hypertrophy and preserved systolic function; Dil, LV dilation and reduced ejection fraction (EF); M, male; F, female; EDD, end-diastolic dimension; ESD, end-systolic dimension; AVA, aortic valve area; NL, normal.
P < 0.05 vs. control.
Figure 1 shows representative phospho (PQD) and total (CBB) protein gels from the three groups of patients (Fig. 1A) and histograms showing relative changes in phosphoprotein staining for each of several contractile proteins, including MyBPC, TnT, TnI, and MLC2 (Fig. 1B). There were no differences in phosphorylation of MyBPC, and TnT in any of the three groups, whereas striking differences were seen in TnI and MLC2. Phosphorylation of TnI was significantly increased in Hyp and decreased in Dil relative to control hearts (P < 0.05), whereas phosphorylation of MLC2 was unchanged in Hyp and significantly increased in Dil (P < 0.05). We have previously demonstrated an association between changes in phosphorylation of MLC2 and TnI (31) and have hypothesized that the combinatorial phosphorylation of the myofilament may be deterministic in regulating contractile function. Here, marked changes in the ratio of TnI phosphorylation to MLC2 phosphorylation underscore the relative changes in phosphorylation of each protein (Fig. 1C). Data were analyzed by one-way ANOVA (P < 0.05) followed by Fisher's least significant difference test; the Hyp group did not quite achieve significant difference from control (P = 0.08) but was highly significantly different from the Dil group (P < 0.001). The Dil group was significantly different from both the control group and the Hyp group.
Fig. 1.

A: representative SDS-PAGE of control, left ventricular (LV) hypertrophy and preserved systolic function (Hyp), and LV dilation and reduced ejection fraction (Dil, failing) samples. Phosphoprotein stain was achieved by staining with ProQ Diamond gel stain (PQD). Total protein stain was with Coomassie brilliant blue (CBB). B: histogram represents the PQD signal for each protein divided by CBB signal for each protein. C: histogram represents the ratio of phospho-troponin I (pTnI) to phospho-myosin light chain 2 (pMLC2). n = 5 (control), 6 (Hyp), and 4 (Dil). *P < 0.05 vs. control; #P < 0.05 vs. Hyp.
Since there are several possible phosphorylation sites in TnI that have distinct functional properties, we utilized TnI phosphoprotein antibodies to determine whether the TnI changes reflected the sites linked to PKA activation at serine 22/23, which are associated with preserved tension development, a decrease in calcium sensitivity of the myofilament, and an increase in rates of fiber shortening, or the sites more commonly associated with PKC activation at serine 43/45, which are associated with a decrease in peak tension development and velocity of fiber shortening as well as decreased calcium sensitivity. As shown in Fig. 2, the Hyp group had increased phosphorylation of the serine 22/23 sites, but phosphorylation at the serine 43 site was not different from the control group. Conversely, in the Dil group, phosphorylation of the serine 22/23 sites was not different from control, and the serine 43 site was significantly reduced. Interestingly, in these samples, TnI was visualized as an immunoreactive doublet. This has been shown previously both in animal models and in human samples (22) and is ascribed to COOH-terminal proteolysis of TnI. However, this has not been confirmed in this study because of the very limited nature of these tissues and may represent either proteolysis or other posttranslational modifications. Both bands demonstrated NH2-terminal immunoreactivity and were included in the quantitation of phosphorylation. We were unable to detect changes at serine 150, a putative AMP kinase site in either group (data not shown). These results together with those from the phosphoprotein stain clearly demonstrate that the patterns of contractile protein phosphorylation differ strikingly in these two phenotypic models of ventricular remodeling.
Fig. 2.

Representative Western blots of phosphorylated TnI. Samples were separated on 12.5% SDS-PAGE and probed with antibodies generated against phosphoserine 22/23 TnI (A) and phosphoserine 43 TnI (C). Blots were reprobed with a pan-TnI antibody (B and D). Phosphorylation and total TnI levels were quantified with ImageJ, and the normalized intensities (phospho signal/total protein signal) for each group are shown in the histograms on right. n = 5 (control), 6 (Hyp), and 4 (Dil). *P < 0.05 vs. control.
Since contractile protein phosphorylation reflects not only selective activation of regulatory kinases but also phosphatase-mediated dephosphorylation, we investigated whether there were differences in the abundance of the major myofilament protein phosphatases, PP1A and PP2A, between Hyp and Dil. These data are shown in Fig. 3, and they demonstrate that relative to control hearts there is a clear reduction in PP1A in Hyp and a reduction in PP2A in Dil.
Fig. 3.

Representative Western blots of protein phosphatase 1 (pp1A) and protein phosphatase 2 (pp2A). Samples were separated on 10.0% SDS-PAGE and probed with antibodies generated against pp1A catalytic subunit (A) and pp2A catalytic subunit (B). After stripping, blots were reprobed with an anti-sarcomeric actinin antibody. Blots were quantified with ImageJ, and the expression of phosphatase (normalized to total cardiac actinin) for each group is shown in the histograms on right. n = 5 (control), 6 (Hyp), and 4 (Dil). *P < 0.05 vs. control.
Finally, since the hallmark of pathological hypertrophy in small animals as well as in humans is a transcriptional shift from expression of the fast ATPase α-MHC gene and protein to the slower β-MHC (V1 to V3), we established the relative abundance of these two protein isoforms. As has been seen previously in human heart disease, the predominant isoform expressed was the slow V3, ββ-homodimer, which accounted for >95% of the total MHC in both control and diseased hearts as shown in Fig. 4. There was no difference among the three groups.
Fig. 4.
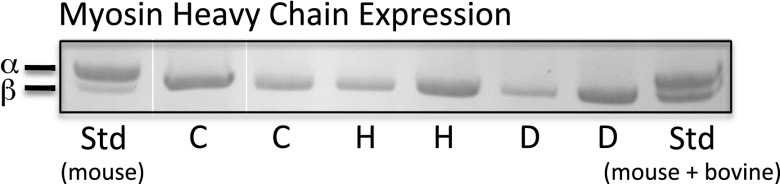
Representative 6% SDS-PAGE gel demonstrating α-,β-separation. Samples were separated by 6% SDS-PAGE and visualized by staining with CBB. Standard samples (Std) of mouse and a mixture of mouse and bovine cardiac extracts were loaded to visualize adequate separation of α- and β-myosin. C, control; H, Hyp; D, Dil.
DISCUSSION
It has long been appreciated that there are two distinct myocardial responses to a sustained pressure overload: The first, which can be viewed as a functional adaptation, consists of ventricular hypertrophy, normalized wall stress, and preserved overall contractility, whereas the second, which is functionally maladaptive, consists of ventricular dilation, increased wall stress, and depressed contractility. While these can be easily defined phenotypically, establishing the nature of the associated biochemical changes and whether they are similar or distinct has remained an open question. This study addresses this issue in the context of clinical aortic stenosis and clearly demonstrates that the patterns of myocardial contractile protein phosphorylation (in particular of TnI and MLC2) in the hypertrophic and dilated patterns of myocardial response are both different and distinct.
Before discussing the specific biochemical changes seen, it is important to comment on the magnitude of overall contractile protein phosphorylation in human heart disease. Several studies have suggested that levels of phosphorylation are low in human heart relative to small animal models and have therefore questioned the importance of these changes in the overall regulation of contraction in human heart disease (13, 18, 36, 37). However, it has also been shown that even modest increases in troponin phosphorylation can have a significant impact on ventricular function (17). Given this, it is important to contrast the patient population and sampling technique employed in this study with these previous reports. In general, prior studies showing low levels of phosphorylation have been done with muscle samples obtained from end-stage myopathic hearts at the time of transplant, hearts that were subjected to extraordinary adrenergic support (likely in the context of β-receptor desensitization) prior to excision. Extrapolating from this to the present circumstance, namely, hearts that were by no means end-stage (and in fact would be predicted to substantially recover after aortic valve replacement) is likely misleading, and indeed the relative robust levels of phosphorylation seen in the present study support this conclusion. Moreover, studies that have taken muscle from nontransplant hearts (for example, during CABG) have generally employed extensive cardioplegia prior to tissue sampling in order to facilitate cell isolation, a strategy that would be predicted to result in substantial protein dephosphorylation. When we have compared samples collected with the strategy of minimal cardioplegia and flash freezing in the OR with these other approaches, we have generally seen far more robust contractile protein phosphorylation (41).
The maintenance of overall TnI phosphorylation in the Hyp group and, more importantly, the apparent redistribution of phosphorylation along the molecule so as to favor the already predominant NH2-terminal serine sites is consistent with other animal models of hypercontractility and hypertrophy without dilation. One model in particular, first described by MacGowan et al. (17a), in which the serines in TnI at 43,45 were mutated to alanines (and rendered nonphosphorylatable) underscores the reciprocal functional relationship between these sites. These animals showed an increase in both +- and −dP/dt that was associated with an apparent increase in phosphorylation of the NH2-terminal serines in concert with the lack of any change in phosphorylation at the serine 43/45 sites (23). Over time, it might be predicted that the adrenergic stimulation would result in β-receptor downregulation and a gradual decline both in contractility and in phosphorylation at the NH2-terminal (PKA dependent) serines (2, 3, 6), which is what is seen in the Dil group. Of course, there are several other potential phosphorylation sites along the molecule that we are unable to evaluate given the limited sample size and biochemical reagents available, and how these contribute to overall contractility is speculative.
The changes in the steady-state phosphorylation levels of TnI in the Hyp group and the Dil group are consistent with those seen both in animal models of heart failure and in other human circumstances in which an overall decline in total phosphorylation has been described (18, 42). The decrease in phosphorylation at the PKA-dependent serine 22,23 sites might well reflect downregulation of β-adrenergic signaling or activation of specific phosphatases (see below) and would certainly be consistent with reduced cross-bridge cycling and slower rates of both contraction and relaxation. The overall decline in phosphorylation at serine 43, the putative PKC site, is somewhat surprising as we have previously shown relative preservation of phosphorylation at this site in end-stage cardiomyopathy patients (1), although these studies employed a different antibody. Indeed, cardiac restricted transgenic overexpression of calcium-independent isoforms of PKC has been shown to be sufficient to induce a dilated cardiomyopathy that is associated with an increase in PKC-dependent, TnI-dependent phosphorylation and a decrease in both maximal force development and the calcium sensitivity of cardiac muscle contraction (11, 32). However, it is striking that the relative ratio of phosphorylation at serine 22/23 and serine 43 is biased toward serine 22/23 in Hyp and toward serine 43 in Dil, clearly supporting the hypothesis that a change in myocyte signaling characterizes the transition from a hypertrophic, hypercontractile phenotype to a dilated hypocontractile phenotype. The findings related to MLC2, and the lack of an effective change in the other surveyed contractile proteins, are both striking and somewhat unexpected. MLC2 has generally been viewed as quite important in regulating contractility in smooth muscle but less important in cardiac muscle. However, phosphorylation of MLC2 (generally felt to occur at serine 15) has been shown to increase cardiac myofilament calcium sensitivity (and transgenic replacement with a nonphosphorylatable MLC2 isoform eliminates the MLCK-mediated left shift in the tension-calcium relationship), with only a modest effect on tension development (20, 21, 30). It has also been shown that the gradient of tension development across the myocardium correlates with an analogous gradient of MLC2 phosphorylation (7). Our group and others have also shown that ablation of MLC2 phosphorylation is associated with a reduction in calcium-activated tension development (but not calcium sensitivity) and also with an increase in phosphorylation of other contractile proteins including TnI and MyBPC (31). Given this, it is attractive to imagine that as the ventricle dilates and wall stress increases across the ventricular wall, there is a parallel increase in MLC2 phosphorylation that would not be seen in the Hyp phenotype, in which wall stress is, if anything, slightly reduced.
The absence of a significant change in TnT and MyBPC phosphorylation is noteworthy. Both molecules have been shown to be phosphorylation targets (of both PKA and PKC), and in reconstituted fibers and in transgenic models phosphorylation of these molecules appears to have significant functional effects (5, 23, 29, 38). However, our data suggest that these biochemical modifications are not relevant in these human disease models, especially not when contrasted with the striking findings related to TnI and MLC2.
A remarkable finding that emerges from this study is the directionally opposite changes in the associated phosphatases PP1A and PP2A, which may well be key players in the distinct phosphoprotein fingerprints and mechanical sequelae that are seen. In mammalian heart, two major phosphatases have been identified, PP1A and PP2A. The former serves as a negative regulator of contractility (4, 24) (and has previously been felt to be functionally significant because of its specificity for phospholamban). Overexpression of inhibitors of PP1A (and subsequent decreases in PP1A activity) results in increases in cardiac function (16, 25), a picture that is completely consistent with that seen in the Hyp group. PP2A, on the other hand, is a positive regulator of contraction, and increased PP2A activity has been shown to increase the calcium sensitivity of cardiac contraction (33), so it would be predicted that a reduction in PP2A, as is seen in Dil, would be associated with reductions in overall muscle contractility. Both PP1A and PP2A can be found in functional complexes with regulatory kinases, and it is likely that their activities are coordinately regulated (27, 34, 43). The precise targets of the action of these phosphatases can only be inferred from the present data, although it is certainly attractive to postulate that they influence the contractile protein alterations described, either directly as myofilament-docked proteins or indirectly via activation or inhibition of the relevant regulatory kinases.
The final biochemical interface assayed in this study was the isoform distribution of MHC. A shift from the fast to the slower ATPase enzyme has long been implicated in the transition from a normal heart to one that is hypertrophic and/or failing, and indeed increased expression of β-MHC mRNA in human hearts strongly correlates both with heart failure and also with clinical response to beta-blocker therapy (8, 26, 35). We did not measure mRNA levels in the present study but were able to quantify protein expression, and our data show that there was a predominance of β-MHC protein expression in control hearts and in both disease groups, but the levels of protein expression were equivalent. This suggests that β-MHC protein expression (and a shift from V1 to V3) is not a primary mediator of mechanical dysfunction in this clinical context.
We acknowledge that there are limitations with this study: a limited number of samples, nonuniform medical regimes within the patient population studied, and control samples taken from patients undergoing CABG surgery. We did examine each sample independently, and although the sample size was small, we were unable to detect any significant effect of beta-blocker therapy on any measured outcome (phosphorylation, phosphatase expression, or myosin isoform) either within groups or between groups. Furthermore, while we cannot exclude the possibility that the presence of coronary artery disease in the control subjects might have contributed to a global cardiomyopathy, none of these patients had significant regional wall motion abnormalities and all had normal overall LV function. We did take care to sample from areas of the heart that were not subserved by critically stenosed vessels.
In conclusion, our data suggest that the phenotypically diverse responses to chronic pressure overload seen in patients with aortic stenosis are reflective of changes in contractile protein phosphorylation (in particular of TnI and MLC2) and parallel changes in phosphatase expression. Given the clear functional consequences of these contractile protein changes, it would seem likely that the characteristic “phosphofingerprints” seen might well be characteristic of acquired human hypertrophic and dilated cardiomyopathy in general. We suggest that the combinatorial pattern of myofilament protein phosphorylation and cardiac function are tightly linked, so that cardiac disease is a continuum of changes within the myocardium rather than distinct disease states. Figure 5 summarizes this idea; the transition from a diastolic dysfunction (with preserved ejection fraction) to a systolic dysfunction (evidencing a reduced ejection fraction) is the result of multiple changes in the myofilament proteins. The combinatorial phosphorylation found in an individual patient may reflect the functional status of the myocardium at a particular stage of cardiac disease. Moreover, it would be of great interest to see whether these patterns could help predict the myocardial response to valve replacement or, more generally, to unloading therapy and/or response to pharmacological therapies. Further investigation will be required to answer these questions; however, tissue samples from patients who have recovered from valve surgery have been difficult to acquire. Our preliminary data in patients with dilated cardiomyopathy do suggest that preserved global levels of TnI phosphorylation are associated with clinical response to beta-blocker therapy.
Fig. 5.

Schematic depicting a continuum of cardiac dysfunction and its relationship to myofilament protein phosphorylation changes. The transition from a diastolic dysfunction to a systolic dysfunction is the result of multiple changes in the myofilament proteins. The combinatorial phosphorylation found in an individual patient may reflect the functional status of the myocardium at a particular stage of cardiac disease. Identification of stage-specific phosphorylation patterns (summations of individual phosphoproteins) that can be tightly correlated with functional consequences will serve to further our understanding of cardiac maladaptation and, ultimately, response to therapy.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-077195 (P. M. Buttrick), HL-62426 (P. M. Buttrick), and HL-104352 (L. A. Walker and P. M. Buttrick).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.A.W., D.A.F., and P.M.B. conception and design of research; L.A.W. and D.A.F. performed experiments; L.A.W. analyzed data; L.A.W. and P.M.B. interpreted results of experiments; L.A.W. prepared figures; L.A.W. and P.M.B. drafted manuscript; L.A.W. and P.M.B. edited and revised manuscript; L.A.W., D.A.F., and P.M.B. approved final version of manuscript.
REFERENCES
- 1. Ambardekar AV, Walker JS, Walker LA, Cleveland JC, Jr, Lowes BD, Buttrick PM. Incomplete recovery of myocyte contractile function despite improvement of myocardial architecture with left ventricular assist device support. Circ Heart Fail 4: 425–432, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bristow MR. Treatment of chronic heart failure with beta-adrenergic receptor antagonists: a convergence of receptor pharmacology and clinical cardiology. Circ Res 109: 1176–1194, 2011 [DOI] [PubMed] [Google Scholar]
- 3. Bristow MR, Ginsburg R, Umans V, Fowler M, Minobe W, Rasmussen R, Zera P, Menlove R, Shah P, Jamieson S. Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ Res 59: 297–309, 1986 [DOI] [PubMed] [Google Scholar]
- 4. Carr AN, Schmidt AG, Suzuki Y, del Monte F, Sato Y, Lanner C, Breeden K, Jing SL, Allen PB, Greengard P, Yatani A, Hoit BD, Grupp IL, Hajjar RJ, DePaoli-Roach AA, Kranias EG. Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol 22: 4124–4135, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chandra M, Montgomery DE, Kim JJ, Solaro RJ. The N-terminal region of troponin T is essential for the maximal activation of rat cardiac myofilaments. J Mol Cell Cardiol 31: 867–880, 1999 [DOI] [PubMed] [Google Scholar]
- 6. Cohn JN, Levine TB, Olivari MT, Garberg V, Lura D, Francis GS, Simon AB, Rector T. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med 311: 819–823, 1984 [DOI] [PubMed] [Google Scholar]
- 7. Davis JS, Hassanzadeh S, Winitsky S, Lin H, Satorius C, Vemuri R, Aletras AH, Wen H, Epstein ND. The overall pattern of cardiac contraction depends on a spatial gradient of myosin regulatory light chain phosphorylation. Cell 107: 631–641, 2001 [DOI] [PubMed] [Google Scholar]
- 8. de Tombe PP. Congestive heart failure: role of cross-bridge cycle kinetics. Cardiovasc Res 40: 440–443, 1998 [DOI] [PubMed] [Google Scholar]
- 9. de Tombe PP, Solaro RJ. Integration of cardiac myofilament activity and regulation with pathways signaling hypertrophy and failure. Ann Biomed Eng 28: 991–1001, 2000 [DOI] [PubMed] [Google Scholar]
- 10. Foronjy RF, Sun J, Lemaitre V, D'Armiento JM. Transgenic expression of matrix metalloproteinase-1 inhibits myocardial fibrosis and prevents the transition to heart failure in a pressure overload mouse model. Hypertens Res 31: 725–735, 2008 [DOI] [PubMed] [Google Scholar]
- 11. Goldspink PH, Montgomery DE, Walker LA, Urboniene D, McKinney RD, Geenen DL, Solaro RJ, Buttrick PM. Protein kinase Cepsilon overexpression alters myofilament properties and composition during the progression of heart failure. Circ Res 95: 424–432, 2004 [DOI] [PubMed] [Google Scholar]
- 12. Gradman AH, Wilson JT. Hypertension and diastolic heart failure. Curr Cardiol Rep 11: 422–429, 2009 [DOI] [PubMed] [Google Scholar]
- 13. Hamdani N, Kooij V, van Dijk S, Merkus D, Paulus WJ, Remedios CD, Duncker DJ, Stienen GJ, van der Velden J. Sarcomeric dysfunction in heart failure. Cardiovasc Res 77: 649–658, 2008 [DOI] [PubMed] [Google Scholar]
- 14. Haq S, Choukroun G, Lim H, Tymitz KM, del Monte F, Gwathmey J, Grazette L, Michael A, Hajjar R, Force T, Molkentin JD. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation 103: 670–677, 2001 [DOI] [PubMed] [Google Scholar]
- 15. Hill JA, Olson EN. Cardiac plasticity. N Engl J Med 358: 1370–1380, 2008 [DOI] [PubMed] [Google Scholar]
- 16. Kirchhefer U, Baba HA, Boknik P, Breeden KM, Mavila N, Bruchert N, Justus I, Matus M, Schmitz W, Depaoli-Roach AA, Neumann J. Enhanced cardiac function in mice overexpressing protein phosphatase inhibitor-2. Cardiovasc Res 68: 98–108, 2005 [DOI] [PubMed] [Google Scholar]
- 17. Kirk JA, MacGowan GA, Evans C, Smith SH, Warren CM, Mamidi R, Chandra M, Stewart AF, Solaro RJ, Shroff SG. Left ventricular and myocardial function in mice expressing constitutively pseudophosphorylated cardiac troponin I. Circ Res 105: 1232–1239, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17a. MacGowan GA, Evans C, Hu TC, Debrah D, Mullet S, Chen HH, McTiernan CF, Stewart AF, Koretsky AP, Shroff SG. Troponin I protein kinase C phosphorylation sites and ventricular function. Cardiovasc Res 63: 245–255, 2004 [DOI] [PubMed] [Google Scholar]
- 18. Messer AE, Jacques AM, Marston SB. Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J Mol Cell Cardiol 42: 247–259, 2007 [DOI] [PubMed] [Google Scholar]
- 19. Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res 86: 386–390, 2000 [DOI] [PubMed] [Google Scholar]
- 20. Morano I. Effects of different expression and posttranslational modifications of myosin light chains on contractility of skinned human cardiac fibers. Basic Res Cardiol 87, Suppl 1: 129–141, 1992 [DOI] [PubMed] [Google Scholar]
- 21. Morano I, Ritter O, Bonz A, Timek T, Vahl CF, Michel G. Myosin light chain-actin interaction regulates cardiac contractility. Circ Res 76: 720–725, 1995 [DOI] [PubMed] [Google Scholar]
- 22. Murphy AM, Kogler H, Georgakopoulos D, McDonough JL, Kass DA, Van Eyk JE, Marban E. Transgenic mouse model of stunned myocardium. Science 287: 488–491, 2000 [DOI] [PubMed] [Google Scholar]
- 23. Nassar R, Malouf NN, Kelly MB, Oakeley AE, Anderson PA. Force-pCa relation and troponin T isoforms of rabbit myocardium. Circ Res 69: 1470–1475, 1991 [DOI] [PubMed] [Google Scholar]
- 24. Nicolaou P, Hajjar RJ, Kranias EG. Role of protein phosphatase-1 inhibitor-1 in cardiac physiology and pathophysiology. J Mol Cell Cardiol 47: 365–371, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pathak A, del Monte F, Zhao W, Schultz JE, Lorenz JN, Bodi I, Weiser D, Hahn H, Carr AN, Syed F, Mavila N, Jha L, Qian J, Marreez Y, Chen G, McGraw DW, Heist EK, Guerrero JL, DePaoli-Roach AA, Hajjar RJ, Kranias EG. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ Res 96: 756–766, 2005 [DOI] [PubMed] [Google Scholar]
- 26. Pette D, Staron RS. Myosin isoforms, muscle fiber types, and transitions. Microsc Res Tech 50: 500–509, 2000 [DOI] [PubMed] [Google Scholar]
- 27. Rajashree R, Blunt BC, Hofmann PA. Modulation of myosin phosphatase targeting subunit and protein phosphatase 1 in the heart. Am J Physiol Heart Circ Physiol 289: H1736–H1743, 2005 [DOI] [PubMed] [Google Scholar]
- 28. Reiser PJ, Portman MA, Ning XH, Schomisch Moravec C. Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am J Physiol Heart Circ Physiol 280: H1814–H1820, 2001 [DOI] [PubMed] [Google Scholar]
- 29. Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW, 2nd, Klevitsky R, Seidman CE, Seidman JG, Robbins J. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ Res 97: 1156–1163, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sanbe A, Fewell JG, Gulick J, Osinska H, Lorenz J, Hall DG, Murray LA, Kimball TR, Witt SA, Robbins J. Abnormal cardiac structure and function in mice expressing nonphosphorylatable cardiac regulatory myosin light chain 2. J Biol Chem 274: 21085–21094, 1999 [DOI] [PubMed] [Google Scholar]
- 31. Scruggs SB, Hinken AC, Thawornkaiwong A, Robbins J, Walker LA, de Tombe PP, Geenen DL, Buttrick PM, Solaro RJ. Ablation of ventricular myosin regulatory light chain phosphorylation in mice causes cardiac dysfunction in situ and affects neighboring myofilament protein phosphorylation. J Biol Chem 284: 5097–5106, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scruggs SB, Walker LA, Lyu T, Geenen DL, Solaro RJ, Buttrick PM, Goldspink PH. Partial replacement of cardiac troponin I with a non-phosphorylatable mutant at serines 43/45 attenuates the contractile dysfunction associated with PKCepsilon phosphorylation. J Mol Cell Cardiol 40: 465–473, 2006 [DOI] [PubMed] [Google Scholar]
- 33. Sheehan KA, Ke Y, Solaro RJ. p21-Activated kinase-1 and its role in integrated regulation of cardiac contractility. Am J Physiol Regul Integr Comp Physiol 293: R963–R973, 2007 [DOI] [PubMed] [Google Scholar]
- 34. Sheehan KA, Ke Y, Wolska BM, Solaro RJ. Expression of active p21-activated kinase-1 induces Ca2+ flux modification with altered regulatory protein phosphorylation in cardiac myocytes. Am J Physiol Cell Physiol 296: C47–C58, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev 79: 215–262, 1999 [DOI] [PubMed] [Google Scholar]
- 36. van der Velden J, Narolska NA, Lamberts RR, Boontje NM, Borbely A, Zaremba R, Bronzwaer JG, Papp Z, Jaquet K, Paulus WJ, Stienen GJ. Functional effects of protein kinase C-mediated myofilament phosphorylation in human myocardium. Cardiovasc Res 69: 876–887, 2006 [DOI] [PubMed] [Google Scholar]
- 37. van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, Burton PB, Goldmann P, Jaquet K, Stienen GJ. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res 57: 37–47, 2003 [DOI] [PubMed] [Google Scholar]
- 38. van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JM, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJ, van der Velden J. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 119: 1473–1483, 2009 [DOI] [PubMed] [Google Scholar]
- 39. Villar AV, Llano M, Cobo M, Exposito V, Merino R, Martin-Duran R, Hurle MA, Nistal JF. Gender differences of echocardiographic and gene expression patterns in human pressure overload left ventricular hypertrophy. J Mol Cell Cardiol 46: 526–535, 2009 [DOI] [PubMed] [Google Scholar]
- 40. Walker LA, MacDonald JA, Liu X, Nakamoto RK, Haystead TA, Somlyo AV, Somlyo AP. Site-specific phosphorylation and point mutations of telokin modulate its Ca2+-desensitizing effect in smooth muscle. J Biol Chem 276: 24519–24524, 2001 [DOI] [PubMed] [Google Scholar]
- 41. Walker LA, Medway AM, Walker JS, Cleveland JC, Jr, Buttrick PM. Tissue procurement strategies affect the protein biochemistry of human heart samples. J Muscle Res Cell Motil 31: 309–314, 2011 [DOI] [PubMed] [Google Scholar]
- 42. Walker LA, Walker JS, Ambler SK, Buttrick PM. Stage-specific changes in myofilament protein phosphorylation following myocardial infarction in mice. J Mol Cell Cardiol 48: 1180–1186, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang F, Aiello DL, Pyle WG. Cardiac myofilament regulation by protein phosphatase type 1alpha and CapZ. Biochem Cell Biol 86: 70–78, 2008 [DOI] [PubMed] [Google Scholar]
- 44. Yin FC, Spurgeon HA, Weisfeldt ML, Lakatta EG. Mechanical properties of myocardium from hypertrophied rat hearts. A comparison between hypertrophy induced by senescence and by aortic banding. Circ Res 46: 292–300, 1980 [DOI] [PubMed] [Google Scholar]