

Abstract
Many survivors of sudden cardiac death (SCD) have normal global ventricular function and severe coronary artery disease but no evidence of symptomatic ischemia or infarction before the development of lethal ventricular arrhythmias, and the trigger for ventricular tachycardia (VT)/ventricular fibrillation (VF) remains unclear. We sought to identify the role of spontaneous ischemia and temporal hemodynamic factors preceding SCD using continuous telemetry of left ventricular (LV) pressure and the ECG for periods up to 5 mo in swine (n = 37) with hibernating myocardium who experience spontaneous VT/VF in the absence of heart failure or infarction. Hemodynamics and ST deviation at the time of VT/VF were compared with survivors with hibernating myocardium as well as sham controls. All episodes of VT/VF occurred during sympathetic activation and were initiated by single premature ventricular contractions, and the VT degenerated into VF in ∼ 30 s. ECG evidence of ischemia was infrequent and no different from those that survived. Baseline hemodynamics were no different among groups, but LV end-diastolic pressure during sympathetic activation was higher at the time of SCD (37 ± 4 vs. 26 ± 4 mmHg, P < 0.05) and the ECG demonstrated QT shortening (155 ± 4 vs. 173 ± 5 ms, P < 0.05). The week before SCD, both parameters were no different from survivors. These data indicate that there are no differences in the degree of sympathetic activation or hemodynamic stress when VT/VF develops in swine with hibernating myocardium. The transiently elevated LV end-diastolic pressure and QT shortening preceding VT/VF raises the possibility that electrocardiographically silent subendocardial ischemia and/or mechanoelectrical feedback serve as a trigger for the development of SCD in chronic ischemic heart disease.
Keywords: hibernating myocardium, subendocardial ischemia, sudden cardiac death, ventricular fibrillation, ventricular tachycardia
the majority of the victims of sudden cardiac death (SCD) have significant coronary artery disease. While some are survivors of myocardial infarction and have heart failure, nearly one in three patients present with arrhythmic death as their first and only manifestation of heart disease (21, 32). While these patients are at risk of acute myocardial infarction, pathological studies (2, 26) have shown that acute coronary artery thrombosis and myocardial infarction are less common in previously asymptomatic patients dying of SCD. This observation is paralleled by the modest frequency of acute myocardial infarction and heart failure found in survivors when cardiac arrest is the initial presentation of heart disease (32). Collectively, these clinical observations have led to the notion that reversible asymptomatic myocardial ischemia may provide a trigger for ventricular tachycardia (VT)/ventricular fibrillation (VF) and is supported by the fact that SCD frequently develops during sympathetic activation. While an imbalance between myocardial supply and demand distal to a stenosis increases with tachycardia, the role of subendocardial ischemia in the pathogenesis of SCD remains unclear (30, 32). Challenging the importance of transient ischemia as a trigger are two observations. First, electrocardiographic evidence of ST segment depression or elevation before a lethal arrhythmic event in monitored ambulatory patients is uncommon (1). Second, while subendocardial ischemia is regularly reproduced during exercise stress testing, cardiac arrest in patients with coronary disease and normal ventricular function is infrequent. Identifying patients at risk of SCD in patients with coronary artery disease without depressed left ventricular (LV) function and heart failure continues to be a major clinical problem due to the poor survival after cardiac arrest and the lack of identifiable substrate factors other than underlying coronary artery disease (21).
Understanding the role of spontaneous ischemia in SCD would be greatly advanced by a suitable preclinical animal model of chronic ischemic heart disease. In this regard, we (3, 24) have previously demonstrated that pigs with a critical impairment in coronary flow reserve from a chronic left anterior descending coronary artery (LAD) stenosis develop hibernating myocardium with a high risk of developing spontaneous SCD during normal activity. The arrhythmic mechanism of death is VT, which rapidly progresses to VF in a fashion similar to patients with ischemic heart disease. This occurs in the absence of any malignant prodromal ventricular arrhythmias. Our previous work (3) confirmed that VT/VF in this model is not associated with an acute myocardial infarction since total coronary occlusion and the physiological features of hibernating myocardium frequently precede SCD and significant pathological scar is absent at postmortem. Implantable loop recorders and telemetry of heart rhythm have documented a diurnal variation in SCD similar to humans as well as evidence of sympathetic activation (sinus tachycardia) when VT/VF develops (24). While a clinical study (25) has also demonstrated diurnal variations in the onset of myocardial infarction and infarct size, acute infarction is absent in these animals. In addition, ambulatory monitoring has not demonstrated a high frequency of ischemia by ST segment changes over a limited 24-h recording period (3), which was also found in a preliminary study (24) using telemetry.
Since the ECG may be relatively insensitive to detect subtle degrees of subendocardial ischemia (7), hemodynamic indexes such as LV end-diastolic pressure (LVEDP) or indexes of LV contractility might provide more sensitive indicators of ischemia since they change quickly after total coronary occlusion (27) and before the development of ST segment changes or angina in patients (5). We therefore performed the present study to determine if LV hemodynamic variables at the onset of a spontaneous episode of VT/VF in swine with hibernating myocardium differ from animals that survive. To accomplish this, unrestrained animals were chronically instrumented for continuous telemetric monitoring of LV pressure and ECG. The results confirmed clinical observations in that SCD from VT/VF develops during fairly brief periods of sympathetic activation. These were preceded by significant elevations in LVEDP indicative of subendocardial ischemia that is “electrocardiographically silent” and may provide a trigger for stretch-induced arrhythmogenesis.
METHODS
Experiments were conducted in a total of 46 farm-bred pigs. A preliminary study (24) in a subgroup of the animals evaluating the ECG at the time of SCD (n = 7) in this model has been previously published. All experimental procedures and protocols were approved and conformed with institutional guidelines for the care and use of animals in research.
Porcine model of hibernating myocardium.
Details of the experimental model and its characterization have been previously published (3, 24). Briefly, animals were sedated with Telazol (tiletamine/zolazepam)-xylazine, intubated, and ventilated with an isoflurane-oxygen mixture to achieve a surgical plane of anesthesia. Through a sterile left thoracotomy, the proximal LAD of juvenile pigs (n = 37, 7.9 ± 0.2 kg, 33 ± 1 days of age) was instrumented with a 1.5 mm (inner diameter) Delrin stenosis. At the time of instrumentation, this stenosis approximated the size of the coronary artery; however, with growth of the animal, progressive stenosis of the LAD leads to the development of a chronic occlusion or high-grade stenosis with collateral-dependent myocardium. As a result, the physiological features of hibernating myocardium regularly develop 3 mo after instrumentation. These include regional anterior dysfunction in the absence of necrosis, a critical reduction in the subendocardial flow reserve, and the absence of metabolic evidence of ischemia at rest or during submaximal stress (8–10). Additional sham control animals with normal flow and function were subjected to dissection of the proximal LAD without placement of a stenosis (n = 9, 8.0 ± 0.3 kg, P = 0.81 vs. instrumented animals, 33 ± 2 days of age, P = 0.59 vs. instrumented animals). After the surgery was complete, the chest was closed in layers, the pneumothorax was evacuated, and the animal received analgesics and antibiotics as previously described.
Telemetry implantation.
Surgical implantation of the telemetry system was delayed due to the small initial size of the animal. Thus, pigs underwent a second sterile surgery for implantation of a telemetry transmitter (model TL11M3-D70-CCP, Data Sciences, St. Paul, MN) ∼1 mo after the initial surgery (29 ± 1 days). Details of the surgical procedure have been previously described (24). Briefly, LV pressure was monitored with a small fluid-filled catheter inserted through the LV apex via a subxiphoid approach, and electrocardiographic signals were monitored with two pairs of biopotential electrodes (Fig. 1A). One pair of electrodes was secured to the epicardial surface of the heart overlying the anteroapical wall for measuring the local epicardial electrogram, and the second set was tunneled subcutaneously and positioned to approximate a lead III configuration (24). Preliminary experiments have shown that lead III of the ECG resulted in the greatest ST segment change during acute transient LAD occlusion in closed-chest swine. Our initial approach for telemetry implants placed the transmitter and battery housing in a subcutaneous pocket in the lateral left chest. Unfortunately, this approach was frequently complicated by pocket erosions through the skin and local infections requiring euthanasia of the animals before experiments were complete. To circumvent this, subsequent implants were placed in the upper ventral abdomen and secured in the subcutaneous fat of the anterior abdominal wall with a very low frequency of infectious complications (detailed below). After telemetry implantation, animals were treated with analgesics as well as antibiotics (1 mg/kg iv gentamycin and 25 mg/kg iv cefazolin).
Fig. 1.

Schematic of telemetry implantation and monitoring. Approximately 1 mo before implantation of the telemetry unit, instrumented animals had a Delrin stenosis secured around the proximal left anterior descending coronary artery (LAD). A, left: orientation of the telemetry unit in the ventral abdominal wall with the pressure catheter and biopential leads advanced through the ventral diaphragm. The catheter was secured in the left ventricular (LV) apex with a purse-string suture. One pair of biopotential leads was positioned in the subcutaneous fat to approximate a lead III configuration, and the second set was secured to the anterior epicardium. Right, representative tracings that were continuously acquired from the telemetry unit. B, schematic of the external monitoring equipment. The radiofrequency signal was monitored by one of four receivers per cage and continuously digitized (1,000 Hz) on a personal computer (PC).
The radiofrequency signal of the telemetry transmitters was detected by receivers (model RMC-1, Data Sciences) attached to the side wall of each animal's cage (area: 3.4 m2). When only two receivers were used, signal drop out would occur at various locations within the cage. This was subsequently circumvented by using 4 receivers/cage with the final configuration shown in Fig. 1B. Each of the three channels of telemetry data was digitized at 1,000 Hz and continuously recorded on a personal computer using the HEM analysis program (Notocord Systems, Croissy sur Seine, France). The stated initial accuracy of pressure measurements with the D70 transmitter was ±3 mmHg with a drift of <2 mmHg/mo (Data Sciences).
Exclusions and duration of telemetry monitoring.
Nine animals were euthanized within 1 mo of telemetry implantation and excluded from any further analysis. These included six animals that developed infected transmitters (three of which had the initial chest wall transmitter location), one animal with a defective telemetry unit, and two animals with serious medical conditions (one developed cyanosis, dyspnea, and lethargy due to a large myocardial infarction and one had severe dehydration secondary to unremitting diarrhea). The remaining 37 animals were continuously monitored with telemetry until the occurrence of spontaneous SCD (n = 13) or until they were censored. SCD did not occur in any sham-instrumented animals. Criteria for ending telemetry data acquisition included planned euthanasia in hibernating animals (n = 11, 110 ± 9 days after telemetry implantation and 141 ± 10 days after initial instrumentation) or sham-instrumented animals (n = 5, 94 ± 3 days after telemetry implantation and 122 ± 3 days after initial instrumentation). Eight other animals were observed for >4 mo and censored at the time they began other protocols (hibernating animals: n = 5, 118 ± 5 days after telemetry implantation and 146 ± 6 days after initial instrumentation; sham-instrumented control animals: n = 3, 118 ± 11 days after telemetry implantation and 143 ± 10 days after initial instrumentation). The total duration of functioning telemetry for animals in each subgroup is shown in Fig. 2.
Fig. 2.

Duration of telemetry monitoring from the initial surgery. The days from initial LAD instrumentation to sudden death (red diamonds) or censoring (open symbols) are shown for each animal. All animals were continuously monitored from the day of telemetry implantation. To account for differences in age among the groups, the day of detailed study chosen for the survivors (solid blue diamonds) and sham-instrumented controls (solid green diamonds) was offset by the mean difference in days of monitoring from the sudden cardiac death (SCD) group. The squares show the mean values for each group. Time 0 is the time of LAD instrumentation, and the time of telemetry initiation in each animal is not reported (average of 29 ± 1 days after LAD instrumentation).
Physiological experiments to assess myocardial perfusion and function.
Physiological experiments to document functional stenosis severity were performed on a subset of the chronically instrumented animals ∼3 mo after the initial surgery (n = 16, 92 ± 1 days after stenosis placement). Animals were sedated with Telazol-xylazine, maintained with propofol, and studied using a previously described percutaneous approach (3). We placed an introducer in a brachial artery through which a 5-Fr end-hole transducer-tipped catheter (Millar Instruments) was advanced into the LV for measurements of systolic pressure, LVEDP, and the first derivative of LV pressure (dP/dt). LV systolic pressure was matched to the arterial systolic pressure measured from the side port of the introducer. In 12 of the animals, hemodynamic parameters at baseline in the propofol-sedated state were also compared with a 5-min basal period from the telemetry data in the conscious unrestrained state on the morning of the physiological experiment to assess the effects of anesthesia. Regional wall thickening (end-systolic wall thickness − end-diastolic wall thickness) was quantified by transthoracic echocardiography using anatomic M-mode (GE Vivid 7 or System 5) and standard American Society of Echocardiography criteria. Fractional shortening {[(LV end-diastolic dimension − LV end-systolic dimension)/LV end-diastolic dimension] × 100} and ejection fraction {[(LV end-diastolic dimension3 − LV end-systolic dimension3)/LV end-diastolic dimension3] × 100} were estimated from M-mode measurements and expressed as percentages to provide indexes of global function. Regional myocardial perfusion (n = 15) was assessed with fluorescent-labeled microspheres (15 μm) injected over 15 s into the LV catheter at rest and during adenosine vasodilation (0.9 mg·kg−1·min−1) with phenylephrine coinfused to maintain systolic pressure (8, 28). Finally, the severity of LAD anatomic stenosis was assessed with selective coronary angiography.
Telemetry monitoring continued until spontaneous SCD or until animals were euthanized under isoflurane anesthesia. The heart was immediately removed postmortem, and the LV was sectioned into concentric rings. Myocardial perfusion was quantified in a midventricular ring that was divided into 12 segments, each of which was subdivided into subendocardial, midmyocardial, and subepicardial thirds. Three thin concentric LV rings were stained with triphenyltetrazolium chloride for the quantification of myocardial fibrosis. Areas of infarction were scanned, digitized, and quantified as a percentage of LV mass (3).
Telemetry data and statistical analyses.
Animals with a chronic LAD stenosis that developed SCD were compared with survivors as well as sham-instrumented controls (shams). We determined the mechanism of arrhythmia at the time of SCD as well as hemodynamic parameters and extent of ST segment deviation that were present immediately before the change in rhythm from sinus to VT/VF. The preliminary analysis of SCD events and our previous study (3) consistently revealed a significant increase in heart rate over baseline values immediately before SCD. Since sympathetic activation appeared to be a prerequisite for the development of SCD, our analysis focused on comparing corresponding episodes among the three groups (sudden death, survivors, and shams). To control for the time at which SCD developed, the day of analysis for survivor and sham animals was matched to approximate the average value of the SCD group. Thus, the day of analysis measured from the initial surgery was 72 ± 9 days for SCD animals, 74 ± 6 days for survivors, and 72 ± 5 days for sham animals (P = 0.75 by ANOVA). We also evaluated whether there were temporal changes in the hemodynamic response to sympathetic activation in each animal. To accomplish this, we evaluated hemodynamics and electrocardiographic changes during a comparable episode of sympathetic activation 1 wk before that corresponding to SCD or the comparison day of analysis for hibernating survivors and sham controls.
Physiological parameters were averaged for 20 beats during the stable baseline immediately preceding sympathetic activation and at peak sympathetic activation. For SCD events, the period of peak sympathetic activation was defined as the 20 beats immediately preceding the onset of VT. All other episodes of sympathetic activation were defined as 20 beats before the maximum heart rate on the primary day of analysis. Heart rate, systolic pressure, LVEDP, peak +dP/dt, and peak −dP/dt were determined from the LV pressure tracings. All physiological values were confirmed with digital caliper measurements of a representative beat. In five animals (1 SCD, 3 survivors, and 1 sham), the LV pressure tracing was of poor quality and heart rate was quantified from the subcutaneous ECG. Since automated computer analysis of ST segment deviation from the software was not always reliable, manual measurements were performed. Due to the rapid heart rate during peak sympathetic activation, ST segment deviation was assessed 40 ms after the J point. ST deviation of ≥0.1 mV from baseline was considered significant.
Reported values are means ± SE. Due to the influence of extreme values for the length of sympathetic activation and VT before SCD, the median values and range are also reported. Differences in continuous parameters among groups were compared with ANOVA, with the Holm-Sidak test used for post hoc comparisons among groups (SigmaStat 3.0). For data that were not normally distributed (e.g., length of sympathetic activation), ANOVA on ranks was used (SigmaStat 3.0). The χ2-test was used to compare the frequency of significant ST segment changes among groups. P values of <0.05 were considered statistically significant.
RESULTS
Pathology, hemodynamics, myocardial perfusion, and myocardial function.
Postmortem triphenyltetrazolium chloride staining revealed minimal chronic irreversible injury in the sudden death group (1.0 ± 0.4% of LV mass), which was not different from the survivors (1.0 ± 0.3%) or shams (0.6 ± 0.4%, P = 0.26 by ANOVA on ranks). Most of this was chronic fibrosis localized around the apex of the heart where the purse string suture was placed for the LV pressure catheter. Coronary angiography at the time of physiological study revealed an average stenosis severity of 99 ± 1% with complete occlusion of the LAD and collateral-dependent myocardium in eight animals (53%). Table 1 shows a comparison of hemodynamic values obtained from telemetry in the resting awake state immediately before the physiological experiments with those obtained using acutely placed catheters during propofol sedation. Hemodynamic measurements using the Millar catheter were similar to those obtained using telemetry. There were no significant differences in heart rate, systolic pressure, or LVEDP in awake versus propofol-sedated animals. Peak LV +dP/dt was decreased after propofol anesthesia, whereas LV relaxation (reflected by −dP/dt) was unchanged.
Table 1.
Baseline LV hemodynamics from telemetry versus closed-chest hemodynamics using the LV Millar catheter
| HR, beats/min | SBP, mmHg | LVEDP, mmHg | dP/dtmax, mmHg/s | dP/dtmin, mmHg/s | |
|---|---|---|---|---|---|
| Telemetry | 108 ± 3 | 121 ± 3 | 25 ± 3 | 2612 ± 106 | −2207 ± 112 |
| Physiological study | 101 ± 4 | 115 ± 4 | 28 ± 2 | 1896 ± 84* | −2199 ± 122 |
Values are means ± SE. HR, heart rate; SBP, systolic blood pressure; LVEDP, left ventricular (LV) end-diastolic pressure; dP/dtmax, peak +dP/dt; dP/dtmin, peak −dP/dt.
P < 0.05 vs. telemetry.
Regional perfusion at rest and during adenosine vasodilation is shown in Fig. 3. In contrast to the 6-fold increase in flow to remote, normally perfused myocardium during adenosine (3.31 ± 0.22 vs. 0.56 ± 0.05 ml·min−1·g−1 at baseline, P < 0.0001), full-thickness flow was only able to increase 1.8-fold in the LAD region (1.04 ± 0.12 vs. 0.60 ± 0.05 ml·min−1·g−1 at baseline, P < 0.01). There was a critical reduction in the LAD subendocardium, with vasodilated flow unable to increase over resting values (0.69 ± 0.07 ml·min−1·g−1 at rest and 0.58 ± 0.10 ml·min−1·g−1 during adenosine, P = 0.45). Regional systolic dysfunction was present with LAD wall thickening reduced compared with the remote, normally perfused regions (regional wall thickening: 2.2 ± 0.2 mm in the LAD region vs. 4.3 ± 0.4 mm in the normal remote myocardium, P < 0.001). Overall LV systolic function was near normal (fractional shortening: 25 ± 2%, ejection fraction: 56 ± 4%). Collectively, the physiological findings confirmed viable dysfunctional hibernating myocardium.
Fig. 3.

Regional perfusion at rest and adenosine vasodilation in animals with hibernating myocardium. In animals instrumented with a chronic stenosis, there was a critical limitation in the LAD region (left) subendocardial flow reserve. Full-thickness perfusion (FT) during adenosine vasodilation (gray circles) was only able to increase 80% over baseline values (black triangles). In contrast, there was a 6-fold increase in flow during adenosine vasodilation in the remote normally perfused myocardium of the same animals (right). These findings are typical of swine with hibernating myocardium. Endo, endocardium; Mid, midmyocardium; Epi, epicardium.
Arrhythmic mechanism of SCD in pigs with hibernating myocardium.
Of the 29 pigs with chronic instrumentation of the LAD, SCD occurred in 13 pigs (SCD group) after 43 ± 9 days of continuous telemetry (72 ± 9 days from the initial instrumentation surgery). The remaining 16 pigs with hibernating myocardium survived until the time that they were censored or euthanized (survivors, 113 ± 6 days of telemetry and 142 ± 7 days after initial instrumentation). Eight sham control pigs were also monitored over a similar time frame (103 ± 6 days of telemetry and 132 ± 5 days after initial instrumentation).
All episodes of SCD occurred during the day, and the average time was 9:23 AM ± 36 min. This was similar to the time that peak heart rate occurred during sympathetic activation in the other groups of animals (survivors: 9:26 AM ± 34 min and shams: 8:33 AM ± 51 min, P = 0.27 by ANOVA on ranks). Telemetry recordings from a representative episode of spontaneous SCD are shown in Fig. 4. In each recorded event, the arrhythmic cause of SCD was VT degenerating into VF. The median length of VT before degeneration into VF was 28 s (mean: 107 s, range: 6–550 s). VT was initiated by a premature ventricular complex (coupling interval: 180 ± 6 ms), which occurred after the end of the preceding T wave.
Fig. 4.

Representative telemetry recording of sudden death. Top: low-speed tracings of digitized electrograms and LV pressure from baseline levels to the sympathetic activation (SA) associated with spontaneous SCD. The LV pressure recording was used to determine LV dP/dt and heart rate. Ventricular tachycardia (VT) was initiated by a late-coupled premature ventricular complex, and, within seconds, the rhythm degenerated into ventricular fibrillation (VF) with a loss of LV pressure. Selected time points of interest are expanded in the high-speed recording at the bottom.
We evaluated whether there were differences in the QT interval at baseline and during sympathetic activation among groups. Since heart rates at baseline and sympathetic activation were similar among groups, we used uncorrected raw QT measurements. There were no significant differences in baseline QT interval at rest (shams: 246 ± 6 ms, survivors: 263 ± 10 ms, and SCD 248 ± 7 ms, P = not significant). In contrast, animals that developed VT/VF had a more pronounced reduction in the QT interval during sympathetic activation (155 ± 4 ms in SCD animals vs. 173 ± 5 ms in survivors, P < 0.05). Figure 5 shows the individual QT-RR relation for animals that developed VT/VF versus those that survived. For any given RR interval, the QT interval was systematically shorter immediately before VT/VF versus animals with hibernating myocardium that survived. Thus, while there were no significant baseline differences in myocardial repolarization, animals that developed SCD were characterized by more pronounced QT shortening immediately before the development of VT/VF.
Fig. 5.

QT-RR data at peak SA. The individual data points immediately preceding the development of VT/VF were compared with survivors at the point of peak heart rate. For any given heart rate, the QT interval was shorter in animals that developed SCD versus those that survived. The slopes of the QT-RR relations were significantly different (P < 0.05).
Hemodynamic parameters at the time of SCD.
The duration of sympathetic activation before the development of VT/VF in swine with SCD was relatively brief (median: 200 s, mean: 681 s, range: 55–2,196 s). This was not different from either survivors (median: 263 s, mean: 422 s, range 14–1,337 s) or sham-instrumented control pigs (median: 393 s, mean: 553 s, range: 108–1,435 s, P = 0.80 by ANOVA on ranks). The main hemodynamic determinants of myocardial O2 consumption and LVEDP at baseline and during sympathetic activation for each of the three groups of animals are shown in Fig. 6. Immediately before SCD, heart rate had increased from a baseline of 130 ± 5 to 245 ± 8 beats/min (P < 0.001), and LV systolic pressure increased from 115 ± 4 to 137 ± 4 mmHg (P < 0.001). The values were similar to heart rate and LV systolic pressure at baseline and peak sympathetic activation in survivors and shams. While baseline values of peak positive dP/dt, peak negative dP/dt, and LVEDP were also similar across the three groups, there were significant differences during sympathetic activation. Myocardial contractility during sympathetic activation (as reflected by peak +dP/dt) was lower in animals that developed SCD (4,237 ± 347 mmHg/s) compared with survivors (5,374 ± 444 mmHg/s, P < 0.05) and shams (5,932 ± 472 mmHg/s, P < 0.05) at a similar LV systolic pressure and heart rate. Peak −dP/dt was also lower in animals that developed SCD, reflecting impaired LV relaxation compared with survivors (−3,004 ± 205 in SCD animals vs. −3,621 ± 282 mmHg/s in survivors, P < 0.05).
Fig. 6.

Recorded hemodynamic parameters at baseline (BL) and during SA. On the day of SCD (red symbols), BL hemodynamic parameters were similar to survivors (blue symbols) and shams (green symbols). The episode of SA immediately before SCD resulted in comparable increases in heart rate and systolic pressure compared with the other groups. However, during SA, the SCD group had a higher LV end-diastolic pressure (LVEDP) and lower LV contractility (peak +dP/dt) compared with shams. Peak −dP/dt in the SCD group was lower than in survivors but comparable with sham controls. Paired BL and SA data were all significant (P < 0.05; not shown for clarity) with the exception of LVEDP in the sham group.
Since LV dP/dt is a load-dependent index of contractility, we evaluated LVEDP in each group. There were no differences at rest. In contrast, LVEDP during sympathetic activation in SCD animals (37 ± 4 mmHg) was significantly higher than in shams (26 ± 4 mmHg, P < 0.05) as well as compared with initial baseline measurements (P < 0.001). LVEDP during sympathetic activation in survivors with hibernating myocardium was higher than the initial baseline (P < 0.001) but intermediate (31 ± 3 mmHg) and not significantly different from either of the other two experimental groups. These data indicate that animals that developed lethal VT/VF had similar baseline hemodynamics as survivors but a higher LVEDP and reduced contractility during the transient sympathetic activation immediately preceding arrhythmic death from VT/VF.
The substantial increase in the determinants of myocardial O2 consumption (heart rate, systolic pressure, and LV contractility) arising after sympathetic activation would be expected to result in acute subendocardial ischemia in the setting of a severe stenosis or collateral-dependent myocardium. Thus, the increase in LVEDP in animals with hibernating myocardium may reflect transient demand-induced subendocardial ischemia. While anticipated based on the critical impairment in subendocardial flow at rest, as shown in Fig. 3, significant ST segment deviation was uncommon. During sympathetic activation, only 5 of 29 animals (17%) with a chronic stenosis had ≥0.1-mV change in the ST segment from baseline levels, and the average absolute change in the ST segment was no different among groups (Fig. 7). Furthermore, there was no correlation between peak heart rate and the absolute change in the ST segment for either the survivor (r2 = 0.10) or SCD (r2 = 0.00) groups. There was also no significant correlation between ST segment deviation and the duration of sympathetic activation, although the SCD event with the greatest ST segment deviation (−0.42 mV) had the second longest duration of sympathetic activation (1,745 s). These data indicate that while the transient elevation in preload during stress in swine with hibernating myocardium likely reflects consequences of acute subendocardial ischemia, it is for the most part electrocardiographically silent.
Fig. 7.
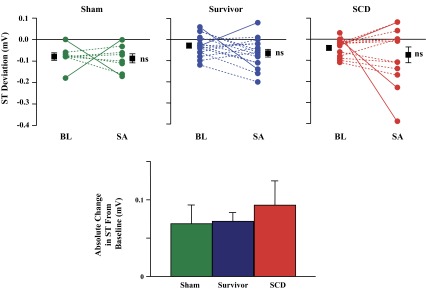
ST segment deviation at BL and during SA. Top: the majority of animals had no significant change in ST segment deviation from BL to SA (dashed lines), and there were no differences in mean values (larger symbols) among groups. A significant change in ST segment deviation (≥0.1 mV from BL, solid lines) occurred in three animals immediately before SCD (red symbols) but also in two survivors (blue symbols) and two sham animals (green symbols). Furthermore, there were no differences among groups in ST segment deviation expressed as the absolute value of deviation from BL (bottom).
Progression of changes in telemetered hemodynamics in swine that developed VT/VF.
Since LV +dP/dt was lower and LVEDP was higher in animals that developed SCD, we tested the hypothesis that the hemodynamic parameters observed at the time VT/VF develops may represent a progression in the severity of LV dysfunction. To evaluate this, telemetered hemodynamic parameters were assessed at rest and during a comparable morning episode of sympathetic activation 1 wk before the time at which SCD developed. Heart rate and LV systolic pressure at baseline and during peak sympathetic activation were similar for each group and time point. Changes in peak LV +dP/dt and LVEDP from baseline to sympathetic activation for SCD animals as well as survivors and shams are shown in Fig. 8. Although peak LV +dP/dt was unchanged between the two time points in each group (Fig. 8, left), it was systematically lower in survivors and animals that developed SCD (P < 0.05). In the sham and survivor groups, the change in LVEDP during sympathetic activation was also similar at both time points (Fig. 8, right). In contrast, the change in LVEDP during sympathetic activation in the SCD group was similar to survivors 1 wk before SCD (+7 ± 3 mmHg) but higher on the day of SCD (+14 ± 3 mmHg, P < 0.05 vs. the week before).
Fig. 8.

Changes in maximum dP/dt and LVEDP from BL to SA. In an effort to determine the temporal progression of the altered hemodynamics immediately before SCD, the change in maximum dP/dt (ΔdP/dt) and LVEDP (ΔLVEDP) during an episode of SA 1 wk before (WB) the primary day of analysis was evaluated. One week before SCD (light red bars), the change in maximum dP/dt from BL to SA (left graph) was comparable with that on the day of SCD (solid red bars) and lower than that in survivors (blue bars) and shams (green bars). In contrast, ΔLVEDP during SA (right graph) 1 wk before SCD was significantly lower than the day of SCD and comparable with ΔLVEDP during SA in survivors. This is consistent with a role for acute myocardial ischemia at the time of SCD. There no were significant differences in the change in peak +dP/dt or LVEDP between days in either the survivor or sham groups.
Table 2 shows a more detailed comparison of the paired telemetered variables at baseline and during sympathetic activation in animals immediately before SCD versus the week before. Baseline parameters were similar at each time point. During sympathetic activation in each group, there were no time-dependent differences in heart rate, LV pressure, or peak LV +dP/dt or LV −dP/dt. There were also no differences in ST segment deviation. In contrast, LVEDP during sympathetic activation increased from 22 ± 3 to 29 ± 4 mmHg 1 wk before SCD and from 23 ± 2 to 37 ± 4 mmHg at the time of SCD. Interestingly, the transient elevation in preload was also accompanied by the development of more pronounced QT shortening during sympathetic activation, but this was of borderline significance (172 ± 9 vs. 155 ± 4 ms at the time of VT/VF, P = 0.15).
Table 2.
Hemodynamics at the time of SCD versus the week before
| Baseline |
Sympathetic Activation |
|||
|---|---|---|---|---|
| Week before | SCD | Week before | SCD | |
| HR, beats/min | 129 ± 7 | 131 ± 5 | 254 ± 9* | 252 ± 9* |
| LV SBP, mmHg | 119 ± 3 | 115 ± 3 | 134 ± 5* | 137 ± 4* |
| LVEDP, mmHg | 22 ± 3 | 23 ± 2 | 29 ± 4* | 37 ± 4*† |
| LV +dP/dt, mmHg/s | 2,766 ± 254 | 2,723 ± 196 | 4,113 ± 413* | 4,237 ± 347* |
| LV −dP/dt, mmHg/s | −2,249 ± 121 | −2,252 ± 136 | −2,698 ± 181* | −3,004 ± 205* |
| ST segment deviation, mV | 0.07 ± 0.01 | 0.05 ± 0.01 | 0.07 ± 0.02 | 0.10 ± 0.03 |
| QT interval, ms | 247 ± 7 | 248 ± 7 | 172 ± 9* | 155 ± 4* |
| Duration of sympathetic activation, s | 308 ± 130 | 681 ± 211 | ||
Values are means ± SE; n = 13 animals. SCD, sudden cardiac death.
P < 0.05 vs. Baseline;
P < 0.05 vs. the week before.
DISCUSSION
Our telemetry study provides novel insights into the physiological parameters that immediately precede the development of spontaneous VT/VF in a model of chronic ischemic heart disease that has preserved global LV function and no significant infarction or heart failure. This may have translational relevance to understanding factors that precipitate SCD as the initial manifestation of underlying coronary artery disease in patients. We found that SCD occurred during sympathetic activation and was initiated by a premature ventricular contraction (PVC) followed by a relatively short period of VT, which degenerated into VF within ∼30 s. Despite a critical impairment in the subendocardial flow reserve, ST segment deviation beyond baseline at the time of VT/VF was uncommon, as it is in patients that develop SCD during Holter monitoring (1). Although most hemodynamic parameters were similar during sympathetic activation, LVEDP was higher immediately before VT/VF and was the only hemodynamic parameter that differed when animals with SCD were evaluated the week before they developed VT/VF. This may reflect electrocardiographically silent subendocardial ischemia and raises the possibility that stretch activated arrhythmias from transient elevations in preload may underlie the development of VT/VF in chronic ischemic heart disease.
Relationship between reversible ischemia and SCD.
Sudden cardiac arrest remains a major public health problem. Clinical studies have largely focused on SCD in high-risk groups that have depressed LV function and myocardial scar where implantable defibrillators are the only intervention that have improved survival. Despite these advances, many patients present with SCD as their initial and only manifestation of heart disease, and they are frequently asymptomatic with no history of infarction or heart failure preceding the presumed lethal ventricular arrhythmia (21, 32). Due to the low survival rate of out of hospital cardiac arrest, nearly all studies of these patients have evaluated the pathological substrate at postmortem. In this regard, while hemodynamically significant coronary stenoses have been identified in the vast majority of patients with out-of-hospital cardiac arrest, occlusive thrombus and significant acute myocardial scar can be absent in many patients (2, 26). These findings have given rise to the notion that acute myocardial ischemia from plaque instability (plaque erosion and plaque rupture) is a primary triggering event. The latter findings are also consistent with the clinical observations showing that many survivors of SCD do not have evidence of an acute myocardial infarction (32).
In the presence of stable coronary plaques, dynamic demand-induced ischemia during sympathetic activation may be an important transient initiating event or “trigger” for SCD (32). The lack of prodromal symptoms of ischemia in these patients could be explained by the fact that most episodes of spontaneous ischemia evaluated using ST segment changes on Holter recording, for example, are not associated with angina (6). Although plausible, asymptomatic or symptomatic electrocardiographic evidence of acute ischemia is usually absent in the unfortunate patients who have developed sudden death during monitoring (1) unless it is transmural as in coronary vasospasm (22).
Our hemodynamic findings raise the possibility that the lack of ST segment changes at the time VT/VF develops may reflect the insensitivity of the ECG, and, as a result, reversible demand-induced subendocardial ischemia may be occurring more frequently than realized and be electrocardiographically silent. We found that pigs with hibernating myocardium regularly developed transient elevations in LVEDP during sympathetic activation that are compatible with ischemia without exhibiting ST changes. Our previous work using 24-h Holter recordings in the same model confirmed infrequent ST segment depression. This was surprising since there was a critical impairment in the subendocardial flow reserve (as shown in Fig. 3) as well as objective pathophysiological consequences of ischemia-induced remodeling characteristic of hibernating myocardium (4). These chronic pathophysiological adaptations include resting myocardial dysfunction, increased 18F-labeled 2-deoxyglucose uptake (10, 19), apoptosis-induced myocyte loss (15), mitochondrial remodeling (13, 20, 23), and regional sympathetic denervation with nerve sprouting (11, 17), among others, and are presumed to arise from spontaneous reversible ischemia. The lack of ECG evidence of ischemia during tachycardia to a heart rate of ∼250 beats/min in these animals raises the possibility that various degrees of subclinical ischemia-induced myocardial remodeling may develop in many patients with asymptomatic as well as symptomatic coronary artery disease. While speculative, this may create myocardial cellular remodeling that leads to a substrate susceptible to lethal arrhythmias and sudden death without heart failure or infarction. Asymptomatic ischemia-induced remodeling and apoptosis-induced myocyte loss may also explain why some patients develop ischemic cardiomyopathy from severe multivessel coronary disease with no prior symptoms of angina or history of myocardial infarction.
The concept of electrocardiographically silent ischemia is supported by invasive hemodynamic studies in humans having spontaneous ischemia during cardiac catheterization. For example, Chierchia et al. (5) studied spontaneous ischemia at rest in six patients with angina. Electrocardiographic evidence of acute ischemia occurred well after a fall in coronary sinus O2 saturation and was frequently only manifested as nonspecific T wave changes as opposed to more specific ST segment depression or elevation on the ECG. They too demonstrated that alterations in hemodynamic parameters including an increase in LVEDP and decrease in LV systolic pressure and LV dP/dt occurred before ST segment changes and chest pain. Animal studies directly measuring myocardial perfusion during acute ischemia have also confirmed the insensitivity of the ECG to detect ischemia that is restricted to the subendocardium. de Chantal et al. (7) found no electrocardiographic evidence of ischemia in swine when subendocardial perfusion was reduced by up to 50% by an acute partial LAD occlusion. Collectively, the findings in humans and animal models of acute ischemia support the notion that electrocardiographically silent myocardial ischemia is common and could explain the development of extensive intrinsic adaptations of the myocardium to chronic repetitive ischemia in the absence of ST segment changes in swine with hibernating myocardium.
Transient preload elevation and the development of VT/VF.
While ischemia-induced remodeling in hibernating myocardium leads to a substrate that facilitates arrhythmogenesis, not all swine develop VT/VF so that a trigger that leads to the development of sudden death is required. We (3) previously demonstrated that PVCs and nonsustained VT occur in a minority of swine with a chronic coronary stenosis using serial Holter monitoring, but they do not presage the development of sustained VT/VF. Likewise, we have never observed self-terminating episodes of sustained VT in the present telemetry study or our previous studies. Although identifying a transient trigger is a bit like looking for a needle in the haystack, our telemetry analysis advances the understanding of this challenging area in several important ways. First, it evaluates spontaneous rather than provoked VT/VF and excludes a number of candidates as triggers. The similarity of hemodynamic variables at rest, including heart rate, LV systolic pressure, LVEDP, and indexes of global contractility, indicate that animals that develop SCD do not in some way have more severe hemodynamic impairment than survivors. This extends the results of our previous study (3), where we demonstrated that animals undergoing physiological experiments before the development of SCD had chronic occlusions with collateral-dependent hibernating myocardium. In that study, regional wall thickening and subendocardial flow were reduced to the same extent in survivors as animals that developed SCD. We also excluded progression of the stenosis as a cause of SCD since coronary angiography demonstrated that most animals had a total coronary occlusion and collateral-dependent myocardium before the development of VT/VF. All recorded episodes of spontaneous VT/VF in the present study developed during brief sympathetic activation, which contrasts with lethal arrhythmias after acute total occlusion that also develop during reperfusion. The telemetry analysis confirms that the extent of sympathetic activation (as reflected by increases in heart rate and LV pressure) or duration of sympathetic activation are no different in animals that develop SCD versus those that survive. Changes in contractility from baseline to peak sympathetic activation were similar over a period of 1 wk in each experimental group, but peak LV dP/dt during sympathetic activation was lower in animals that developed VT/VF versus survivors. This probably does not reflect a transient trigger since the level of LV dP/dt was similar 1 wk before SCD but could indicate a substrate factor (Table 2). One possibility is that it reflects more severe ischemia in animals that develop sudden death. We believe that this is unlikely since LVEDP (a sensitive index of ischemia) was not elevated the week before SCD despite a similar peak dP/dt during sympathetic activation. Likewise, our previous study (3) showed no difference in coronary flow at rest or vasodilation in animals that subsequently developed SCD versus survivors. The lower values of LV dP/dt could also reflect a larger area at risk of ischemia, but this too would also be anticipated to be accompanied by a similar elevation in preload the week before. There was no clear correlation between the development of VT/VF and any particular activity. While no specific event could be correlated with increased sympathetic activation, feeding, turning lights on, and cage washing all occurred in the morning hours. Nevertheless, similar episodes of sympathetic activation occurred throughout waking hours of the day and were not confined to the early morning.
While the vast majority of hemodynamic and electrocardiographic parameters in animals that developed SCD were similar to both survivors and shams, two parameters differed and demonstrated a temporal progression compared with values obtained in the same animal 1 wk before. First, we found that LVEDP during sympathetic activation was higher in animals at the time of SCD than shams. It was also significantly higher using paired analysis with the values obtained the week before. Second, the QT interval immediately before the development of VT/VF was significantly shorter than survivors at similar heart rates. Likewise, QT during sympathetic activation tended to be shorter than paired values obtained the week before in the same animals. These two observations could conceivably be interrelated. For example, QT shortening occurs during acute ischemia, and this could reflect more severe subendocardial ischemia in animals that develop SCD. While not predicted from analysis of the conventional indexes of demand, the differences could arise from time-dependent differences in coronary resistance adjustments during sympathetic activation. The temporal course of subendocardial perfusion would need to be assessed to evaluate this and is not currently technically possible.
An alternative explanation is that QT shortening reflects mechanical electrical feedback (29). A previous study (14) has demonstrated that increases in preload produce QT shortening. Another study (18) has confirmed that demand-induced ischemia is associated with an upward shift in the end-diastolic pressure-segment length relation as well as an increase in end-diastolic segment length and LVEDP that could elicit stretch-induced arrhythmias. It is plausible that this dynamic leads to the subsequent PVC that initiates VT/VF. Other electrophysiological factors that may promote the development of VF during sympathetic activation include steepening of the action potential duration restitution slope, triggered activity secondary to early and delayed afterdepolarizations, and inhomogeneity in cellular repolarization arising from regional sympathetic denervation and altered β-adrenergic receptor density (16). Further studies will be required to define the electrophysiological substrate at rest and during sympathetic activation in this model.
Methodological limitations.
We used heart rate and systolic blood pressure as surrogates of generalized sympathetic activity. Nevertheless, these are indirect hemodynamic indexes that reflect global sympathetic activation, and they may not always correlate with alterations in myocardial sympathetic nerve activity, which have been measured using in vivo recordings in dogs (12, 31). The latter studies demonstrate that increased sympathetic nerve activity precedes the development of ventricular arrhythmias and displays a diurnal variation resulting in myocardial sympathovagal imbalance that is maximal in the morning (8 AM to 12 PM) and correlates with the time frame during which spontaneous VT/VF developed in our swine model. Further studies will be required to determine whether the temporal characteristics of myocardial sympathetic nerve activity differ in swine with and without VT/VF.
Conclusions.
In summary, our data indicate that pigs with hibernating myocardium develop VT/VF in association with a transiently elevated LVEDP during sympathetic activation. The degree of LVEDP elevation is higher at the time of SCD and most likely reflects a subtle variation in the degree of subendocardial ischemia that is electrocardiographically silent. The subclinical episodes of repetitive ischemia stimulate substantial chronic remodeling of the hibernating myocardium without overt ST depression. Thus, the physiological stimuli required to elicit cellular remodeling may develop in the asymptomatic stage of coronary artery disease and could lead to a substrate that presents with SCD from VT/VF in the absence of an acute myocardial infarction as the first and only manifestation of heart disease. Based on this, approaches to identify myocardial cellular remodeling using circulating biomarkers or molecular imaging may afford an approach to identify ischemia-induced substrate remodeling and vulnerability to VT/VF before a lethal arrhythmic event.
GRANTS
This work was supported by the Department of Veterans Affairs, National Heart, Lung, and Blood Institute Grants HL-55324 and HL-61610, the Albert and Elizabeth Rekate Fund, the Mae Stone Goode Trust, and the John R. Oishei Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.F.P., G.S., M.D.B., and B.M.H. performed experiments; M.F.P., G.S., M.D.B., B.M.H., J.A.F., and J.M.C. analyzed data; M.F.P., J.A.F., and J.M.C. prepared figures; M.F.P. and J.A.F. drafted manuscript; M.F.P., G.S., M.D.B., B.M.H., J.A.F., and J.M.C. approved final version of manuscript; G.S., M.D.B., B.M.H., J.A.F., and J.M.C. interpreted results of experiments; J.A.F. and J.M.C. edited and revised manuscript; J.M.C. conception and design of research.
ACKNOWLEDGMENTS
The authors acknowledge the technical assistance of Deana Gretka and Elaine Granica in conducting the surgical instrumentation and assisting with the experimental aspects of the study. The authors also thank Anne Coe for assistance with the preparation of the manuscript and figures.
REFERENCES
- 1. Bayes de Luna A, Coumel P, Leclercq JF. Ambulatory sudden cardiac death: mechanisms of production of fatal arrhythmia on the basis of data from 157 cases. Am Heart J 117: 151–159, 1989 [DOI] [PubMed] [Google Scholar]
- 2. Burke AP, Kolodgie FD, Farb A, Weber DK, Malcom GT, Smialek J, Virmani R. Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation 103: 934–940, 2001 [DOI] [PubMed] [Google Scholar]
- 3. Canty JM, Jr, Suzuki G, Banas MD, Verheyen F, Borgers M, Fallavollita JA. Hibernating myocardium: chronically adapted to ischemia but vulnerable to sudden death. Circ Res 94: 1142–1149, 2004 [DOI] [PubMed] [Google Scholar]
- 4. Canty JM, Jr, Suzuki G. Myocardial perfusion and contraction in acute ischemia and chronic ischemic heart disease. J Mol Cell Cardiol 52: 822–831, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chierchia S, Brunelli C, Simonetti I, Lazzari M, Maseri A. Sequence of events in angina at rest: primary reduction in coronary flow. Circulation 61: 759–68, 1980 [DOI] [PubMed] [Google Scholar]
- 6. Cohn PF, Fox KM, Daly C. Silent myocardial ischemia. Circulation 108: 1263–1277, 2003 [DOI] [PubMed] [Google Scholar]
- 7. de Chantal M, Diodati JG, Nasmith JB, Amyot R, LeBlanc AR, Schampaert E, Pharand C. Progressive epicardial coronary blood flow reduction fails to produce ST-segment depression at normal heart rates. Am J Physiol Heart Circ Physiol 291: H2889–H2896, 2006 [DOI] [PubMed] [Google Scholar]
- 8. Fallavollita JA, Logue M, Canty JM., Jr Stability of hibernating myocardium in pigs with a chronic left anterior descending coronary artery stenosis: absence of progressive fibrosis in the setting of stable reductions in flow, function and coronary flow reserve. J Am Coll Cardiol 37: 1989–1995, 2001 [DOI] [PubMed] [Google Scholar]
- 9. Fallavollita JA, Malm BJ, Canty JM., Jr Hibernating myocardium retains metabolic and contractile reserve despite regional reductions in flow, function, and oxygen consumption at rest. Circ Res 92: 48–55, 2003 [DOI] [PubMed] [Google Scholar]
- 10. Fallavollita JA, Perry BJ, Canty JM., Jr 18F-2-deoxyglucose deposition and regional flow in pigs with chronically dysfunctional myocardium: evidence for transmural variations in chronic hibernating myocardium. Circulation 95: 1900–1909, 1997 [DOI] [PubMed] [Google Scholar]
- 11. Fernandez SF, Ovchinnikov V, Canty JM, Jr, Fallavollita JA. Hibernating myocardium results in partial sympathetic denervation and nerve sprouting. Am J Physiol Heart Circ Physiol 304: H318–H327, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Han S, Kobayashi K, Joung B, Piccirillo G, Maruyama M, Vinters HV, March K, Lin SF, Shen C, Fishbein MC, Chen PS, Chen LS. Electroanatomic remodeling of the left stellate ganglion after myocardial infarction. J Am Coll Cardiol 59: 954–961, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu Q, Suzuki G, Young RF, Page BJ, Fallavollita JA, Canty JM., Jr Reductions in mitochondrial O2 consumption and preservation of high-energy phosphate levels after simulated ischemia in chronic hibernating myocardium. Am J Physiol Heart Circ Physiol 297: H223–H232, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lerman BB, Burkhoff D, Yue DT, Franz MR, Sagawa K. Mechanoelectrical feedback: independent role of preload and contractility in modulation of canine ventricular excitability. J Clin Invest 76: 1843–1850, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lim H, Fallavollita JA, Hard R, Kerr CW, Canty JM., Jr Profound apoptosis-mediated regional myocyte loss and compensatory hypertrophy in pigs with hibernating myocardium. Circulation 100: 2380–2386, 1999 [DOI] [PubMed] [Google Scholar]
- 16. Lopshire JC, Zipes DP. Sudden cardiac death: better understanding of risks, mechanisms, and treatment. Circulation 114: 1134–1136, 2006 [DOI] [PubMed] [Google Scholar]
- 17. Luisi AJ, Jr, Suzuki G, deKemp R, Haka MS, Toorongian SA, Canty JM, Jr, Fallavollita JA. Regional 11C-hydroxyephedrine retention in hibernating myocardium: Chronic inhomogeneity of sympathetic innervation in the absence of infarction. J Nucl Med 46: 1368–1374, 2005 [PubMed] [Google Scholar]
- 18. Maehara K, Maruyama Y, Yoshikata S, Takishima T. Effects of exercise stress on left ventricular end diastolic pressure-length strain relations in dogs with and without coronary stenosis. Cardiovasc Res 26: 770–778, 1992 [DOI] [PubMed] [Google Scholar]
- 19. McFalls EO, Baldwin D, Palmer B, Marx D, Jaimes D, Ward HB. Regional glucose uptake within hypoperfused swine myocardium as measured by positron emission tomography. Am J Physiol Heart Circ Physiol 272: H343–H349, 1997 [DOI] [PubMed] [Google Scholar]
- 20. McFalls EO, Sluiter W, Schoonderwoerd K, Manintveld OC, Lamers JM, Bezstarosti K, van Beusekom HM, Sikora J, Ward HB, Merkus D, Duncker DJ. Mitochondrial adaptations within chronically ischemic swine myocardium. J Mol Cell Cardiol 41: 980–988, 2006 [DOI] [PubMed] [Google Scholar]
- 21. Myerburg RJ, Junttila MJ. Sudden cardiac death caused by coronary heart disease. Circulation 125: 1043–1052, 2012 [DOI] [PubMed] [Google Scholar]
- 22. Myerburg RJ, Kessler KM, Mallon SM, Cox MM, deMarchena E, Interian A, Jr, Castellanos A. Life-threatening ventricular arrhythmias in patients with silent myocardial ischemia due to coronary-artery spasm. N Engl J Med 326: 1451–1455, 1992 [DOI] [PubMed] [Google Scholar]
- 23. Page B, Young R, Iyer V, Suzuki G, Lis M, Korotchkina K, Patel M, Blumenthal K, Fallavollita JA, Canty JM., Jr Persistent regional downregulation in mitochondrial enzymes and upregulation of stress proteins in swine with chronic hibernating myocardium. Circ Res 102: 103–112, 2008 [DOI] [PubMed] [Google Scholar]
- 24. Pizzuto MF, Valverde AM, Heavey BM, Banas MD, Michelakis N, Suzuki G, Fallavollita JA, Canty JM., Jr Brief sympathetic activation precedes the development of ventricular tachycardia and ventricular fibrillation in hibernating myocardium. J Electrocardiol 39: S140–S145, 2006 [DOI] [PubMed] [Google Scholar]
- 25. Reiter R, Swingen C, Moore L, Henry TD, Traverse JH. Circadian dependence of infarct size and left ventricular function after ST elevation myocardial infarction. Circ Res 110: 105–110, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roberts WC, Kragel AH, Gertz SD, Roberts CS. Coronary arteries in unstable angina pectoris, acute myocardial infarction, and sudden coronary death. Am Heart J 127: 1588–1593, 1994 [DOI] [PubMed] [Google Scholar]
- 27. Serruys PW, Wijns W, van den Brand M, Meij S, Slager C, Schuurbiers JC, Hugenholtz PG, Brower RW. Left ventricular performance, regional blood flow, wall motion, and lactate metabolism during transluminal angioplasty. Circulation 70: 25–36, 1984 [DOI] [PubMed] [Google Scholar]
- 28. Suzuki G, Lee TC, Fallavollita JA, Canty JM., Jr Adenoviral gene transfer of FGF-5 to hibernating myocardium improves function and stimulates myocytes to hypertrophy and reenter the cell cycle. Circ Res 96: 767–775, 2005 [DOI] [PubMed] [Google Scholar]
- 29. Taggart P, Lab M. Cardiac mechano-electric feedback and electrical restitution in humans. Prog Biophys Mol Biol 97: 452–460, 2008 [DOI] [PubMed] [Google Scholar]
- 30. Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res 95: 754–763, 2004 [DOI] [PubMed] [Google Scholar]
- 31. Zhou S, Jung BC, Tan AY, Trang VQ, Gholmieh G, Han SW, Lin SF, Fishbein MC, Chen PS, Chen LS. Spontaneous stellate ganglion nerve activity and ventricular arrhythmia in a canine model of sudden death. Heart Rhythm 5: 131–139, 2008 [DOI] [PubMed] [Google Scholar]
- 32. Zipes DP, Wellens HJ. Sudden cardiac death. Circulation 98: 2334–2351, 1998 [DOI] [PubMed] [Google Scholar]