

Abstract
Endothelial cells (ECs) are constantly subjected to cyclic strain that arises from periodic change in vessel wall diameter as a result of pulsatile blood flow. Application of physiological levels of cyclic strain inhibits EC apoptosis; however, the underlying mechanism is not known. Since heme oxygenase-1 (HO-1) is a potent inhibitor of apoptosis, the present study investigated whether HO-1 contributes to the antiapoptotic action of cyclic strain. Administration of physiological cyclic strain (6% at 1 Hz) to human aortic ECs stimulated an increase in HO-1 activity, protein, and mRNA expression. The induction of HO-1 was preceded by a rise in reactive oxygen species (ROS) and Nrf2 protein expression. Cyclic strain also stimulated an increase in HO-1 promoter activity that was prevented by mutating the antioxidant responsive element in the promoter or by overexpressing dominant-negative Nrf2. In addition, the strain-mediated induction of HO-1 and activation of Nrf2 was abolished by the antioxidant N-acetyl-l-cysteine. Finally, application of cyclic strain blocked inflammatory cytokine-mediated EC death and apoptosis. However, the protective action of cyclic strain was reversed by the HO inhibitor tin protoporphyrin-IX and was absent in ECs isolated from HO-1-deficient mice. In conclusion, the present study demonstrates that a hemodynamically relevant level of cyclic strain stimulates HO-1 gene expression in ECs via the ROS-Nrf2 signaling pathway to inhibit EC death. The ability of cyclic strain to induce HO-1 expression may provide an important mechanism by which hemodynamic forces promote EC survival and vascular homeostasis.
Keywords: vascular biology, hemodynamic forces, endothelium
blood vessels are constantly exposed to hemodynamic forces in the form of shear stress and circumferential mechanical strain. These biomechanical forces act on vascular cells and play a critical role in regulating vascular structure and function. While the effect of fluid shear stress on the properties of endothelial cells has been extensively investigated, less is known regarding the action of cyclic mechanical strain on endothelial cell function. However, accumulating evidence indicates that cyclic strain, which arises from periodic change in vessel wall diameter as a result of pulsatile blood flow, is also a key modulator of endothelial cell function. Cyclic strain alters endothelial cell morphology and orientation (21) and plays a fundamental role in modulating blood flow by stimulating the release of humoral factors from the endothelium (3, 41). In addition, cyclic strain may promote angiogenesis by stimulating endothelial cell proliferation, migration, and tube formation and blood vessel remodeling by regulating matrix metalloproteinase expression and activation (10, 34, 45, 50). Moreover, studies from our laboratory and others indicate that physiologically relevant levels of cyclic strain are capable of inhibiting endothelial cell apoptosis (26, 29, 34). However, the underlying mechanism by which cyclic strain regulates endothelial cell survival is poorly defined.
Heme oxygenase-1 (HO-1) is a highly inducible enzyme that catalyzes the degradation of heme into equimolar amounts of carbon monoxide (CO), biliverdin, and free iron (see Refs. 14, 15, 31). Biliverdin is subsequently metabolized to bilirubin by the enzyme biliverdin reductase, while iron is rapidly sequestered by ferritin. The oxidation of heme by HO-1 is inhibited by various metalloporphyrins, including tin protoporphyrin-IX (SnPP). The induction of HO-1 by various forms of cellular stress provides an important defense mechanism against tissue injury. The cytoprotection afforded by HO-1 is mediated by several distinct mechanisms. In particular, HO-1 catabolizes prooxidant heme to the bile pigments biliverdin and bilirubin, which are effective antioxidants capable of scavenging peroxy radicals and inhibiting lipid peroxidation (40). Furthermore, the coordinated induction of ferritin with HO-1 exerts an additional antioxidant effect by chelating free iron (44). Finally, HO-1 generates the diatomic gas CO, which possesses potent antiapoptotic properties (4, 28, 48). Interestingly, the application of cyclic strain stimulates HO-1 expression in vascular cells, but the functional significance of this induction remains unclear (9, 35, 47). In the present study, we investigated the mechanism by which a hemodynamically relevant regimen of cyclic strain induces HO-1 expression in human vascular endothelium. In addition, we determined whether the induction of HO-1 by cyclic strain contributes to its ability to modulate endothelial cell survival.
MATERIALS AND METHODS
Materials.
M199 medium, streptomycin, penicillin, gelatin, ascorbic acid, uric acid, Trypan blue, sulfanilamide, dithiothreitol, trypsin, naphthyl ethylenediamine dihydrochloride, ceramide, methyl-l-arginine, heparin, formaldehyde, chloroform, sodium dodecyl sulfate (SDS), Nonidet P-40 (NP-40), NaCl, EDTA, glucose-6-phosphate, sucrose, glucose-6-phosphate dehydrogenase, hemin, HEPES, Tris, and N-acetyl-l-cysteine were purchased from Sigma-Aldrich (St. Louis, MO). Phenylmethylsulfonyl fluoride, aprotinin, leupeptin, and pepstatin A were from Roche Applied Biosciences (Indianapolis, IN). SnPP was from Frontier Scientific (Logan, UT), and Dowex 50W-X8 was from Bio-Rad Laboratories (Hercules, CA). Tumor necrosis factor-α (TNF-α) was from Genzyme (Boston, MA). An antibody against HO-1 was from Assay Designs (Ann Arbor, MI), antibodies directed against β-actin and nuclear factor erythroid-derived 2-related factor (Nrf)2 were from Santa Cruz Biotechnologies (Santa Cruz, CA), antibodies against Nrf1, glutamate-cysteine ligase modifier subunit (GCLM), and NAD(P)H dehydrogenase quinone 1 (NQO1) were from GeneTex (Irvine, CA), an antibody against endothelial nitric oxide synthase (eNOS) was from BD Transduction Laboratories (San Jose, CA), and an antibody against platelet-endothelial cell adhesion molecule-1 (PECAM-1) was from BD Biosciences (San, Jose, CA). MG-132 was from EMD Millipore (Billerica, MA). 5,6-Chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) was from Invitrogen (Carlsbad, CA). [32P]dCTP (3,000 Ci/mmol) was from Amersham (Arlington Heights, IL), and l-[3H]arginine (43 Ci/mmol) was from Perkin Elmer (Boston, MA).
Cell culture.
Human aortic endothelial cells (HAECs) and human umbilical vein endothelial cells (HUVECs) were purchased from Lonza Incorporated (Allendale, NJ) and propagated in M199 medium supplemented with 20% bovine calf serum, 2 mM l-glutamine, 50 μg/ml endothelial cell growth factor, 90 μg/ml heparin, and 100 U/ml of penicillin and streptomycin (36). Murine aortic endothelial cells (MAECs) from wild-type (HO-1+/+) and HO-1-deficient (HO-1−/−) mice were initially provided by Dr. Anupam Agarwal (Division of Nephrology, University of Alabama at Birmingham) and subsequently isolated from mice by plating thoracic aortas on Matrigel-coated plates and purifying MAEC outgrowth with a PECAM-1 antibody, as we previously described (23). Cells were characterized as endothelial cells by both positive staining for PECAM-1 and uptake of acetylated low-density lipoprotein by >95% of cells. MAECs were grown in endothelial basal medium supplemented with 10% fetal bovine serum, 50 μg/ml gentamicin, 50 μg/ml amphotericin, 1 μg/ml hydrocortisone, 10 ng/ml human epidermal growth factor, and 6 μg/ml bovine extract (Lonza Incorporated). Endothelial cells were serially cultured on gelatin-coated dishes and propagated in an atmosphere of 95% air-5% CO2.
Cyclic strain.
Cells were subjected to cyclic strain with the Flexercell 4000T Tension Plus strain unit (Flexercell, McKeesport, PA). This system consists of baseplates equipped with 25-mm loading posts connected to a tension controller and vacuum pump with attached pressure and vacuum reservoir. This system provides uniform radial and circumferential strain across a membrane surface along all radii. Cells were plated onto six-well BioFlex dishes coated with type I collagen, and these flexible-bottom dishes were placed into the baseplate assembly. Vacuum was repetitively applied to the bottom of the dishes via the baseplate, which was placed in a humidified incubator with 5% CO2 at 37°C. Endothelial cells were exposed to a physiologically relevant strain of 6% at a frequency of 1 Hz. Cells grown on BioFlex plates and simultaneously placed in a cell culture incubator served as static controls.
Western blotting.
Cells were lysed in sample buffer [125 mM Tris (pH 6.8), 12.5% glycerol, 2% SDS, 50 mM sodium fluoride, and trace bromophenol blue], sonicated, boiled at 100°C for 5 min, and centrifuged at 13,000 g for 10 min. Proteins were separated by SDS-polyacrylamide gel electrophoresis and electrophoretically transferred to nitrocellulose membranes. Blots were blocked with nonfat milk (5%) in PBS for 1 h and then incubated overnight at 4°C with specific antibodies directed against HO-1 (1:1,500), Nrf2 (1:100), Nrf1 (1:400), eNOS (1:200), PECAM-1 (1:200), GCLM (1:400), NQO1 (1:800), or β-actin (1:200). Membranes were washed in PBS, incubated with horseradish peroxidase-conjugated secondary antibodies, and developed with commercial chemoluminescence reagents (Amersham). Protein expression was quantified by scanning densitometry and normalized with respect to β-actin.
Northern blotting.
Total RNA was loaded onto 1.2% agarose gels, fractionated by electrophoresis, and blot transferred to Gene Screen Plus membranes (Perkin Elmer Life Sciences, Waltham, MA). Membranes were prehybridized for 4 h at 68°C in rapid hybridization buffer (Amersham) and then incubated overnight at 68°C in hybridization buffer containing [32P]DNA probes (1 × 108 cpm) for HO-1, Nrf2, GAPDH, or 18S rRNA. DNA probes were generated by reverse transcriptase-polymerase chain reaction (RT-PCR) and labeled with [32P]dCTP with a random priming kit (Amersham) (48). After hybridization, membranes were washed and exposed to X-ray film at −70°C, and HO-1 expression was quantified by scanning densitometry and normalized with respect to GAPDH or 18S rRNA.
Real-time polymerase chain reaction.
Total RNA was extracted with TRIzol reagent (Life Technologies, Grand Island, NY) and transcribed to cDNA with a reverse transcription kit (Bio-Rad Laboratories). Real-time PCR was performed with SYBR Green Supermix in a SYBR Green Cycler iQ 5 RT-PCR detection system (Bio-Rad Laboratories) with appropriate primers (1, 38). The primer sequences are as follows: Nox2 forward 5′-AGCTATGAGGTGGTGACCA-3′, reverse 5′-CACAATATTTGTACCAGACAGACTTGAG-3′; Nox4 forward 5′-tgttgggcctaggattgtgtt-3′, reverse 5′-AGGGACCTTCTGTGATCCTCG-3′, p22phox forward 5′-GTACTTTGGTGCCTACTCCA-3′, reverse 5′-CGGCCCGAACATAGTAATTC-3′; p47phox forward 5′-TTGAGAAGCGCTTCGTACCC-3′, reverse 5′-CGTCAAACCACTTGGGAGCT-3′; and p67phox forward 5′-CAGTTCAAGCTGTTTGCCTG-3, reverse 5′-TTCTTGGCCAGCTGAGCCAC. Relative expression of target genes was analyzed with the comparative threshold cycle (2−ΔΔCt) method, normalized to 18S rRNA, and given as a fraction compared with control, static conditions.
HO activity.
Cells were sonicated in MgCl2 (2 mM) phosphate (100 mM) buffer (pH 7.4) and centrifuged at 18,000 g for 15 min at 4°C. Supernatants were then added to a reaction mixture containing NADPH (0.8 mM), glucose-6-phosphate (2 mM), glucose-6-phosphate dehydrogenase (0.2 U), rat liver cytosol (2 mg), and hemin (20 μM). The reaction was conducted for 1 h at 37°C in the dark and terminated by the addition of chloroform. The extracted bilirubin was calculated by the difference in absorption between 464 and 530 nm with an extinction coefficient of 40 mM−1cm−1.
HO-1 small interference RNA and HO-1 gene transfer.
HO-1 expression was silenced by transfecting endothelial cells with HO-1 small interference RNA (siRNA) (100 nM) or nontargeting (NT) siRNA (100 nM) that was purchased from Dharmacon (Lafayette, CO) (48). Gene transfer of HO-1 was achieved by infecting endothelial cells with a replication-defective adenovirus expressing HO-1 (AdHO-1), as we previously reported (42). Cells were infected with AdHO-1 or an adenovirus expressing green fluorescent protein (AdGFP) at a multiplicity of infection of 20.
Reactive oxygen species measurement.
Cell monolayers were incubated with CM-H2DCFDA (5 μM) for 30 min at 37°C prior to collection. The cells were then washed, detached, and centrifuged at 1,000 g for 5 min. Precipitated cells were washed and resuspended in PBS containing 1% fetal bovine serum, and reactive oxygen species (ROS) production was determined by measuring the intracellular oxidation of the dye to 2,7-dichlorofluorescein by flow cytometry using excitation at 488 nm and emission at 530 nm (Dickinson FACScan, Franklin Lakes, NJ).
eNOS activity.
eNOS activity was determined by measuring the conversion of l-[3H]arginine to [3H]citrulline, as we previously described (16). Cells were incubated with l-[3H]arginine (2 μCi)-containing culture medium for 4 h, culture medium was then removed, and cells were washed three times with PBS. Cells were then collected in ice-cold Tris lysis buffer (50 mM, pH 7.4) containing Triton X-100 (0.1%) and centrifuged at 13,000 g for 5 min at 4°C. Supernatants were then applied to the cation exchange resin Dowex 50W-X8, and neutrally charged citrulline was eluted and quantified by liquid scintillation counting.
Nrf2 activation.
Cells were scraped into ice-cold lysis buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 0.5 mM dithiothreitol, and 0.4% NP-40), incubated for 10 min, and centrifuged at 14,000 g for 5 min at 4°C. The resulting nuclear pellet was resuspended in extraction buffer (20 mM HEPES, pH 7.9, 0.4 M NaCl, 1.0 mM EDTA, 1 mM dithiothreitol, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, and 10% glycerol), kept on ice for 15 min, and centrifuged at 14,000 g for 5 min at 4°C. The supernatant containing nuclear protein was collected and stored at −70°C. Nrf2 activity was determined with the TransAM Nrf2 assay (Active Motif, Carlsbad, CA). Nuclear extracts (5 μg) were incubated with ARE consensus site oligonucleotides (5′-GTCACAGTGACTCAGCAGAATCTG-3′) immobilized to 96-well plates. Bound protein was detected with an antibody specific to DNA-bound Nrf2 and visualized by colorimetric reaction catalyzed by horseradish peroxidase-conjugated secondary antibody, and absorbance was measured at 405 nm.
Chromatin immunoprecipitation assay.
Chromatin immunoprecipitation (ChIP) assays were performed with the ChIP assay kit from Upstate Cell Signaling (Charlottesville, VA) as previously described (30). Briefly, cells were fixed with formaldehyde and sonicated, and sheared chromatin was immunocleared with protein agarose A. A fraction of the precleared chromatin was stored and labeled as “input DNA.” The remaining chromatin was immunoprecipitated with IgG or Nrf2 antibodies, protein-DNA complexes were eluted with elution buffer (1% SDS, 0.1 NaHCO3), and formaldehyde cross-links were reversed by addition of NaCl (5 M) and heating at 65°C for 4 h. DNA was purified, and PCR was performed with a primer pair that spanned the HO-1 E1 enhancer. The primers used were E1 forward 5′-AAGAGCTCCACCCCCACCCA-3′ and reverse 5′-GGGCTAGCATGCGAAGTGAG-3′. A 1.5% agarose gel with ethidium bromide was used to separate and examine the PCR products.
Cell viability and caspase-3 activation.
Cell viability was assessed by measuring the uptake of the membrane-impermeant dye Trypan blue, as we previously described (28, 29, 48). Caspase-3 activity was determined by monitoring the cleavage of the p-nitroanilide-conjugated caspase-3 substrate DEVD. Briefly, cells were trypsinized, pooled with detached cells, washed in PBS, and suspended in lysis buffer (50 mM HEPES, pH 7.4, 10% sucrose, 0.1% Triton X-100) on ice for 10 min. After centrifugation at 13,000 g for 5 min at 4°C, supernatants were incubated with 50 μM DEVD and absorbance was measured at 405 nm with a μQuant spectrophotometer (Bio-Tek Instruments, Winooski, VT).
Statistical analysis.
Results are expressed as means ± SE. Statistical differences between groups were evaluated with a Student's two-tailed t-test or by ANOVA with Tukey's post hoc test when multiple groups were compared. P values <0.05 were considered statistically significant.
RESULTS
Application of a physiological level of cyclic strain (6% at 1 Hz) to HAECs stimulated a time-dependent increase in HO-1 mRNA and protein (Fig. 1, A and B). A significant and progressive elevation in HO-1 mRNA was first detected after 4 h of mechanical strain. The induction of HO-1 protein expression by cyclic strain was delayed, with a significant increase in HO-1 protein detected 8 h after mechanical strain, and levels increased further after 24 h. The induction of HO-1 by cyclic strain was associated with an approximately twofold increase in HO activity (Fig. 1C). Application of cyclic strain also stimulated a time-dependent rise in HO-1 mRNA and protein expression and HO activity in HUVECs (Fig. 2).
Fig. 1.
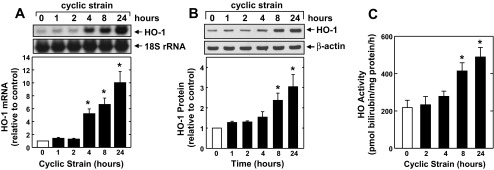
Cyclic strain stimulates heme oxygenase-1 (HO-1) expression and HO activity in human aortic endothelial cells (HAECs). Application of cyclic strain to HAECs increased HO-1 mRNA (A) and protein (B) and HO activity (C) in a time-dependent manner. HO-1 protein or mRNA was quantified by scanning densitometry, normalized with respect to β-actin and 18 S rRNA, respectively, and expressed relative to that of control, static cells. Results are means ± SE (n = 3–5). *Statistically significant effect of cyclic strain.
Fig. 2.
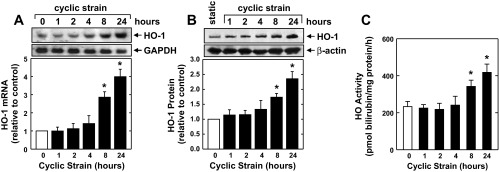
Cyclic strain stimulates HO-1 expression and HO activity in human umbilical vein endothelial cells (HUVECs). Application of cyclic strain to HUVECs increased HO-1 mRNA (A) and protein (B) and HO activity (C) in a time-dependent manner. HO-1 protein or mRNA was quantified by scanning densitometry, normalized with respect to β-actin or GAPDH, respectively, and expressed relative to that of control, static cells. Results are means ± SE (n = 3–6). *Statistically significant effect of cyclic strain.
In the next series of experiments the molecular mechanism by which cyclic strain induces HO-1 expression was examined. To evaluate whether stabilization of HO-1 mRNA contributes to the induction of HO-1 by cyclic strain, HAECs were treated with actinomycin D and the decay of HO-1 mRNA followed. These actinomycin D chase experiments revealed that HO-1 transcripts have a half-life of ∼1.5 h. However, the stability of HO-1 mRNA was unaffected by cyclic strain (Fig. 3A). To determine whether increases in HO-1 expression in response to cyclic strain involve the transcriptional activation of the gene, cells were transiently transfected with a HO-1 reporter construct and promoter activity was monitored. Application of cyclic strain stimulated a nearly fourfold increase in promoter activity (Fig. 3B). Interestingly, mutation of the antioxidant responsive elements (AREs) attenuated basal promoter activity and abolished the response to cyclic strain, suggesting that cyclic strain activates HO-1 gene transcription via the AREs (Fig. 3B). Because the transcription factor Nrf2 plays a predominant role in ARE-mediated gene transcription, we examined whether Nrf2 mediates the activation of HO-1 by cyclic strain. Transfection of endothelial cells with a dominant-negative mutant of Nrf2 that had its transactivation domain deleted inhibited basal promoter activity and the cyclic strain-mediated increase in promoter activity (Fig. 3B). In addition, cyclic strain resulted in a rapid, time-dependent rise in Nrf2 protein expression, independent of any change in Nrf1 protein expression (Fig. 3C). However, cyclic strain failed to elevate Nrf2 mRNA expression (Fig. 3D). Cyclic strain also stimulated the activation of Nrf2, as reflected by the increase in binding of nuclear Nrf2 to the ARE (Fig. 3E). To further assess the involvement of Nrf2 in HO-1 transcription, we determined the binding of Nrf2 to the HO-1 promoter in its native chromatin environment. ChIP assays with an antibody directed against Nrf2 revealed modest binding of Nrf2 to the HO-1 E1 enhancer that was substantially enriched after exposure to cyclic strain (Fig. 3F). Cyclic strain was also able to increase the expression of other ARE-responsive genes, including GCLM and NQO1 (Fig. 3G).
Fig. 3.
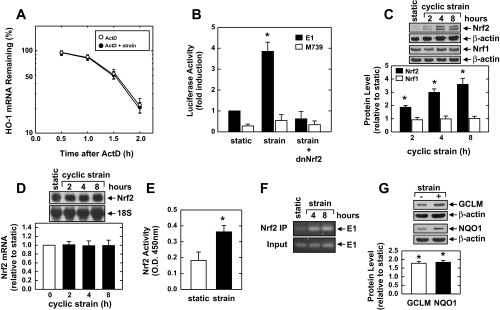
Cyclic strain stimulates HO-1 promoter activity and nuclear factor erythroid-derived 2-related factor (Nrf)2 activation without affecting HO-1 mRNA stability in HAECs. A: effect of cyclic strain on HO-1 mRNA stability. Cells were subjected to cyclic strain for 24 h and then treated with actinomycin D (ActD; 2 μg/ml) in the presence or absence of cyclic strain. B: cyclic strain stimulates HO-1 promoter activity. Cells were transfected with a HO-1 promoter construct (E1) or a mutated HO-1 promoter construct (M739) and a Renilla luciferase construct, subjected to cyclic strain for 8 h, and then analyzed for luciferase activity. In some experiments, a dominant-negative Nrf2 (dnNrf2) construct was cotransfected into cells. C: cyclic strain stimulates Nrf2 but not Nrf1 protein expression. Cells were subjected to cyclic strain for various times (0–8 h), and Nrf2 and Nrf1 protein were quantified by scanning densitometry, normalized with respect to β-actin, and expressed relative to those of control, static cells. D: cyclic strain does not stimulate Nrf2 mRNA expression. Cells were subjected to cyclic strain for various times (0–8 h), and Nrf2 mRNA was quantified by scanning densitometry, normalized with respect to 18S rRNA, and expressed relative to that of control, static cells. E: cyclic strain stimulates Nrf2 activity. Cells were subjected to cyclic strain for 8 h, and nuclear extracts were analyzed for Nrf2 binding to the antioxidant responsive element by ELISA. O.D., optical density. F: chromatin immunoprecipitation (ChIP) assay demonstrating Nrf2 binding to the HO-1 E1 enhancer following exposure to cyclic strain for 4 and 8 h. G: cyclic strain stimulates glutamate-cysteine ligase modifier subunit (GCLM) and NAD(P)H dehydrogenase quinone 1 (NQO1) protein expression. Cells were subjected to cyclic strain for 24 h, and GCLM and NQO1 protein were quantified by scanning densitometry, normalized with respect to β-actin, and expressed relative to those of control, static cells. Results are means ± SE (n = 3–6). *Statistically significant effect of cyclic strain.
In subsequent experiments, we determined the upstream signaling pathway that stimulated HO-1 expression. Since cyclic strain activates eNOS and releases nitric oxide (NO) (3), which is a well-established inducer of HO-1 (30, 33), we initially determined the role of this enzyme in mediating the induction of HO-1. The application of cyclic strain resulted in a significant increase in eNOS expression and activity (Fig. 4, A and B). Although the eNOS blocker methyl-l-arginine inhibits the activation of eNOS by cyclic strain, it failed to repress the induction of HO-1, indicating that cyclic strain-mediated increases in HO-1 expression are independent of NO (Fig. 4, B and C). Furthermore, the peroxynitrite scavenger uric acid (37) had no effect on the induction of HO-1 by cyclic strain (Fig. 4C). Since oxidative stress has also been implicated in the induction of HO-1 by Nrf2 (2, 22, 51), the involvement of ROS in the induction of HO-1 was investigated. Application of cyclic strain resulted in a time-dependent increase in the level of ROS that peaked 8 h after the onset of strain and returned to near control levels after 24 h (Fig. 5A). The induction of oxidative stress by cyclic strain was not accompanied by any changes in the expression of Nox2 and Nox4 or their associated regulatory proteins (Fig. 5B). Interestingly, the glutathione donor N-acetyl-l-cysteine blocked the increase in ROS (Fig. 5C) and the induction of HO-1 expression (Fig. 5D) evoked by cyclic strain. In addition, N-acetyl-l-cysteine abolished the cyclic strain-mediated activation of Nrf2 (Fig. 5E).
Fig. 4.
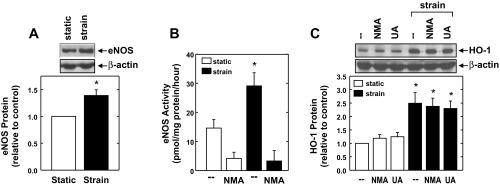
Cyclic strain-induced HO-1 expression is not dependent on nitrosative stress. A and B: cyclic strain for 24 h stimulates endothelial nitric oxide synthase (eNOS) protein expression (A) and eNOS activity (B) in HAECs. C: cyclic strain-mediated HO-1 expression is independent of eNOS activity or peroxynitrite formation. HAECs were subjected to cyclic strain for 24 h in the absence or presence of methyl-l-arginine (NMA; 2 mM) or uric acid (UA; 300 μM). Results are means ± SE (n = 3–5). *Statistically significant effect of cyclic strain.
Fig. 5.
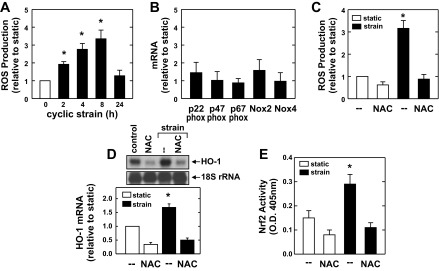
Cyclic strain-mediated HO-1 expression is dependent on oxidative stress. A: cyclic strain stimulates reactive oxygen species (ROS) production in a time-dependent manner. B: cyclic strain does not stimulate NAD(P)H oxidase expression. HAECs were subjected to cyclic strain for 8 h, and the expression of Nox2 and Nox4 or their associated regulatory proteins was determined by real-time PCR. C–E: effect of N-acetyl-l-cysteine (NAC) on cyclic strain-induced oxidative stress (C), HO-1 mRNA expression (D), and Nrf2 activation (E). HAECs were subjected to cyclic strain in the presence and absence of NAC (10 mM) for 8 h. HO-1 mRNA was quantified by scanning densitometry, normalized with respect to 18 S rRNA, and expressed relative to that of control, static cells. Results are means ± SE of 4–6 experiments. *Statistically significant effect of cyclic strain.
In the following experiments, the functional significance of the induction of HO-1 by cyclic strain was investigated. Treatment of HAECs with the combination of TNF-α (100 ng/ml) and ceramide (25 μM) resulted in a significant decline in the number of viable cells and stimulated a marked increase in the activity of the proapoptotic effector caspase-3 (Fig. 6, A and B, respectively). Interestingly, cyclic strain attenuated the cytokine-mediated loss of cell viability (Fig. 6A). However, the HO inhibitor SnPP increased cytokine-mediated cell death and reversed the cytoprotection afforded by cyclic strain. In the absence of cytokines, the application of cyclic strain or SnPP, either alone or in combination, had no effect on cell viability (Fig. 6A) or caspase-3 activity (data not shown). Cyclic strain also blocked cytokine-induced caspase-3 activation, and this was prevented by SnPP (Fig. 6B).
Fig. 6.

Cyclic strain inhibits endothelial cell apoptosis in a HO-1-dependent manner. A: cyclic strain blocks the cytokine-mediated loss of cell viability. HAECs were subjected to cyclic strain for 24 h, treated with tumor necrosis factor-α (TNF-α; 100 ng/ml) and ceramide (Cer; 25 μM) in the absence or presence of tin protoporphryin-IX (SnPP; 10 μM), and then exposed to an additional 24 h of cyclic strain. B: cyclic strain inhibits cytokine-mediated activation of caspase-3. HAECs were subjected to cyclic strain for 24 h, treated with TNF-α (100 ng/ml) and Cer (25 μM) in the absence or presence of SnPP (10 μM), and then exposed to an additional 24 h of cyclic strain. Results are means ± SE (n = 5–7). *Statistically significant effect of TNF-α and Cer; †statistically significant effect of cyclic strain.
The cytoprotective role of HO-1 in endothelial cells was further corroborated with two distinct molecular approaches. Infection of HAECs with AdHO-1 resulted in a pronounced increase in HO-1 expression, and this was associated with a decrease in cell death following cytokine exposure (Fig. 7, A and B). The control adenovirus AdGFP had no effect on HO-1 protein expression or cell survival. Alternatively, transfection of HAECs with HO-1 siRNA for 3 days reduced HO-1 protein expression by >80%, whereas the NT siRNA minimally affected protein expression (Fig. 7C). Although HO-1 siRNA had no adverse effect on cell survival under basal conditions, it potentiated cell death in response to the inflammatory mediators (Fig. 7D). In contrast, NT siRNA failed to modulate cell survival. The effect of cyclic strain on endothelial cell HO-1 expression and function was mimicked by the proteasome inhibitor MG-132. Treatment of HAECs with MG-132 caused a time-dependent increase in Nrf2 protein that preceded a rise in HO-1 expression (Fig. 8A). A significant elevation in Nrf2 protein was detected 2 h after MG-132-treatment, and levels peaked between 4 and 8 h. A significant rise in HO-1 protein was first observed 4 h after MG-132 exposure, and levels increased further after 24 h. Interestingly, treatment of HAECs with MG-132 reduced the cytokine-mediated loss in cell viability (Fig. 8B).
Fig. 7.

HO-1 promotes endothelial cell survival. A: infection of cells with adenovirus expressing HO-1 (AdHO-1), but not adenovirus expressing green fluorescent protein (AdGFP), for 2 days stimulates HO-1 protein expression. B: HO-1 overexpression inhibits cytokine-mediated cell death. Cells were infected with AdHO-1 [20 multiplicity of infection (MOI)] and AdGFP (20 MOI) for 2 days and then treated with TNF-α (100 ng/ml) and ceramide (Cer; 25 μM) for 24 h. C: HO-1 small interference RNA (siRNA) inhibits HO-1 protein expression in HAECs. Cells were treated with HO-1 siRNA (100 nM) or nontargeting (NT) siRNA (100 nM) for 3 days. D: HO-1 silencing enhances cytokine-mediated loss of cell viability. HAECs were transfected with HO-1 siRNA (100 nM) or NT siRNA (100 nM) for 3 days and then treated with TNF-α (100 ng/ml) and Cer (25 μM) for 24 h. HO-1 protein was quantified by scanning densitometry, normalized with respect to β-actin, and expressed relative to that of control cells. Results are means ± SE (n = 3–5). *Statistically significant effect of AdHO-1 or HO-1 siRNA; †statistically significant effect of TNF-α and Cer.
Fig. 8.

The proteasome inhibitor MG-132 mimics the actions of cyclic strain. A: MG-132 stimulates Nrf2 and HO-1 protein expression in HAECs in a time-dependent fashion. Cells were treated with MG-132 (2 μM) for various times (0–24 h), and Nrf2 and HO-1 protein were quantified by scanning densitometry, normalized with respect to β-actin, and expressed relative to those of control cells. B: MG-132 blocks the cytokine-mediated loss of endothelial cell viability. HAECs were treated with MG-132 (2 μM) for 8 h and then exposed to TNF-α (100 ng/ml) and Cer (25 μM) for an additional 24 h. Results are means ± SE (n = 3–5). *Statistically significant effect of MG-132; †statistically significant effect of TNF-α and Cer.
To further investigate the role of HO-1 in cell protection, we exposed MAECs isolated from HO-1+/+ and HO-1−/− mice to cyclic strain. Cyclic strain induced a significant increase in HO-1 protein expression in MAECs derived from HO-1+/+ mice (Fig. 9A). In contrast, HO-1 expression was absent in HO-1−/− MAECs and the application of cyclic strain failed to induce the expression of HO-1. Cyclic strain also evoked an increase in oxidative stress in MAECs, and this was enhanced in cells taken from HO-1−/− mice (Fig. 9B). Finally, treatment of HO-1+/+ MAECs with TNF-α and ceramide induced cell death that was attenuated by the application of cyclic strain (Fig. 9C). Cytokine exposure also promoted cell death in MAECs obtained from HO-1−/− mice. However, the degree of cell death was greater in HO-1−/− endothelial cells, and cyclic strain failed to protect these endothelial cells from cytokine-mediated cell death.
Fig. 9.
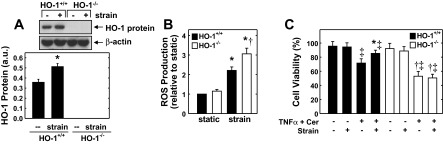
HO-1 gene deletion abolishes the cytoprotection afforded by cyclic strain in murine aortic endothelial cells (MAECs). A: HO-1 protein expression in MAECs derived from wild-type (HO-1+/+) or HO-1-deficient (HO-1−/−) mice in the absence or presence of cyclic strain. a.u., Arbitrary units. B: cyclic strain for 8 h stimulates ROS production in MAECs. C: cyclic strain-mediated cytoprotection was absent in HO-1−/− MAECs. MAECs were subjected to cyclic strain for 24 h, treated with TNF-α (100 ng/ml) and Cer (25 μM), and then exposed to an additional 24 h of cyclic strain. HO-1 protein was quantified by scanning densitometry, normalized with respect to β-actin, and expressed relative to that of control, static cells. Results are means ± SE (n = 3–8). *Statistically significant effect of cyclic strain; †statistically significant effect of HO-1 gene deletion; ‡statistically significant effect of TNF-α and Cer.
DISCUSSION
The present study demonstrates that a physiologically relevant level of cyclic strain is able to stimulate the expression of HO-1 in endothelial cells. The induction of HO-1 by cyclic strain is observed in endothelial cells derived from both the arterial and venous circulation and occurs in an eNOS-independent fashion. The stimulation of HO-1 expression by mechanical strain is dependent on the production of ROS and is mediated by Nrf2. Significantly, we found that cyclic strain inhibits cytokine-mediated endothelial cell apoptosis and that HO-1 underlies this cytoprotective effect. Thus the ability of cyclic strain to induce HO-1 gene expression may provide a novel mechanism by which hemodynamic forces promote endothelial cell survival and vascular homeostasis.
The application of cyclic strain stimulates a time-dependent increase in HO-1 protein and mRNA expression in endothelial cells. A significant elevation in HO-1 protein and activity is detected at 8 h, and this progressively increases during 24 h of cyclic strain exposure. The level of cyclic strain (6%) required to stimulate HO-1 expression is physiologically relevant since the maximum stretch of large systemic vessels during the cardiac cycle has been reported to vary between 5% and 12% under normotensive conditions (6, 13, 43). We found that cyclic strain stimulates HO-1 expression in endothelial cells derived from both the human and mouse aorta and human umbilical vein, extending previous work showing cyclic strain-mediated induction of HO-1 in rat pulmonary endothelial cells (35). Thus the induction of HO-1 by cyclic strain is observed in endothelial cells derived from various blood vessels across multiple species. Cyclic strain also stimulates the expression of HO-1 in vascular smooth muscle cells and ventricular myocytes, demonstrating that biomechanical strain induces HO-1 expression in diverse cardiovascular cells (17, 47).
The induction of HO-1 by cyclic strain does not involve alterations in mRNA stabilization and is likely due to the transcriptional activation of the gene since transient luciferase reporter assays demonstrate that cyclic strain directly stimulates HO-1 promoter activity. Interestingly, the induction of HO-1 gene transcription requires the presence of AREs, since mutation of this responsive element abolishes the stimulation of promoter activity by cyclic strain. Although several transcription factors can bind to AREs, Nrf2 plays the predominant role in ARE-dependent HO-1 gene expression (2). Consistent with this, we found that cyclic strain stimulates Nrf2 protein expression and nuclear Nrf2 binding to the AREs. However, the increase in Nrf2 protein by cyclic strain is not associated with a rise in Nrf2 mRNA expression, suggesting that cyclic strain stimulates Nrf2 protein expression via a posttranscriptional mechanism that may involve protein stabilization (22, 51). Furthermore, transfection of endothelial cells with a dominant-negative Nrf2 construct abrogates the activation of HO-1 promoter activity in response to cyclic strain. Thus the mobilization of Nrf2 plays an integral role in mediating HO-1 gene transcription by cyclic strain. In contrast, cyclic strain did not stimulate Nrf1 expression, indicating a selective activation of Cap'n'Collar transcription factors by cyclic strain. Our finding that a hemodynamically relevant regimen of cyclic strain activates Nrf2 in HAECs builds on earlier work showing that mechanical pulmonary ventilation-associated stretch and fluid shear stress induce Nrf2 activation in endothelial cells (19, 34), highlighting the mechanosensitive nature of this transcription factor.
The induction of HO-1 by cyclic strain is dependent on the production of ROS. Consistent with earlier reports (18, 20), we found that cyclic strain stimulates a time-dependent increase in oxidative stress that peaks ∼8 h after the application of cyclic strain. Interestingly, the generation of ROS returns toward baseline 24 h after the application of cyclic strain. This likely reflects the ability of cyclic strain to induce a battery of Nrf2-responsive antioxidant genes including HO-1, GCLM, NQO1, and others to combat cyclic strain-mediated increases in ROS generation (35, 46). Consistent with this notion, we found that cyclic strain-mediated elevations in ROS are greater in HO-1-deficient endothelial cells. Although NAD(P)H oxidase plays a pivotal role in cyclic strain-induced endothelial ROS production (18, 20), a physiological level of cyclic strain has no effect on the expression of the major endothelial catalytic subunits Nox2 and Nox4 or any of the regulatory subunits, suggesting that cyclic strain stimulates ROS formation by promoting the assembly of a functional NAD(P)H oxidase complex. However, the cyclic strain-mediated increase in oxidative stress is prevented by the glutathione donor N-acetyl-l-cysteine. Moreover, N-acetyl-l-cysteine blocks the activation of Nrf2 and the induction of HO-1 by cyclic strain. In contrast, treatment of endothelial cells with the NOS inhibitor methyl-l-arginine or the peroxynitrite scavenger uric acid fails to inhibit cyclic strain-mediated HO-1 expression. Thus oxidative, but not nitrosative, stress is responsible for the induction of HO-1 by cyclic strain. Although the mechanism by which oxidative stress activates Nrf2 is not fully known, the oxidation of cysteine residues in Kelch-like erythroid cell-derived protein 1 (Keap1) is likely involved, since several cysteine residues in Keap1 are able to undergo redox-dependent changes that result in the release and/or inhibition of Keap1-dependent ubiquitination and degradation of Nrf2 (22, 51).
Recent studies indicate that cyclic strain serves a critical survival function in endothelial cells. Application of physiologically relevant levels of cyclic strain inhibits apoptosis in bovine aortic endothelial cells and in a human endothelial cell line (26, 29, 34). In the present study, we now show that cyclic strain also promotes the survival of primary HAECs and MAECs. Moreover, we are the first to demonstrate that HO-1 plays an important role in the cytoprotection mediated by cyclic strain. In particular, the application of a physiological level of cyclic strain attenuates endothelial cell death and caspase-3 activation in response to TNF-α and ceramide. The cytoprotection afforded by cyclic strain is mediated by HO-1, since inhibition of HO-1 activity or genetic ablation of the enzyme abolishes the salutary effect of cyclic strain. Interestingly, cell death in response to inflammatory stress is augmented in endothelial cells derived from HO-1-deficient mice. This later finding is in agreement with an earlier report showing increased susceptibility of HO-1-null endothelial cells to oxidized lipid-induced cell injury (8). We further corroborated the cytoprotective property of the enzyme by showing that adenovirus-mediated gene transfer of HO-1 retards, while HO-1 silencing augments, cytokine-mediated endothelial cell death. Moreover, the induction of HO-1 expression following MG-132 exposure confers endothelial cell protection. The antiapoptotic action of HO-1 is likely mediated by CO since scavenging of this gas with hemoglobin reverses the antiapoptotic action of HO-1 in endothelial cells while the exogenous administration of CO reproduces the protection induced by HO-1 in response to numerous apoptotic stimuli, including TNF-α (4, 32). While HO-1 and CO elicit numerous antiapoptotic actions, their ability to activate the prosurvival kinase Akt (5, 7, 25) may be particularly relevant since inhibition of endothelial cell apoptosis by cyclic strain is dependent on Akt (29, 34).
The ability of cyclic strain to stimulate HO-1 gene expression may provide an important mechanism by which hemodynamic forces preserve vascular homeostasis. Aside from preventing endothelial apoptosis, the induction of HO-1 in endothelial cells may inhibit vascular smooth muscle tone and platelet aggregation by releasing CO (15, 24, 47). In addition, HO-1 exerts potent anti-inflammatory effects by blocking the activation of endothelial cells and the recruitment of immune cells into the vessel wall (27, 39). Furthermore, HO-1 restores endothelial integrity of injured blood vessels by stimulating the proliferation and migration of endothelial cells from adjacent border regions of the injured artery and by recruiting circulating endothelial progenitor cells to the site of damage (11, 12, 49). Thus the induction of HO-1 by physiological levels of cyclic strain may serve an important homeostatic role to maintain blood flow and fluidity and initiate endothelial repair at sites of arterial injury.
In conclusion, the present study demonstrates that a hemodynamically relevant regime of cyclic strain induces HO-1 gene expression in endothelial cells derived from various vascular beds by activating the ROS-Nrf2 signaling pathway. In addition, it found that cyclic strain inhibits endothelial cell death in a HO-1-dependent fashion. The ability of cyclic strain to stimulate HO-1 expression may provide a critical mechanism by which hemodynamic forces promote endothelial cell survival and vascular homeostasis.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01 HL-59976 and R01 HL-74966.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: X.-m.L., K.J.P., and W.D. conception and design of research; X.-m.L. and K.J.P. performed experiments; X.-m.L., K.J.P., and W.D. analyzed data; X.-m.L., K.J.P., and W.D. interpreted results of experiments; X.-m.L. and K.J.P. edited and revised manuscript; X.-m.L., K.J.P., and W.D. approved final version of manuscript; K.J.P. prepared figures; W.D. drafted manuscript.
REFERENCES
- 1. Ago T, Kitazono T, Ooboshi H, Iyama T, Han YH. Nox 4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation 109: 227–233, 2004 [DOI] [PubMed] [Google Scholar]
- 2. Alam J, Cook JL. Transcriptional regulation of the heme oxygenase-1 gene via the stress response pathway. Curr Pharm Des 9: 2499–2511, 2003 [DOI] [PubMed] [Google Scholar]
- 3. Awolesi MA, Sessa WC, Sumpio BE. Cyclic strain upregulates nitric oxide synthase in cultured bovine aortic endothelial cells. J Clin Invest 96: 1449–1454, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, Soares MP. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med 192: 1015–1026, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brunt KR, Fenrich KK, Kiani G, Tse MY, Pang SC, Ward CA, Melo LG. Protection of human vascular smooth muscle cells from H2O2-induced apoptosis through functional codependence between HO-1 and AKT. Arterioscler Thromb Vasc Biol 26: 2027–2034, 2006 [DOI] [PubMed] [Google Scholar]
- 6. Buntin CM, Silver FH. Noninvasive assessment of mechanical properties of peripheral arteries. Ann Biomed Eng 18: 549–566, 1990 [DOI] [PubMed] [Google Scholar]
- 7. Busserolles J, Megias J, Terencio MC, Alcaraz MJ. Heme oxygenase-1 inhibits apoptosis in Caco-2 cells via activation of Akt pathway. Int J Biochem Cell Biol 38: 1510–1517, 2006 [DOI] [PubMed] [Google Scholar]
- 8. Chen S, Segal M, Agarwal A. “Lumen digestion” technique for isolation of aortic endothelial cells from heme oxygenase-1 knockout mice. Biotechniques 37: 84–89, 2010 [DOI] [PubMed] [Google Scholar]
- 9. Cheng JJ, Wung BS, Chao YJ, Hsieh HJ, Wang CL. Cyclic strain induces redox changes in endothelial cells. Chin J Physiol 42: 103–111, 1999 [PubMed] [Google Scholar]
- 10. Cummins PM, von Offenberg Sweeney N, Killeen MT, Birney YA, Redmond EM, Cahill PA. Cyclic strain-mediated matrix metalloproteinase regulation within the vascular endothelium: a force to be reckoned with. Am J Physiol Heart Circ Physiol 292: H28–H42, 2007 [DOI] [PubMed] [Google Scholar]
- 11. Deramaudt BM, Braunstein S, Remy P, Abraham NG. Gene transfer of human heme oxygenase into coronary endothelial cells potentially promotes angiogenesis. J Cell Biochem 68: 121–127, 1998 [DOI] [PubMed] [Google Scholar]
- 12. Deshane J, Chen S, Caballero S, Grochot-Przeczek A, Was H, Li Calzi S, Lach R, Hock TD, Chen B, Hill-Kapturczak N, Siegal GP, Dulak J, Jozkowicz A, Grant MB, Agarwal A. Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J Exp Med 204: 605–618, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dobrin PB. Mechanical properties of arteries. Physiol Rev 58: 397–460, 1978 [DOI] [PubMed] [Google Scholar]
- 14. Durante W. Targeting heme oxygenase-1 in vascular disease. Curr Drug Targets 11: 1504–1516, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Durante W, Johnson FK, Johnson RA. Role of carbon monoxide in cardiovascular function. J Cell Mol Med 10: 672–686, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Durante W, Liao L, Iftikhar I, O'Brien WE, Schafer AI. Differential regulation of l-arginine transport and nitric oxide production by vascular smooth muscle and endothelium. Circ Res 78: 1075–1082, 1996 [DOI] [PubMed] [Google Scholar]
- 17. Frank D, Kuhn C, Brors B, Hanselmann C, Ludde M, Katus HA, Frey N. Gene expression patterns in biomechanically stretched cardiomyocytes: evidence for a stretch-specific gene program. Hypertension 51: 1–10, 2008 [DOI] [PubMed] [Google Scholar]
- 18. Hishikawa K, Luscher TF. Pulsatile stretch stimulates superoxide production in human aortic endothelial cells. Circulation 96: 3610–3616, 1997 [DOI] [PubMed] [Google Scholar]
- 19. Hosoya T, Maruyama A, Kang MI, Kawatani Y, Shibata T, Uchida K, Itoh K, Yamamato M. Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stress in endothelial cells. J Biol Chem 280: 27244–27250, 2005 [DOI] [PubMed] [Google Scholar]
- 20. Howard AB, Alexander RW, Nerem RM, Griendling KK, Taylor WR. Cyclic strain induces an oxidative stress in endothelial cells. Am J Physiol Cell Physiol 272: C421–C427, 1997 [DOI] [PubMed] [Google Scholar]
- 21. Iba T, Sumpio BE. Morphological response of human endothelial cells subjected to cyclic strain in vitro. Microvasc Res 42: 245–254, 1991 [DOI] [PubMed] [Google Scholar]
- 22. Itoh K, Wakabayashi N, Katoh Y, Ishii T, O'Connor Y, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuffling and degradation of Nrf2 in response to electrophiles. Genes Cells 8: 379–391, 2003 [DOI] [PubMed] [Google Scholar]
- 23. Jiang X, Yang F, Tan H, Liao D, Bryan RM, Jr, Randhawa JK, Rumbaut RE, Durante W, Schafer AI, Yang X, Wang H. Hyperhomocysteinemia impairs endothelial function and eNOS activity via PKC activation. Arterioscler Thromb Vasc Biol 25: 2515–2521, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson RA, Lavesa M, Askari B, Abraham NG, Nasjletti A. A heme oxygenase product, presumably carbon monoxide, mediates a vasodepressor function in rats. Hypertension 25: 166–169, 1996 [DOI] [PubMed] [Google Scholar]
- 25. Kim HS, Loughran PA, Rao J, Billiar TR, Zuckerbraun BS. Carbon monoxide activates NF-kappaB via ROS generation and Akt pathways to protect against cell death of hepatocytes. Am J Physiol Gastrointest Liver Physiol 295: G146–G152, 2008 [DOI] [PubMed] [Google Scholar]
- 26. Kou B, Zhang J, Singer DR. Effects of cyclic strain on endothelial cell apoptosis and tubulogenesis are dependent on ROS production via NAD(P)H subunit p22phox. Microvasc Res 77: 125–133, 2009 [DOI] [PubMed] [Google Scholar]
- 27. Lin CC, Liu XM, Peyton KJ, Wang H, Yang WC, Lin SJ, Durante W. Far infrared therapy inhibits vascular endothelial inflammation via the induction of heme oxygenase-1. Arterioscler Thromb Vasc Biol 28: 739–745, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu XM, Chapman GB, Peyton KJ, Schafer AI, Durante W. Carbon monoxide inhibits apoptosis in vascular smooth muscle cells. Cardiovasc Res 55: 396–405, 2002 [DOI] [PubMed] [Google Scholar]
- 29. Liu XM, Ensenat D, Wang H, Schafer AI, Durante W. Physiologic cyclic stretch inhibits apoptosis in vascular endothelium. FEBS Lett 541: 52–56, 2003 [DOI] [PubMed] [Google Scholar]
- 30. Liu XM, Peyton KJ, Ensenat D, Wang H, Hannink M, Alam J, Durante W. Nitric oxide stimulates heme oxygenase-1 gene transcription via the Nrf2/ARE complex to promote vascular smooth muscle cell survival. Cardiovasc Res 75: 381–389, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maines CD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J 2: 2557–2568, 1988 [PubMed] [Google Scholar]
- 32. Morse D, Lin L, Choi AM, Ryter SW. Heme oxygenase-1: a critical arbitrator of cell death pathways in lung injury and disease. Free Radic Biol Med 47: 1–12, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Motterlini R, Foresti R, Intaglietta M, Winslow RM. NO-mediated activation of heme oxygenase: endogenous cytoprotection against oxidative stress to the endothelium. Am J Physiol Heart Circ Physiol 270: H107–H114, 1996 [DOI] [PubMed] [Google Scholar]
- 34. Nishimura K, Li W, Hoshino Y, Kadohama T, Asada H, Ohgi S, Sumpio BE. Role of AKT in cyclic strain-induced endothelial cell proliferation and survival. Am J Physiol Cell Physiol 290: C812–C821, 2006 [DOI] [PubMed] [Google Scholar]
- 35. Papaiahgari S, Yerrapureddy A, Hassoun PM, Garcia JG, Birukov KG, Reddy SP. EGFR-activated signaling and actin remodeling regulate cyclic stretch-induced Nrf2-ARE activation. Am J Respir Cell Mol Biol 36: 304–312, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peyton KJ, Liu XM, Yu Y, Yates B, Durante W. Activation of AMP-activated protein kinase inhibits the proliferation of human endothelial cells. J Pharmacol Exp Ther 342: 827–834, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Santos CX, Anjos EI, Augusto O. Uric acid oxidation by peroxynitrite: multiple reactions, free radical formation, and amplification of lipid oxidation. Arch Biochem Biophys 372: 285–294, 1999 [DOI] [PubMed] [Google Scholar]
- 38. Schroder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmler S, Shah AM, Brandes RP. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res 110: 1217–1225, 2012 [DOI] [PubMed] [Google Scholar]
- 39. Soares MP, Seldon MP, Gregoire IP, Vassilevskaia T, Berberat PO, Yu J, Tsui TY, Bach FH. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J Immunol 172: 3553–3563, 2004 [DOI] [PubMed] [Google Scholar]
- 40. Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science 235: 1043–1046, 1987 [DOI] [PubMed] [Google Scholar]
- 41. Sumpio BE, Banes AJ. Prostacyclin synthetic activity in cultured aortic endothelial cells undergoing cyclic mechanical deformation. Surgery 104: 383–389, 1988 [PubMed] [Google Scholar]
- 42. Tulis DA, Durante W, Liu XM, Evans AJ, Peyton KJ, Schafer AI. Adenovirus-mediated heme oxygenase-1 gene delivery inhibits injury-induced vascular neointima formation. Circulation 104: 2710–2715, 2001 [DOI] [PubMed] [Google Scholar]
- 43. Van Merode T, Brands PJ, Hoeks AP, Reneman RS. Different effects of aging on elastic and muscular arterial bifurcations in men. J Vasc Res 33: 47–52, 1996 [DOI] [PubMed] [Google Scholar]
- 44. Vile GF, Tyrrell RM. Oxidative stress resulting from ultraviolet A irradiation of human skin fibroblasts leads to a heme oxygenase dependent increase in ferritin. J Biol Chem 268: 14678–14681, 1993 [PubMed] [Google Scholar]
- 45. von Offenberg Sweeney N, Cummins PM, Cotter EJ, Fitzpatrick PA, Birney YA, Redmond EM, Cahill PA. Cyclic strain-mediated regulation of vascular endothelial cell migration and tube formation. Biochem Biophys Res Commun 329: 573–582, 2005 [DOI] [PubMed] [Google Scholar]
- 46. Wagner AH, Kautz O, Fricke K, Zerr-Fouineau M, Demicheva E, Guldenzorph B, Bermejo JL, Korff T, Hecker M. Regulation of glutathione peroxidase offsets stretch-induced proatherogenic gene expression in human endothelial cells. Arterioscler Thromb Vasc Biol 29: 1894–1901, 2009 [DOI] [PubMed] [Google Scholar]
- 47. Wagner CT, Durante W, Christodoulides N, Hellums JD, Schafer AI. Hemodynamic forces induce the expression of heme oxygenase in cultured vascular smooth muscle cells. J Clin Invest 100: 589–596, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wei Y, Liu XM, Peyton KJ, Wang H, Johnson FK, Johnson RA, Durante W. Hypochlorous acid-induced heme oxygenase-1 gene expression promotes human endothelial cell survival. Am J Physiol Cell Physiol 297: C907–C915, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu BJ, Midwinter RG, Cassano C, Beck K, Wang Y, Changsiri D, Gamble JR, Stocker R. Heme oxygenase-1 increases endothelial progenitor cells. Arterioscler Thromb Vasc Biol 29: 1537–1542, 2009 [DOI] [PubMed] [Google Scholar]
- 50. Yung YC, Chae J, Buehler MJ, Hunter CP, Mooney DJ. Cyclic tensile strain triggers a sequence of autocrine and paracrine signaling to regulate angiogenic sprouting in human vascular cells. Proc Natl Acad Sci USA 106: 15279–15284, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang DD, Hannink M. Distinct cysteine residues in keap1 are required for keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol 23: 8137–8151, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]