

Abstract
In patients with hypertension, volitional slowing of the respiratory rate has been purported to reduce arterial pressure via withdrawal of sympathetic tone. We examined the effects of paced breathing at 7, 14, and 21 breaths/min, with reciprocal changes in tidal volume, on muscle sympathetic nerve activity, forearm blood flow, forearm vascular conductance, and blood pressure in 21 men and women, 8 of whom had modest elevations in systemic arterial pressure. These alterations in breathing frequency and volume did not affect steady-state levels of sympathetic activity, blood flow, vascular conductance, or blood pressure (all P > 0.05), even though they had the expected effect on sympathetic activity within breaths (i.e., increased modulation during low-frequency/high-tidal volume breathing) (P < 0.001). These findings were consistent across subjects with widely varied baseline levels of sympathetic activity (4-fold), mean arterial pressure (78–110 mmHg), and vascular conductance (15-fold), and those who became hypocapnic during paced breathing vs. those who maintained normocapnia. These findings challenge the notion that slow, deep breathing lowers arterial pressure by suppressing steady-state sympathetic outflow.
Keywords: respiration, blood pressure, hypertension
powerful within-breath respiratory modulation of sympathetic vasoconstrictor activity has been well documented in humans and experimental animals (7, 10, 17, 26, 40). Based primarily on correlational studies, previous investigators have proposed that altered within-breath modulation of sympathetic activity might be causally linked with heightened sympathoexcitation and hypertension in the steady state (6, 15, 30, 31, 45, 50). However, evidence gathered from hypothesis-driven research in animal models and humans is conflicting. For example, in anesthetized or decorticate rodents with spontaneous (6, 45) angiotensin II- and salt- (50) dependent, or chronic intermittent hypoxia-induced (28) hypertension, sympathetic activity in the steady state was shown to be positively correlated with enhanced within-breath modulation of sympathetic outflow. However, the within-breath link between phrenic nerve activity and sympathetic outflow in these debuffered animals, especially the dominant modulatory role of central respiratory motor output, differs qualitatively from that in the intact human (also see the discussion). The high muscle sympathetic nerve activity (MSNA) observed in humans with congestive heart failure has also been linked to respiratory pattern, but paradoxically, compared with the rodent, to reduced within-breath modulation of MSNA attributed to the tachypneic, low-tidal volume breathing pattern in these patients (15, 31). In contrast, humans with essential hypertension and elevated MSNA burst frequency were reported to have normal within-breath modulation of MSNA (13).
In several recent clinical studies, daily training with voluntary reductions in breathing frequency (fB) in patients with hypertension has produced mixed effects on blood pressure and/or MSNA (3, 11, 16, 19, 25, 27, 37, 38). Reductions in MSNA were reported with acute voluntary slowing of fB in two studies, but blood pressure was unchanged and blood flow was not measured (19, 33). Our previous study found no effect of marked increases in fB and tidal volume (VT) on steady-state MSNA burst frequency (9, 40), but we did not consider whether the currently proposed idea of slowing fB below eupneic levels might reduce steady-state MSNA, nor did we determine whether respiratory pattern affected blood pressure or vascular conductance in the steady state. We interpret this compilation of diverse findings to mean that the issue of whether the magnitude of within-breath modulation of MSNA affects steady-state MSNA, vascular conductance, and blood pressure in the human remains unresolved. To determine whether voluntary alterations in fB might be useful in the treatment of hypertension, controlled studies that include measures of both sympathetic activity and the hemodynamic response are necessary.
We have now undertaken a novel, comprehensive study of whether acute alterations in breathing pattern influence autonomic control of the circulation in the steady state. Subjects voluntarily varied their breathing patterns across a threefold change in fB and a twofold change in VT, and we determined the effects on within-breath modulation of MSNA as well as steady-state MSNA, systemic blood pressure, and forearm blood flow (FBF). We examined these effects of a changing respiratory pattern across subjects who varied markedly in their baseline levels of MSNA, blood pressure, and vascular conductance and also in the magnitude of their within-breath modulation of MSNA. Our findings confirm that modulation of MSNA within a breath is markedly dependent upon breathing pattern and lung volume; however, these transient, within-breath influences are not manifested as significant effects on MSNA, vascular conductance, or blood pressure in the steady state.
MATERIALS AND METHODS
Ethical approval.
The experiments reported herein conformed to the Declaration of Helsinki. All subjects provided informed, written consent, and the experimental protocol was approved by the University of Wisconsin Health Sciences Institutional Review Board.
Subjects.
Eight women and 13 men served as subjects. The mean age was 36 ± 14 (SD) yr, with a range of 20–73. All subjects were nonsmokers, were free from overt cardiovascular disease and neurological disorders, and only one subject was taking antihypertensive medications (amlodipine and losartan) and a lipid-lowering medication (simvastatin). All but two of the subjects were participants in two larger studies investigating the effects of metabolic syndrome on MSNA and vascular function (24). Ten of the subjects met the criteria (2) for metabolic syndrome: 10 for waist circumference, 10 for low high-density lipoprotein cholesterol, 7 for high triglycerides, 7 for elevated blood pressure (systolic pressure ≥130 and/or diastolic pressure ≥85), and none for high fasting glucose. Two of these subjects were overweight [body mass index (BMI) 25–29.9], and eight were obese (BMI ≥30). The mean BMI for the entire sample was 28.3 ± 8.0 kg/m2. Premenopausal female subjects were studied during the early follicular phase of the menstrual cycle (placebo phase if taking oral contraceptives) and had a negative urine pregnancy test.
General procedure.
Subjects were studied in the supine position during wakefulness after a 10-h fast. They were instructed to refrain from exercise, nonsteroidal anti-inflammatory drugs, alcohol, and caffeine for 24 h before the study visit. Room temperature was controlled at 22°C.
Cardiorespiratory variables.
Heart rate was measured from the electrocardiogram. Beat-by-beat arterial pressure was measured by finger pulse photoplethysmography (Finapres model 2300; Ohmeda, Englewood, CO) and corrected using automated arm cuff sphygmomanometry (Datex-Ohmeda, Helsinki, Finland). Mean arterial pressure (MAP) was calculated as one-third pulse pressure plus diastolic pressure. Subjects breathed through a mouthpiece with the nose occluded. Expired air was sampled from the mouthpiece to measure end-tidal CO2 tension (PetCO2) (S-3A/I and CD-3A; Ametek, Berwyn, PA). In 10 subjects, VT was estimated using a respiratory belt transducer (MLT1132; ADInstruments, Colorado Springs, CO). In the remaining 11 subjects, VT was measured using a heated pneumotach (Hans Rudolph, Shawnee, KS) and spirometer (ML311 Spirometer Pod; ADInstruments).
Brachial artery flow velocity and diameter.
A 12-MHz linear array Doppler ultrasound probe (Vivid 7; General Electric, Milwaukee, WI) was placed over the brachial artery with an insonation angle of ≤60°, and the sample volume was adjusted to cover the width of the artery. An interface unit (Multigon Industries, Yonkers, NY) processed the angle-corrected, intensity-weighted Doppler audio information from the GE Vivid ultrasound system into a flow velocity signal sampled in real time with signal-processing software (PowerLab; ADInstruments). Brachial artery diameters were obtained from B-mode video images, and measurements resulted in a 15-s loss of pulse wave signal. Artery diameter was measured off-line in a longitudinal section of the brachial artery and was identified by strong wall signals, and results were reported as the median of five measurements in late diastole. FBF was calculated by multiplying mean blood velocity (cm/s) by cross-sectional area (π × radius2), and values were multiplied by 60 to convert from milliliters per second to milliliters per minute. Forearm vascular conductance (ml·min−1·100 mmHg−1) was calculated (FBF divided by MAP × 100).
MSNA.
The technique of Vallbo et al. (51) was used to record postganglionic MSNA from the right fibular nerve as described previously (22). Neural signals were passed to a differential preamplifier, an amplifier (total gain = 100,000), a band-pass filter (700–2,000 Hz), and an integrator (time constant = 100 ms). Placement of the recording electrode within a muscle nerve fascicle was confirmed by 1) the presence of muscle twitches, but not paresthesias, in response to electrical stimulation; 2) the pulse-synchronous nature of the nerve activity; 3) the appearance of afferent activity in response to tapping or stretching of muscle, but not gentle stroking of the skin, in the appropriate receptive fields; and 4) the absence of neural activation in response to arousal stimuli. Once an acceptable nerve recording was obtained, the subject was instructed to maintain the leg in a relaxed position for the duration of the study. Acceptable neurograms (signal-to-noise ratio >3:1) were obtained in 20 of 21 subjects.
Experimental protocol.
Data collection commenced ∼1.5 h after placement of the nerve recording electrode (after completion of the parent protocol). To ensure stability of the neurogram, the subject rested quietly for at least 10 additional minutes. After a 3-min baseline data collection period during which the subject breathed spontaneously, s/he was asked to make graded alterations in fB using auditory cues that signaled inspiration and expiration. Data were recorded during 3-min periods of paced breathing at three frequencies (7, 21, and 14 breaths/min), each separated by 2 min of spontaneous breathing. A duty cycle (inspiratory time/total time) of 0.50 was maintained throughout paced breathing. In 16 subjects, verbal cues were given to adjust VT as necessary to maintain baseline levels of PetCO2. In the remaining subjects, PetCO2 was allowed to fluctuate. Data collection concluded with a 2- to 3-min period of spontaneous breathing. All hemodynamic and respiratory data were digitized, stored on a computer at 400 Hz, and analyzed off-line using LabChart (ADInstruments).
Data analysis.
Sympathetic bursts were identified by computer-assisted inspection of the mean voltage neurogram. Briefly, data were sampled in real time with signal-processing software (PowerLab, LabChart7; ADinstruments) and analyzed off-line. This program detected deviations from baseline voltage within a 0.5-s search window and an expected burst latency of 1.3 s from the preceding R-wave. A voltage deviation would be identified as a burst if it exceeded a noise “threshold” (typically 20% of the maximal deviation from zero of the recorded data). A single human observer would then review the record and manually add or remove bursts as appropriate using visual appraisal of the raw neurogram for confirmation. MSNA was quantified as burst frequency (bursts/min), burst incidence (bursts/100 cardiac cycles), and total minute activity (bursts/min × mean burst amplitude).
For time domain analysis, MSNA and respiratory variables were averaged over the 3-min periods of spontaneous or paced breathing, whereas blood velocity and arterial pressure were averaged over the last 30 s of each minute. For frequency domain analysis, power spectra for each 3-min breathing period were obtained by fast-Fourier transform (LabChart; ADInstruments). Power of the integrated MSNA signal at the respiratory frequency, our measure of magnitude of within-breath modulation of MSNA, and at the cardiac frequency was normalized within and between subjects by expressing it as standard normal variance (power at frequency of interest minus mean power for the entire trial divided by the SD of power for that trial) (36).
Statistics.
One-way, repeated-measures ANOVAs were used to compare cardiorespiratory variables and MSNA across the range of breathing frequencies. Scheffé's post hoc tests were applied when omnibus F statistics were significant. To compare the effects of fB in specified subgroups (i.e., subjects with high vs. low MSNA burst incidence based on an arbitrary cut point of 40% and those who maintained normocapnia during paced breathing vs. those who did not), group × frequency interactions were sought using two-way, repeated-measures ANOVAs. P values <0.05 were considered statistically significant. Data are presented as means ± SE.
RESULTS
Within-breath modulation of MSNA.
Figure 1 contains polygraph recordings from one representative subject showing the effects of altered fB and VT on within-breath modulation of MSNA. The pattern of inspiratory inhibition and expiratory excitation of MSNA was evident at all breathing frequencies but was most pronounced and consistent during the trial with the lowest fB (7 breaths/min) and highest VT (194% of baseline). MSNA burst frequency was comparable across the three conditions.
Fig. 1.
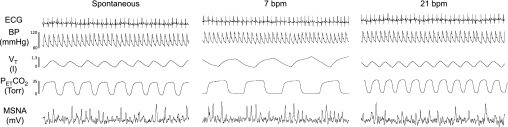
Original polygraph records showing cardiorespiratory responses to a voluntary increase and decrease in breathing frequency (fB) [with corresponding decrease and increase in tidal volume (VT)] in a representative subject. In this example, steady-state values for muscle sympathetic nerve activity (MSNA) burst frequency remained unchanged over the range of fB and VT (39, 41, and 38 bursts/min for spontaneous breathing, 7 breaths/min, and 21 breaths/min). PetCO2, end-tidal CO2 tension.
Figure 2A shows power spectra of integrated MSNA across all respiratory frequencies and VT in a typical subject. Note the tendency for the power of the MSNA signal to be greatest at the cardiac and respiratory frequencies. With reduced fB and increased VT, the power of the MSNA signal at the cardiac frequency remained unchanged, whereas the power at the respiratory frequency was increased.
Fig. 2.

A: MSNA power spectra during spontaneous breathing and voluntary decreases and increases in fB with reciprocal changes in VT in one subject. Note that power at the respiratory frequency was greatly increased by slowing fB to 7 breaths/min and diminished by increasing fB to 21 breaths/min. B: mean values for power at the respiratory frequency in subjects divided into two groups: those with relatively high baseline MSNA (burst incidence >40%; filled bars) vs. those with relatively low baseline MSNA (burst incidence <40%; hatched bars). Two-way repeated-measures ANOVA revealed a significant main effect of frequency (P < 0.001) and a significant frequency-by-group interaction (P = 0.027). Sp1, initial spontaneous breathing period; Sp2, final spontaneous breathing period.
Group mean values for MSNA power at the prevailing respiratory frequency, our measure of the magnitude of within-breath modulation of MSNA, are shown in Table 1. MSNA power was greatest during the slowest fB/highest VT trials (156% of power at the spontaneous fB) (P < 0.001) and least at the fastest fb/lowest VT trials (70% of spontaneous). In contrast, MSNA power at the cardiac frequency was unaffected by changes in fB and VT (P > 0.05).
Table 1.
Respiratory and cardiac frequencies during spontaneous and paced breathing and power of the MSNA signal, expressed as standard normal variance (power at frequency of interest minus mean power for the entire trial divided by the SD of power for that trial), at these frequencies
| Targeted Breathing Frequency, breaths/min |
|||||
|---|---|---|---|---|---|
| Variable | Spontaneous | 7 | 14 | 21 | Spontaneous |
| Respiratory frequency, Hz | 0.24 ± 0.01 | 0.12 ± 0.01 | 0.24 ± 0.00 | 0.35 ± 0.00 | 0.22 ± 0.02 |
| Cardiac frequency, Hz | 1.05 ± 0.04 | 1.07 ± 0.03 | 1.09 ± 0.04 | 1.09 ± 0.04 | 1.06 ± 0.04 |
| Power at respiratory frequency | 1.99 ± 0.24 | 3.11 ± 0.32* | 1.82 ± 0.28 | 1.43 ± 0.22 | 2.08 ± 0.30 |
| Power at cardiac frequency | 4.28 ± 0.44 | 3.74 ± 0.45 | 4.03 ± 0.44 | 3.95 ± 0.52 | 3.57 ± 0.44 |
Values are means ± SE; n = 20 experiments. MSNA, muscle sympathetic nerve activity.
P < 0.05 vs. all other frequencies by Scheffé's pairwise comparisons following one-way ANOVA.
Figure 2B shows group mean values for normalized power at the respiratory frequency in subjects who had relatively high levels of MSNA (i.e., burst incidence >40% of cardiac cycles) during baseline spontaneous breathing vs. those that had relatively low MSNA (burst incidence <40%). Within-breath modulation of MSNA was enhanced by 119 ± 39% at low fB and raised VT (P < 0.001 to P = 0.021 vs. all other conditions). This increase occurred in subjects with relatively low or relatively high levels of baseline MSNA. Interestingly, two-way repeated-measures ANOVA revealed a statistically significant frequency-by-group interaction (P = 0.027) such that subjects with MSNA burst incidence <40% had greater increases in power at low fB and greater decreases at high fB than those with burst incidence >40%.
Effects of respiratory rate and VT on steady-state MSNA.
Group mean values for MSNA expressed as burst frequency, burst incidence, and total minute activity (percent of eupneic control condition) are shown in Table 2. There were no significant effects of fB on any of these measures of steady-state MSNA (P = 0.335–0.999). To determine whether MSNA responses to alterations in fB and VT differed according to baseline levels of MSNA, we again partitioned subjects into groups with relatively high and relatively low baseline MSNA. Figure 3, A–C, shows the effects of altering breathing pattern on MSNA in each subject and also the mean values for the two groups. Under control conditions of spontaneous eupneic breathing, there was marked interindividual variability in MSNA, both in terms of burst frequency (8–82 bursts/min) and burst incidence (10–93% of cardiac cycles). Across the threefold change in fB and twofold variation in VT, none of the indexes of steady-state MSNA showed any systematic variation. This lack of effect of breathing pattern on steady-state MSNA occurred across all subjects, despite up to fourfold interindividual differences in their baseline levels of steady-state MSNA.
Table 2.
Steady-state values for cardiorespiratory variables during spontaneous and paced breathing
| Targeted Breathing Frequency, breaths/min |
|||||
|---|---|---|---|---|---|
| Variable | Spontaneous (15 ± 1) | 7 | 14 | 21 | Spontaneous (14 ± 1) |
| MSNA | |||||
| Frequency, bursts/min | 28 ± 4 | 28 ± 4 | 28 ± 4 | 28 ± 4 | 28 ± 4 |
| Incidence, bursts/100 cardiac cycles | 44 ± 6 | 43 ± 6 | 42 ± 5 | 42 ± 6 | 43 ± 6 |
| Total activity, %baseline | 100 | 94 ± 5 | 90 ± 5 | 91 ± 3 | 99 ± 6 |
| Systolic pressure, mmHg | 125 ± 3 | 127 ± 3 | 131 ± 4 | 128 ± 4 | 129 ± 4 |
| Diastolic pressure, mmHg | 78 ± 2 | 78 ± 3 | 80 ± 2 | 79 ± 2 | 79 ± 2 |
| Mean arterial pressure, mmHg | 94 ± 2† | 94 ± 3 | 96 ± 3 | 94 ± 3 | 97 ± 3* |
| Forearm blood flow, ml/min | 92 ± 16 | 105 ± 17 | 106 ± 19 | 106 ± 19 | 93 ± 16 |
| Vascular conductance, ml · min−1 · mmHg−1 | 97 ± 16 | 109 ± 17 | 110 ± 20 | 111 ± 20 | 94 ± 17 |
| Tidal volume, liters | 0.77 ± 0.05 | 1.38 ± 0.07*† | 0.79 ± 0.05 | 0.65 ± 0.04† | 0.85 ± 0.07 |
| Ti/TTOT | 0.47 ± 0.02 | 0.44 ± 0.01 | 0.43 ± 0.01 | 0.45 ± 0.01 | 0.47 ± 0.01 |
| PetCO2, Torr | 36.5 ± 1.2 | 34.9 ± 1.3 | 35.3 ± 1.2 | 34.2 ± 1.5* | 35.6 ± 1.3 |
Values are means ± SE; n = 11 for tidal volume and 20 for MSNA; otherwise, n = 21. Ti/TTOT, inspiratory time as a fraction of total breath time; PetCO2, end-tidal CO2 tension. The nos. in parentheses are the means ± SE for the actual breathing frequencies during the initial and final periods of spontaneous breathing.
P < 0.05 vs. initial spontaneous breathing period;
P < 0.05 vs. final spontaneous breathing period by Scheffé's pairwise comparisons following one-way ANOVA.
Fig. 3.

Effects of voluntary alterations in breathing pattern on MSNA expressed as burst frequency (A), burst incidence (B), and total minute activity (%baseline) (C) in individual subjects. In A–C, subjects are partitioned into two groups: those with MSNA burst incidence >40% (group mean indicated by filled circles and individual subjects by solid lines) and those with burst incidence <40% (open circles, dashed lines). Two-way repeated-measures ANOVA revealed no frequency-by-group interaction (P = 0.605–0.936).
A small but statistically significant decrease from the baseline PetCO2 was observed during the 21 breaths/min trial (−2 ± 3 mmHg; P = 0.008) (Table 2). Across the entire range of fB and VT, 16 of the 21 subjects showed random changes in PetCO2 of less than ±1 mmHg. In the remaining five subjects, PetCO2 fell 2–7 mmHg between the lowest and highest breathing frequencies. The same lack of effect of breathing pattern on MSNA was observed in subjects who maintained strict normocapnia and in those with these noted reductions in PetCO2 (P > 0.05; data not shown).
Effects of respiratory rate and VT on MAP and vascular conductance.
Group mean values for MAP, FBF, and vascular conductance are shown in Table 2. MAP during paced breathing was unchanged from the initial period of spontaneous breathing; however, MAP during the final period of spontaneous breathing was higher than in the initial period of spontaneous breathing (+4 ± 2 mmHg; P = 0.035). We observed small increases in FBF and conductance during all levels of paced breathing that did not reach statistical significance (P values 0.095–0.121). These increases occurred during both decreases and increases in respiratory rate; thus, they were associated with the voluntary act of paced breathing, per se, rather than with changes in fB and VT.
In Fig. 4, A–C, we have again partitioned the subjects into two groups of relatively high and low baseline MSNA to show the effects of changes in respiratory pattern on MAP, limb blood flow, and vascular conductance. Note first that the average MAP was higher in subjects with relatively high MSNA vs. those with low MSNA (P = 0.014), although there were many individual exceptions. There was a significant difference in FBF (P = 0.04) but no difference in conductance (P = 0.069) in subjects with high MSNA vs. low MSNA. As was the case with MSNA, we found no systematic effect of alterations in fB and VT on steady-state values for MAP, FBF, or limb vascular conductance. To the contrary (and similarly to MSNA; see Fig. 3), the great majority of individual steady-state values for MAP, blood flow, and vascular conductance remained unaltered from eupneic control values, as fB and VT were voluntarily changed over the five different experimental conditions. The two repeat conditions of spontaneous eupnea before and after paced breathing produced nearly identical levels of fB, MAP, conductance, and MSNA in all subjects. The effects of breathing pattern on hemodynamic variables were not different in subjects who maintained strict normocapnia during paced breathing vs. those whose PetCO2 fell (P > 0.05; data not shown).
Fig. 4.

Effects of voluntary alterations in breathing pattern on mean arterial pressure (A), forearm blood flow (B), and forearm vascular conductance (C) in individual subjects. In A–C, subjects are partitioned into two groups: those with MSNA burst incidence >40% (group mean indicated by filled circles and individual subjects by solid lines) and those with burst incidence <40% (open circles, dashed lines). There were no frequency-by-group interactions (P = 0.100–0.274).
In Fig. 5, A–C, we have partitioned the subjects into two groups according to baseline blood pressure: those below the median pressure and those at or above the median pressure. Steady-state vascular responses to alterations in fB and VT were negligible and virtually identical in the two groups (P > 0.05).
Fig. 5.

Effects of voluntary alterations in breathing pattern on mean arterial pressure (MAP, A), forearm blood flow (B), and forearm vascular conductance (C) in individual subjects. In A–C, subjects are partitioned into two groups: those with mean arterial pressures greater than or equal to the median for the group (group mean indicated by filled circles and individual subjects by solid lines) and those with pressures less than the group median (open circles, dashed lines). Two-way repeated-measures ANOVA revealed no frequency-by-group interactions (P = 0.710–0.866).
DISCUSSION
The major finding of this study is that manipulation of respiratory rate with reciprocal changes in VT above and below eupneic levels had no effect on steady-state blood pressure, FBF, vascular conductance, or MSNA. Nevertheless, we observed robust within-breath respiratory modulation of MSNA in our subjects, the power of which changed significantly with alterations in fB and VT (see Fig. 2, A and B). These effects were consistent across subjects who varied markedly in their steady-state levels of MSNA, blood pressure, and vascular conductance. The present results confirm our previous negative findings on steady-state MSNA at increased fB and VT (9, 40, 42, 47). Importantly, the present data also extend our previous findings by demonstrating a similar lack of effect on steady-state MSNA of slowing of the respiratory rate below eupneic levels with reciprocal increases in VT. Moreover, we documented a lack of effect of variations in fB and VT, above and below eupneic levels, on steady-state blood flow, vascular conductance, and blood pressure.
Mechanisms of within-breath modulation of MSNA.
In intact humans, MSNA is inhibited by inspiration, i.e., from midinspiration to midexpiration, with MSNA activation occurring during late expiration (7, 10, 26, 40). Changes in lung volume, baroreceptor feedback, and feedforward central respiratory motor output all have been implicated as causes of this respiratory modulation. Clearly, lung volume is a contributor because within-breath modulation is more pronounced when VT is increased, either voluntarily or via hypercapnia, or when inspiratory time is prolonged. Furthermore, MSNA inhibition begins earlier during inspiration when end-expiratory lung volume is voluntarily elevated (40, 41). This inspiratory-phase inhibition of MSNA is equally effective under control, eupneic conditions and when baseline MSNA is elevated during such conditions as lower-body negative pressure, ischemic handgrip, or induced hypercapnia (40). Nevertheless, in humans, chronic denervation of the lungs reduces, but does not eliminate, lung volume-dependent modulation of MSNA (41). Arterial pressure and aortic arch transmural pressure fluctuate across the respiratory cycle, changes that are consistent with a baroreceptor-mediated influence on within-breath MSNA (4). However, evidence against a primary role for baroreceptors in within-breath modulation of MSNA includes: 1) no effect of positive vs. negative pressure ventilation on within-breath MSNA modulation (26, 41) and 2) the observation that variations in MSNA occur with changes in lung volume even at equal levels of within-breath diastolic blood pressure (40, 41). Reducing (via mechanical ventilation) or increasing the magnitude of central respiratory motor output, by itself, does not influence within-breath MSNA modulation independent of respiratory pattern in intact humans (48).
Effects of breathing pattern alterations on blood pressure and MSNA.
Previous investigators have studied the effects of daily training with volitional slowing of the respiratory rate on blood pressure in patients with hypertension (3, 12, 16, 19, 25, 27, 37, 38). In these studies, an auditory device cued subjects to reduce respiratory rates to <10 breaths/min for 10–15 min/day for 8–9 wk. Control subjects received usual care (19, 37), performed self-monitoring of blood pressure and heart rate (11, 27) or, in four studies, listened to music through headphones for an amount of time equal to that spent in slow breathing by the experimental subjects (3, 16, 25, 38). In all four of these studies with the most appropriate experimental designs (i.e., music as a placebo control), blood pressure was reduced in both the treatment and control groups; however, the between-group difference in blood pressure lowering (∼4 mmHg) was statistically significant in only two of the four studies (16, 38).
Acute reductions in blood pressure have also been reported in patients with hypertension during short periods (∼2 min) of slow, deep breathing (21), and the presumed mechanism for this effect is a reduction in sympathetic vasoconstrictor outflow (21, 44). The acute effects of slow, deep breathing on MSNA in mildly hypertensive subjects were reported in two recent studies (19, 33). Slowing of breathing rate below 10/min for 15 min resulted in mean decreases in MSNA burst frequency of 8 (33) and 10 (19) bursts/min. Neither study reported a significant acute effect of slow breathing on blood pressure.
In contrast to these results (19, 33), we observed no effects of fB alterations on steady-state MSNA. Breathing rates were slowed to the same extent in all three studies (6–7 breaths/min); nevertheless, there are several possible reasons for the discordant findings. First, slow breathing was maintained for 15 min in the previous studies, whereas we observed 3-min trials at each of the breathing frequencies. We consider it unlikely that a longer observation period would have yielded different results because, in our previous studies, steady-state MSNA and blood pressure were unchanged during 10- to 15-min periods in which both fB and VT were volitionally increased two to three times baseline (40, 47, 48) or when VT was increased twofold with normal fB and respiratory duty cycle substantially prolonged (47, 48). These latter two studies, like the present one, showed a marked effect of breathing pattern on within-breath modulation of MSNA (47, 48). In addition, reduced PaCO2 has an inhibitory effect on the carotid chemoreceptor (46) and is an important vasoconstrictor, most notably in the cerebral circulation (20). Hypocapnia may have occurred in the previous studies (19, 33) (VT and Pco2 were not reported) which, if sufficient, could have reduced MSNA. Finally, our subjects appeared to be on average healthier than the mildly hypertensive subjects of Oneda et al.(33) and Hering et al.(19); however, our findings were consistent across subjects who varied markedly in MSNA burst incidence (4-fold), forearm conductance (15-fold), and blood pressure (3 subjects >140/90 mmHg). Although we cannot fully explain the differences in our findings vs. those of the previous studies, it should be noted that the 18 and 34% reductions in MSNA observed in the previous studies were not associated with reductions in blood pressure.
Does altered within-breath respiratory modulation contribute to heightened sympathoexcitation in the clinical setting?
Several mechanisms have been postulated to underlie elevated sympathetic activity in patients with hypertension, e.g., decreased baroreceptor sensitivity (34), increased carotid chemoreceptor sensitivity (49), elevated pulmonary vascular pressures (14, 23), increased density of sympathetic innvervation (1), and altered noradrenergic transmission and reuptake (1, 5). Recently, several authors (28–30, 45, 50) suggested, based on correlative observations in vagally denervated, decerebrate animals, that hypertension and sympathoexcitation in hypertensive rats are attributable to augmented modulation of sympathetic activity by central respiratory motor output. Our present data in humans are not consistent with this hypothesis because we observed that changing the power of within-breath respiratory modulation did not alter steady-state MSNA burst frequency, incidence, or total activity and that this dissociation between within-breath MSNA modulation and steady-state MSNA held for subjects with substantial differences in baseline MSNA (see Fig. 3B). Consistent with our findings, Fatouleh and Macefield (13) recently reported normal levels of within-breath modulation of MSNA in patients with chronic obstructive pulmonary disease and those with essential hypertension, all of whom had markedly elevated MSNA burst frequencies in the steady state.
With reference to the above discussion contrasting current with previous findings, we would like to point out some important differences in respiratory-sympathetic coupling between the debuffered, anesthetized, or decerebrate rodent and the intact human. First, inhibition of sympathetic activity occurs primarily during inspiration in the human vs. during expiration in the debuffered rodent. Second, unlike the debuffered rodent or cat, in the intact human neither within-breath nor steady-state MSNA is influenced by central inspiratory motor output, per se, whether it is augmented severalfold via voluntary respiratory efforts or eliminated via mechanical ventilation (48). We speculate that these differences reflect a dominance of feedback inhibitory influences on MSNA in the intact human vs. a dominant feed-forward effect of central respiratory motor output in the debuffered animal. These differences clearly limit the inferences that can be made from the animal model to the human. Finally, our current findings relating within-breath to steady-state MSNA in humans showed, unlike the debuffered rat preparation (6, 45), that heightened within-breath modulation was associated with reduced steady-state burst incidence (see Fig. 2B).
The current findings would also not support the notion that the tachypneic breathing pattern, per se, observed in patients with heart failure determines their high levels of steady-state MSNA (15, 31). Instead, based on the present findings, we would postulate that breathing pattern and steady-state MSNA are correlated in heart failure because they are both driven by common influences, e.g., sensitization of carotid chemoreceptors (39), elevated cardiac filling and pulmonary vascular pressures (14, 23), and/or chemoreceptor stimulation via increased levels of circulating catecholamines (8, 18).
Limitations.
While our findings provide consistent evidence against the concept that alterations in fB and VT will alter steady-state MSNA or its vascular consequences, there are several limitations to the broader implications of these data. First, our sample included subjects who demonstrated a 5- to 10-fold variation in MSNA burst frequency and severalfold variations in limb vascular conductance and even some individuals who met three of the criteria for “metabolic syndrome”; however, less than half of our subjects were in the borderline hypertensive range. So, it is important that our experimental protocol be extended to patients with greater elevations in blood pressure, although Fatouleh and Macefield recently reported that patients with essential hypertension exhibited high MSNA burst frequencies in the face of normal within-breath modulation (13). Second, our protocols imposed only acute changes in breathing pattern, and the lack of effects we observed on steady-state MSNA and vascular responses might not apply to protocols that employ sessions of “breath training” over prolonged periods. Nevertheless, it is important to note that, in one recent study, an 8-wk intervention with slow breathing failed to reduce MSNA burst frequency (19). Third, our findings do not apply to alterations in fB and VT that require substantial and sustained increases in the work of breathing because fatiguing contractions of inspiratory and expiratory muscles have been shown to increase MSNA (9, 47) and reduce limb vascular conductance (42, 43). Furthermore, our findings also might not apply to nonfatiguing conditions where VT and inspiratory time are markedly elevated even in the face of unchanged fB, resulting in significant steady-state increases in limb vascular conductance with no change in MAP (42).
In conclusion, subjects in the present study demonstrated robust within-breath modulation of MSNA. In contrast, there was no evidence that alterations in breathing rate with reciprocal changes in VT influenced MSNA, blood flow, or vascular conductance in the steady state. This incongruity between within-breath modulation and steady-state levels of MSNA held across subjects with a fourfold difference in baseline MSNA burst frequency. Our findings call into question the supposition that blood pressure reduction observed after daily training with slow, deep breathing (16, 38) is caused by reduced sympathetic vasoconstrictor outflow. The question of whether or not training with simple slowing of respiratory rate causes long-term reductions in blood pressure, as has been claimed for other forms of biofeedback (32, 35), is unresolved. Our data would suggest that, if such training is to be clinically useful, the effective combination of VT and respiratory duty cycle must first be established.
GRANTS
This work was funded by American Heart Association Predoctoral Fellowships (nos. 0815622G and 10PRE3870000 to J. K. Limberg) and National Heart, Lung, and Blood Institute Grants RO-1-HL-15469 (to J. A. Dempsey) and RO-1-HL-105820 (to W. G. Schrage).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: J.K.L., B.J.M., W.G.S., and J.A.D. performed experiments; J.K.L., B.J.M., and W.G.S. analyzed data; J.K.L., B.J.M., W.G.S., and J.A.D. interpreted results of experiments; J.K.L., B.J.M., and W.G.S. prepared figures; J.K.L., B.J.M., and W.G.S. drafted manuscript; J.K.L., B.J.M., W.G.S., and J.A.D. edited and revised manuscript; J.K.L., B.J.M., W.G.S., and J.A.D. approved final version of manuscript; J.A.D. conception and design of research.
ACKNOWLEDGMENTS
We are grateful to Anthony Jacques, David Pegelow, Anne Bolgert, John Harrell, Edward McKenna, and Keelin O'Neil for assistance with data collection and analysis and to Meghan Crain for help with subject recruitment.
REFERENCES
- 1. Adams MA, Bobik A, Korner PI. Differential development of vascular and cardiac hypertrophy in genetic hypertension. Relation to sympathetic function Hypertension 14: 191–202, 1989 [DOI] [PubMed] [Google Scholar]
- 2. Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart JC, James WP, Loria CM, Smith SC, International Diabetes Federation Task Force on Epidemiology and Prevention, National Heart, Lung, and Blood Institute, American Heart Association, World Heart Federation, International Atherosclerosis Society, International Association for the Study of Obesity Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 120: 1640–1645, 2009 [DOI] [PubMed] [Google Scholar]
- 3. Altena MR, Kleefstra N, Logtenberg SJ, Groenier KH, Houweling ST, Bilo HJ. Effect of device-guided breathing exercises on blood pressure in patients with hypertension: a randomized controlled trial. Blood Press 18: 273–279, 2009 [DOI] [PubMed] [Google Scholar]
- 4. Angell James JE. The effects of changes of extramural, ‘intrathoracic’, pressure on aortic arch baroreceptors. J Physiol 214: 89–103, 1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cabassi A, Vinci S, Quartieri F, Moschini L, Borghetti A. Norepinephrine reuptake is impaired in skeletal muscle of hypertensive rats in vivo. Hypertension 37: 698–702, 2001 [DOI] [PubMed] [Google Scholar]
- 6. Czyzyk-Krzeska MF, Trzebski A. Respiratory-related discharge pattern of sympathetic nerve activity in the spontaneously hypertensive rat. J Physiol 426: 355–368, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Burgh Daly M, Hazzledine JL, Ungar A. The reflex effects of alterations in lung volume on systemic vascular resistance in the dog. J Physiol 188: 331–351, 1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dempsey JA, Gledhill N, Reddan WG, Forster HV, Hanson PG, Claremont AD. Pulmonary adaptation to exercise: effects of exercise type and duration, chronic hypoxia and physical training. Ann NY Acad Sci 301: 243–261, 1977 [DOI] [PubMed] [Google Scholar]
- 9. Derchak PA, Sheel AW, Morgan BJ, Dempsey JA. Effects of expiratory muscle work on muscle sympathetic nerve activity. J Appl Physiol 92: 1539–1552, 2002 [DOI] [PubMed] [Google Scholar]
- 10. Eckberg DL, Nerhed C, Wallin BG. Respiratory modulation of muscle sympathetic and vagal cardiac outflow in man. J Physiol 365: 181–196, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Elliot WJ, Izzo JL, White WB, Rosing DR, Snyder CS, Alter A, Gavish B, Black HR. Graded blood pressure reduction in hypertensive outpatients associated with use of a device to assist with slow breathing. J Clin Hypertens (Greenwich) 6: 553–559, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Elliott WJ, Izzo JL. Device-guided breathing to lower blood pressure: case report and clinical overview (Abstract). MedGenMed 8: 23, 2006 [PMC free article] [PubMed] [Google Scholar]
- 13. Fatouleh R, Macefield VG. Respiratory modulation of muscle sympathetic nerve activity is not increased in essential hypertension or chronic obstructive pulmonary disease. J Physiol 589: 4997–5006, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferguson DW, Berg WJ, Sanders JS. Clinical and hemodynamic correlates of sympathetic nerve activity in normal humans and patients with heart failure: evidence from direct microneurographic recordings. J Am Coll Cardiol 16: 1125–1134, 1990 [DOI] [PubMed] [Google Scholar]
- 15. Goso Y, Asanoi H, Ishise H, Kameyama T, Hirai T, Nozawa T, Takashima S, Umeno K, Inoue H. Respiratory modulation of muscle sympathetic nerve activity in patients with chronic heart failure. Circulation 104: 418–423, 2001 [DOI] [PubMed] [Google Scholar]
- 16. Grossman E, Grossman A, Schein MH, Zimlichman R, Gavish B. Breathing-control lowers blood pressure. J Hum Hypertens 15: 263–269, 2001 [DOI] [PubMed] [Google Scholar]
- 17. Hagbarth KE, Vallbo AB. Pulse and respiratory grouping of sympathetic impulses in human muscle-nerves. Acta Physiol Scand 74: 96–108, 1968 [DOI] [PubMed] [Google Scholar]
- 18. Heistad DD, Wheeler RC, Mark AL, Schmid PG, Abboud FM. Effects of adrenergic stimulation on ventilation in man. J Clin Invest 51: 1469–1475, 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hering D, Kucharska W, Kara T, Somers VK, Parati G, Narkiewicz K. Effects of acute and long-term slow breathing exercise on muscle sympathetic nerve activity in untreated male patients with hypertension. J Hypertens 31: 739–746, 2013 [DOI] [PubMed] [Google Scholar]
- 20. Ide K, Eliasziw M, Poulin MJ. Relationship between middle cerebral artery blood velocity and end-tidal PCO2 in the hypocapnic-hypercapnic range in humans. J Appl Physiol 95: 129–137, 2003 [DOI] [PubMed] [Google Scholar]
- 21. Joseph CN, Porta C, Casucci G, Casiraghi N, Maffeis M, Rossi M, Bernardi L. Slow breathing improves arterial baroreflex sensitivity and decreases blood pressure in essential hypertension. Hypertension 46: 714–718, 2005 [DOI] [PubMed] [Google Scholar]
- 22. Khayat RN, Przybylowski T, Meyer KC, Skatrud JB, Morgan BJ. Role of sensory input from the lungs in control of muscle sympathetic nerve activity during and after apnea in humans. J Appl Physiol 97: 635–640, 2004 [DOI] [PubMed] [Google Scholar]
- 23. Leimbach WN, Wallin BG, Victor RG, Aylward PE, Sundlöf G, Mark AL. Direct evidence from intraneural recordings for increased central sympathetic outflow in patients with heart failure. Circulation 73: 913–919, 1986 [DOI] [PubMed] [Google Scholar]
- 24. Limberg JK, Morgan BJ, Sebranek JJ, Proctor LT, Walker BJ, Eldridge MW, Schrage WG. Altered neurovascular control of the resting circulation in human metabolic syndrome. J Physiol 590: 6109–6119, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Logtenberg SJ, Kleefstra N, Houweling ST, Groenier KH, Bilo HJ. Effect of device-guided breathing exercises on blood pressure in hypertensive patients with type 2 diabetes mellitus: a randomized controlled trial. J Hypertens 25: 241–246, 2007 [DOI] [PubMed] [Google Scholar]
- 26. Macefield VG, Wallin BG. Effects of static lung inflation on sympathetic activity in human muscle nerves at rest and during asphyxia. J Auton Nerv Syst 53: 148–156, 1995 [DOI] [PubMed] [Google Scholar]
- 27. Meles E, Giannattasio C, Failla M, Gentile G, Capra A, Mancia G. Nonpharmacologic treatment of hypertension by respiratory exercise in the home setting. Am J Hypertens 17: 370–374, 2004 [DOI] [PubMed] [Google Scholar]
- 28. Molkov YI, Zoccal DB, Moraes DJ, Paton JF, Machado BH, Rybak IA. Intermittent hypoxia-induced sensitization of central chemoreceptors contributes to sympathetic nerve activity during late expiration in rats. J Neurophysiol 105: 3080–3091, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moraes DJ, Dias MB, Cavalcanti-Kwiatkoski R, Machado BH, Zoccal DB. Contribution of the retrotrapezoid nucleus/parafacial respiratory region to the expiratory-sympathetic coupling in response to peripheral chemoreflex in rats. J Neurophysiol 108: 882–890, 2012 [DOI] [PubMed] [Google Scholar]
- 30. Moraes DJ, Zoccal DB, Machado BH. Medullary respiratory network drives sympathetic overactivity and hypertension in rats submitted to chronic intermittent hypoxia. Hypertension, 2012 [DOI] [PubMed] [Google Scholar]
- 31. Naughton MT, Floras JS, Rahman MA, Jamal M, Bradley TD. Respiratory correlates of muscle sympathetic nerve activity in heart failure. Clin Sci (Lond) 95: 277–285, 1998 [PubMed] [Google Scholar]
- 32. Nidich SI, Rainforth MV, Haaga DA, Hagelin J, Salerno JW, Travis F, Tanner M, Gaylord-King C, Grosswald S, Schneider RH. A randomized controlled trial on effects of the Transcendental Meditation program on blood pressure, psychological distress, and coping in young adults. Am J Hypertens 22: 1326–1331, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oneda B, Ortega KC, Gusmão JL, Araújo TG, Mion D. Sympathetic nerve activity is decreased during device-guided slow breathing. Hypertens Res 33: 708–712, 2010 [DOI] [PubMed] [Google Scholar]
- 34. Parati G, Esler M. The human sympathetic nervous system: its relevance in hypertension and heart failure. Eur Heart J 33: 1058–1066, 2012 [DOI] [PubMed] [Google Scholar]
- 35. Pramanik T, Pudasaini B, Prajapati R. Immediate effect of a slow pace breathing exercise Bhramari pranayama on blood pressure and heart rate. Nepal Med Coll J 12: 154–157, 2010 [PubMed] [Google Scholar]
- 36. Randolph TW. Scale-based normalization of spectral data. Cancer Biomark 2: 135–144, 2006 [DOI] [PubMed] [Google Scholar]
- 37. Schein MH, Gavish B, Baevsky T, Kaufman M, Levine S, Nessing A, Alter A. Treating hypertension in type II diabetic patients with device-guided breathing: a randomized controlled trial. J Hum Hypertens 23: 325–331, 2009 [DOI] [PubMed] [Google Scholar]
- 38. Schein MH, Gavish B, Herz M, Rosner-Kahana D, Naveh P, Knishkowy B, Zlotnikov E, Ben-Zvi N, Melmed RN. Treating hypertension with a device that slows and regularises breathing: a randomised, double-blind controlled study. J Hum Hypertens 15: 271–278, 2001 [DOI] [PubMed] [Google Scholar]
- 39. Schultz HD, Li YL, Ding Y. Arterial chemoreceptors and sympathetic nerve activity: implications for hypertension and heart failure. Hypertension 50: 6–13, 2007 [DOI] [PubMed] [Google Scholar]
- 40. Seals DR, Suwarno NO, Dempsey JA. Influence of lung volume on sympathetic nerve discharge in normal humans. Circ Res 67: 130–141, 1990 [DOI] [PubMed] [Google Scholar]
- 41. Seals DR, Suwarno NO, Joyner MJ, Iber C, Copeland JG, Dempsey JA. Respiratory modulation of muscle sympathetic nerve activity in intact and lung denervated humans. Circ Res 72: 440–454, 1993 [DOI] [PubMed] [Google Scholar]
- 42. Sheel AW, Derchak PA, Morgan BJ, Pegelow DF, Jacques AJ, Dempsey JA. Fatiguing inspiratory muscle work causes reflex reduction in resting leg blood flow in humans. J Physiol 537: 277–289, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sheel AW, Derchak PA, Pegelow DF, Dempsey JA. Threshold effects of respiratory muscle work on limb vascular resistance. Am J Physiol Heart Circ Physiol 282: H1732–H1738, 2002 [DOI] [PubMed] [Google Scholar]
- 44. Sica DA. Device-guided breathing and hypertension: a yet to be determined positioning. Cardiol Rev 19: 45–46, 2011 [DOI] [PubMed] [Google Scholar]
- 45. Simms AE, Paton JF, Pickering AE, Allen AM. Amplified respiratory-sympathetic coupling in the spontaneously hypertensive rat: does it contribute to hypertension? J Physiol 587: 597–610, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smith CA, Harms CA, Henderson KS, Dempsey JA. Ventilatory effects of specific carotid body hypocapnia and hypoxia in awake dogs. J Appl Physiol 82: 791–798, 1997 [DOI] [PubMed] [Google Scholar]
- 47. St. Croix CM, Morgan BJ, Wetter TJ, Dempsey JA. Fatiguing inspiratory muscle work causes reflex sympathetic activation in humans. J Physiol 529: 493–504, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. St. Croix CM, Satoh M, Morgan BJ, Skatrud JB, Dempsey JA. Role of respiratory motor output in within-breath modulation of muscle sympathetic nerve activity in humans. Circ Res 85: 457–469, 1999 [DOI] [PubMed] [Google Scholar]
- 49. Tan ZY, Lu Y, Whiteis CA, Simms AE, Paton JF, Chapleau MW, Abboud FM. Chemoreceptor hypersensitivity, sympathetic excitation, and overexpression of ASIC and TASK channels before the onset of hypertension in SHR. Circ Res 106: 536–545, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Toney GM, Pedrino GR, Fink GD, Osborn JW. Does enhanced respiratory-sympathetic coupling contribute to peripheral neural mechanisms of angiotensin II-salt hypertension? Exp Physiol 95: 587–594, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vallbo AB, Hagbarth KE, Torebjörk HE, Wallin BG. Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiol Rev 59: 919–957, 1979 [DOI] [PubMed] [Google Scholar]