

Abstract
Cardiovascular disease risk factors, such as diabetes, hypertension, dyslipidemia, obesity, and physical inactivity, are all correlated with impaired endothelial nitric oxide synthase (eNOS) function and decreased nitric oxide (NO) production. NO-mediated regulation of mitochondrial biogenesis has been established in many tissues, yet the role of eNOS in vascular mitochondrial biogenesis and dynamics is unclear. We hypothesized that genetic eNOS deletion and 3-day nitric oxide synthase (NOS) inhibition in rodents would result in impaired mitochondrial biogenesis and defunct fission/fusion and autophagy profiles within the aorta. We observed a significant, eNOS expression-dependent decrease in mitochondrial electron transport chain (ETC) protein subunits from complexes I, II, III, and V in eNOS heterozygotes and eNOS null mice compared with age-matched controls. In response to NOS inhibition with NG-nitro-l-arginine methyl ester (l-NAME) treatment in Sprague Dawley rats, significant decreases were observed in ETC protein subunits from complexes I, III, and IV as well as voltage-dependent anion channel 1. Decreased protein content of upstream regulators of mitochondrial biogenesis, cAMP response element-binding protein and peroxisome proliferator-activated receptor-γ coactivator-1α, were observed in response to 3-day l-NAME treatment. Both genetic eNOS deletion and NOS inhibition resulted in decreased manganese superoxide dismutase protein. l-NAME treatment resulted in significant changes to mitochondrial dynamic protein profiles with decreased fusion, increased fission, and minimally perturbed autophagy. In addition, l-NAME treatment blocked mitochondrial adaptation to an exercise intervention in the aorta. These results suggest that eNOS/NO play a role in basal and adaptive mitochondrial biogenesis in the vasculature and regulation of mitochondrial turnover.
Keywords: endothelial nitric oxide synthase, vascular mitochondria
the vasculature is a complex tissue with a cellular architecture that permits specific contractile profiles for optimal tissue perfusion and adaptation to physiological demands. One of the primary regulators of physiological vasomotion is nitric oxide (NO), generated enzymatically by a family of nitric oxide synthases (NOS): endothelial NOS (eNOS), neuronal NOS (nNOS), and inducible NOS. In addition to the regulation of blood flow, NO modulates vascular structure and function through direct signaling to vascular endothelium, vascular smooth muscle cells, and inflammatory and adventitial cells (reviewed in Ref. 11). Endothelium-derived NO is a physiologically significant vasodilator and inhibitor of platelet aggregation and adhesion. Vascular NO also prevents leukocyte adhesion to the endothelium and inhibits proliferation of vascular smooth muscle cells (11). Cardiovascular disease (CVD) risk factors, such as diabetes, hypertension, dyslipidemia, obesity, and physical inactivity, are all correlated with impaired eNOS function and NO production (48).
Both eNOS and NO play an important role in signaling to mitochondria; Nisoli and colleagues demonstrated that NO is an important regulator of mitochondrial biogenesis in numerous target tissues (34). eNOS null mice have reductions in skeletal muscle mitochondrial biogenesis, and deletion of eNOS in brown adipocytes interferes with cGMP induction of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) and mitochondrial biogenesis in response to calorie restriction (35). Recently, examination of specific vascular segments revealed that physiological pulsatile flow led to the activation of sirtuin 1 (SIRT1) and AMP-activated protein kinase (AMPK), each contributing to the activation of eNOS, PGC-1α, and mitochondrial biogenesis (7). Baseline determinants of vascular mitochondrial content and function have not been directly examined, but eNOS/NO are plausible determinants.
Vascular mitochondrial dysfunction, increased mitochondrial reactive oxygen species (ROS), and decreased respiration have recently emerged as risks for vascular dysfunction (40, 53, 25). Mitochondrial diseases, such as mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes syndrome (MELAS), have increased vascular stiffness and decreased NO-dependent vasodilatation (32, 19). Our group recently reported that aortic mitochondrial content is decreased in rat models of hypertension, metabolic syndrome, and diabetes (18). We further demonstrated that exposure of control rats to exercise led to an adaptive increase in aortic mitochondrial protein. In rodent models of diabetes, hypertension, and endothelial dysfunction, this mitochondrial adaptation is absent; reports also suggest a similar blunted response to exercise in the skeletal muscle (22, 33).
New information on mitochondrial dynamics indicates that mitochondrial turnover determines mitochondrial quality and may affect vasomotion (61, 39, 10). Mitochondrial fusion is an important process for maintenance of mitochondrial quality, preserving mitochondrial shape (41), ensuring organelle mixing (46), and maintaining mitochondrial integrity and membrane potential (3). In contrast, fission is an important function in regulating the quality of mitochondria by splitting off defunct sections for degradation (52). Fission and fusion coordinate to maintain optimal mitochondrial and quality. Mitochondrial fragmentation was reported in endothelial cells in human subjects and rodent models of vascular disease (43, 58). Perturbations that increase fission [dynamin-related protein 1 (DRP-1)] or decrease fusion (mitofusin 2) are correlated with endothelial dysfunction and increased smooth muscle cell proliferation (27, 68). Inhibition of fission leads to greater mitochondrial networks and decreased ROS production (13). Autophagy plays an important role in recycling of organelles and is thought to play a role in maintenance of mitochondrial quality through selective degradation (17). The roles of eNOS or NO in mitochondrial biogenesis, fission, fusion, and autophagy have been little examined in the vasculature in health or disease.
A common feature of vascular disease is endothelial dysfunction, including impaired function of eNOS. We hypothesized that NO supports vascular mitochondrial biogenesis, potentially through a cAMP response element-binding protein (CREB)/PGC-1α mediated mechanism, and also via modulation of mitochondrial dynamics. To evaluate this hypothesis, we examined vascular mitochondrial protein expression and markers of mitochondrial turnover in rodent models. Specifically, we used a genetic deletion of eNOS (chronic) and 3-day NG-nitro-l-arginine methyl ester (l-NAME) treatment for NOS inhibition (acute) to assess the importance of the eNOS/NOS for vascular mitochondrial regulation.
MATERIALS AND METHODS
Materials.
We used l-NAME (Cayman Chemicals, Ann Arbor, MI) and 1× Mammalian Lysis Buffer [150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 5 mM sodium pyrophosphate, 1 mM sodium orthovandadate, 20 mM sodium fluoride, 500 mM okadaic acid, protease inhibitor cocktail p8340 (Sigma-Aldrich, St. Louis, MO), and M-PER mammalian protein extraction reagent (Thermo Scientific, Rockford, IL)]. Rabbit monoclonal CREB (catalog no. 9197, 1:1,000), polyclonal pCREB (catalog no. 9191, 1:1,000), microtubule-associated protein light chain 3β (LC3B) (catalog no. 2775, 1:1,000), Beclin1 (catalog no. 3738, 1:1,000), BCL2/adenovirus E1B 19-kDa protein-interacting protein 3 (BNIP3) (catalog no. 3769, 1:1,000), anti-rabbit IgG secondary (catalog no. 7054, 1:1,000), and anti-mouse IgG secondary (catalog no. 7056, 1:1,000) antibodies were purchased from Cell Signaling Technology (Danvers, MA). Mouse monoclonal eNOS (catalog no. 610296, 1:1,000), optic atrophy 1 (OPA1) (catalog no. 612606, 1:1,000), and DRP-1 (catalog no. 611738, 1:1,000) antibodies were purchased from BD Transduction Laboratories (San Jose, CA). Mouse monoclonal mitofusin 2 (catalog no. ab56889, 1:1,000) and rabbit polyclonal uncoupling protein 3 (UCP3) (catalog no. ab3477, 1:1,000), voltage-dependent anion channel 1 (VDAC1; catalog no. ab14734, 1:1,000), PGC-1α (catalog no. ab54481, 1:1,000), and SIRT1 (catalog no. ab28170, 1:1,000) antibodies were purchased from Abcam (Cambridge, UK). Rabbit polyclonal manganese superoxide dismutase (MnSOD) antibody (catalog no. 06–984, 1:1,000) was purchased from Millipore (Bedford, MA). Mouse monoclonal β-actin antibody (catalog no. A5441, 1:1,000 or 1:10,000) and rabbit polyclonal p62 (catalog no. P0067, 1:1,000) were purchased from Sigma-Aldrich. Mouse monoclonal mitochondrial complex cocktail antibodies (catalog no. MS603, 1:1,000) were purchased from MitoSciences (Eugene, OR). Mouse monoclonal fission 1 (Fis1) antibody (catalog no. IMG-5113A, 1:1,000) was purchased from Imgenex (San Diego, CA). Mouse monoclonal mitofusin 1 antibody (catalog no. NBP1–51841SS, 1:1,000) was purchased from NOVUS Biologicals (Littleton, CO). CDP-Star reagent kit was purchased from New England BioLabs (Ipswich, MA). The Precision Plus Protein Kaleidoscope Molecular Weight Marker was purchased from Bio-Rad (catalog no. 161-0375).
Animals.
Twelve-week-old male Sprague Dawley rats (Harlan Laboratories) and ∼18-wk-old male wild-type, partial eNOS null heterozygotes and eNOS null (homozygotes) mice on C57/B6 background (gift from Dr. Steve Abman, University of Colorado AMC) were used as animal models. Sprague Dawley rats were used for l-NAME and exercise intervention experiments. Animals were fed ad libitum and housed with a 12:12-h light cycle. The use of animals, experimental interventions, and euthanization procedures used in this study received prior approval from the Institutional Animal Care and Use Committees of Denver Veterans Affairs Medical Center and the University of Colorado Denver (Denver, CO).
l-NAME treatment.
Sprague Dawley rats were subjected to daily intraperitoneal injection of 50 mg/kg l-NAME in PBS or PBS (controls). After the last treatment (24 h), the animals were killed with pentobarbital sodium at 100 mg/kg, and aorta tissue was removed. The aorta tissue was immediately frozen in liquid nitrogen.
Exercise intervention.
Rats were subjected to 8 days of exercise on a Columbus Instrument Exer 3/6 treadmill with shock grid following acclimation. Acclimation took place over the course of 1 wk and began by placing rats on a stationary treadmill (0% grade) for 5–15 min on day 1 with progressively increased speed to 10 m/min on the treadmill by the end of the week. Exercised animals were run on the treadmill (0% grade) at 15 m/min for 30 min on days 1 and 2 and for 45 min on days 3–8. l-NAME and control PBS injections began on the first day of exercise (before exercise bout), and injections and exercise continued for 8 days total (injections given after exercise bout after initial day). After last exercise bout and injection (24 h), animals were killed using pentobarbital sodium at 100 mg/kg, and aorta tissue was removed. The aorta tissue was immediately frozen in liquid nitrogen and stored at −80°C until further analysis.
Aorta tissue lysate.
Frozen aortas from rats and mice were pulverized in 1× Mammalian Lysis Buffer in a nitrogen-cooled mortar. After thaw on ice, the lysates where homogenized with an Ultra-Turrax T8 homogenizer (Fisher Scientific, Pittsburgh, PA) and centrifuged at 4°C at 14,000 rpm. Supernatants were moved to a separate tube to prevent contamination from cellular debris. Bradford protein assay was performed on the supernatant to determine the protein concentration in the lysates. Samples were diluted to an equal amount of protein in 5× Laemmli sample buffer with DTT, boiled, and stored at −80°C until Western blot analysis.
Western blot analysis.
Equal amounts of protein (20–40 μg) were electrophoretically separated by 12% SDS-PAGE and transferred to a polyvinylidene fluoride membrane (Millipore). Subsequently, protein loading was assessed by staining of membrane-bound proteins by Ponceau stain and then blocked with 5% milk powder in 1× Tris-buffered saline (pH 7.5) with 0.1% Tween 20 (TBST) for 1 h. Next, the membrane was incubated overnight at 4°C with all the previously listed primary antibodies (diluted in 5% BSA in TBST). Identification of protein by antibody means was assessed by incubation with alkaline phosphatase-conjugated, species-specific IgG diluted in 5% milk in TBST at room temperature for 1 h. Bands were visualized using CDP-Star Enhanced Chemiluminescence on film, with bands picked based on molecular weight when more than one band was apparent. Finally, densitometric analysis was performed using QuantityOne Software (Bio-Rad, Richmond, CA). When reprobing was needed, the blots were stripped between antibodies for 20 min at 55°C in TBST with 0.008% 2-mercaptoethanol. To control for actual protein loading, blots were last probed with β-actin antibody.
Statistics.
Two-tailed Student's t-tests were used to compare control groups with treatment or null groups using an a priori null hypothesis. Results were considered significant with a P value <0.05.
RESULTS
eNOS deletion results in decreased mitochondrial electron transport chain subunit content.
As a first step in understanding the role of NOS on vascular mitochondrial content, we looked at electron transport chain (ETC) content in the aortas of partial eNOS null heterozygotes and eNOS null mice compared with age-matched C57/B6 controls. We observed a significant, eNOS expression-dependent decrease in ETC content in aortic lysates (Fig. 1). eNOS manipulation resulted in a decrease in subunits from complexes I, II, III, and V. For complex I subunit NDUFA9, heterozygotes showed a 50% decrease (P = 0.0176) while eNOS null animals showed a 65% decrease (P = 0.0008). For complex II subunit succinate dehydrogenase complex, subunit A (SDHA), heterozygotes showed a 22% decrease (P = 0.0048) while eNOS null animals showed a 33% decrease (P = 0.0037). For complex III core protein, heterozygotes showed a 61% reduction (P = 0.0006) while eNOS null animals showed a 90% reduction (P < 0.0001). For complex IV subunit 4, no significant decreases were observed. For complex V α-subunit, heterozygotes showed a 43% decrease (P = 0.0048) while eNOS null animals showed a 63% decrease (P < 0.0001).
Fig. 1.

Endothelial nitric oxide synthase (eNOS) null aortas have reduced mitochondrial electron transport chain (ETC). Aortas were taken from 18-wk-old eNOS partial null heterozygotes and eNOS null mice and submitted to SDS-PAGE. Western blotting was performed for mitochondrial ETC subunits representing complexes I–V as follows: I, complex I subunit NDUFA9; II, complex II subunit succinate dehydrogenase complex, subunit A (SDHA); III, complex III core protein; IV, complex IV subunit 4; V, complex V α-subunit. A and B: representative blots (A) and compiled data (B) from Western analysis. Graph shown is the mean ± SE from 6–7 animals/group. *P < 0.05, **P < 0.01, and ***P < 0.001.
Acute NOS inhibition results in decreased mitochondrial ETC subunit content.
Genetic models with targeted deletion of eNOS have the limitation that compensation may occur in redundant or complimentary pathways (45, 15). To assess the effect of total NOS inhibition, we treated Sprague Dawley rats with 50 mg·kg−1·day−1 l-NAME, a nonselective NOS inhibitor, for 3 days. Complex I subunit NDUFA9 showed a 42% reduction (P = 0.0216), complex II subunit SDHA showed an 18% reduction [not significant (NS)], complex III core protein showed a 45% reduction (P = 0.0059), complex IV subunit 4 showed a 51% reduction (P = 0.0126), and complex V α-subunit showed a 17% reduction (NS) (Fig. 2A). We further observed a 29% reduction in VDAC1 protein content (P = 0.0023), a proxy marker of cellular mitochondrial content (Fig. 2B).
Fig. 2.
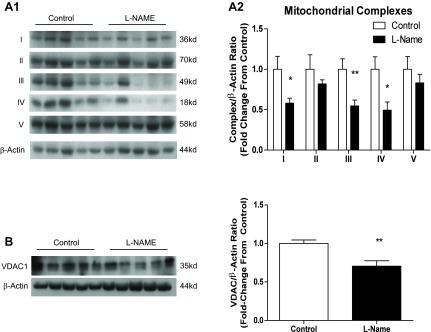
NG-nitro-l-arginine methyl ester (l-NAME) injection decreases mitochondrial content. Twelve-week-old male Sprague Dawley rats were injected with 50 mg·kg−1·day−1 l-NAME or vehicle (PBS) for 3 days. Aortas were harvested 24 h following the last injection and subjected to SDS-PAGE. Western blotting was performed for mitochondrial ETC subunits representing complexes I–V. A: representative blots (A1) and compiled data (A2) from Western analysis of ETC complexes. B: representative blots and compiled data for voltage-dependent acnion channel 1 (VDAC1). Graphs shown are means ± SE from 10 animals/group. *P < 0.05 and **P < 0.01.
NOS inhibition decreases signaling to mitochondrial biogenesis.
Based upon the established link between NOS and signaling to CREB and PGC-1α, we next explored the effect of l-NAME on signaling to those targets. We observed a 33% reduction in CREB content (P = 0.0129) and a 38% reduction in CREB activity (P = 0.0054), as measured by phosphorylation at Ser133, in response to NOS inhibition (Fig. 3A). We observed a 54% reduction in PGC-1α content (P < 0.0001, Fig. 3B) but did not observe any change in PGC-1α's deacetylating regulator SIRT1 (Fig. 3C).
Fig. 3.
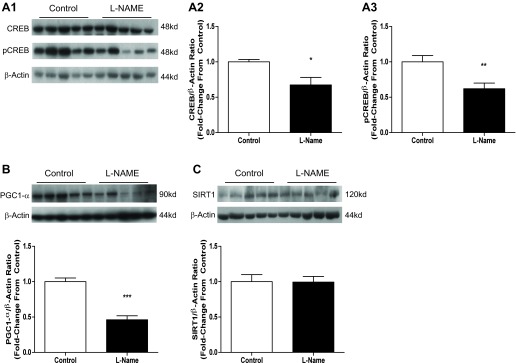
The effect of 3-day l-NAME injection on mitochondrial biogenesis signaling. Twelve-week-old male Sprague Dawley rats were injected with 50 mg·kg−1·day−1 l-NAME or vehicle (PBS) for 3 days. Aortas were harvested 24 h following the last injection and subjected to SDS-PAGE. Western blotting was done for cAMP response element-binding protein (CREB) content and activity, peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) content, and silent mating type information regulation 2 homolog 1 (SIRT1) content. A: representative blots for CREB and pCREB. B and C: compiled data from Western analysis of CREB and pCREB. D: representative blots and compiled data from Western analysis of PGC-1α. E: representative blots and compiled data from Western analysis of SIRT1. Graphs shown are means ± SE from 10 animals/group. *P < 0.05, **P < 0.01, and ***P < 0.001.
eNOS manipulation and NOS inhibition perturb mitochondrial ROS handling.
Uncoupling electron transport from ATP generation via UCP3 is an important mitochondrial homeostatic mechanism used to prevent/counter ROS generation (51), whereas superoxide dismutases convert superoxide radicals to hydrogen peroxide. MnSOD is the enzyme responsible for such conversion in mitochondria (62). We assessed the effect of eNOS deletion on MnSOD content (data not shown) and observed a 24 ± 11.63% (SE) decrease (P = 0.0829). NOS inhibition by l-NAME resulted in a 58 ± 13.74% decrease in MnSOD content (P = 0.0006) and a 40 ± 22.12% reduction in SIRT3 (P = 0.0874), a mitochondrial NAD-dependent deacetylase with MnSOD regulatory function. A 110 ± 28.05% increase (P = 0.0011) in UCP3 was also observed in response to NOS inhibition.
l-NAME treatment decreases mitochondrial fusion and increases mitochondrial fission.
Changes in fusion/fission profiles were observed following 3-day NOS inhibition. Fusion markers OPA1, mitofusin 1, and mitofusin 2 were significantly decreased in rat aortas exposed to l-NAME treatment [OPA1: 25% decrease (P = 0.0011, Fig. 4C), mitofusin 1: 47% decrease (P = 0.0002, Fig. 4A), mitofusin 2: 41% decrease (P = 0.0027, Fig. 4B)]. Fis1 content was increased 41% (P = 0.043, Fig. 4D), and DRP-1 content was increased by 20% (P = 0.0023, Fig. 4E).
Fig. 4.
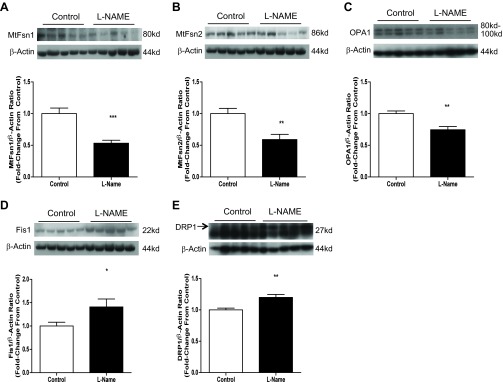
Three-day l-NAME injection decreases markers of mitochondrial fusion and increases markers of mitochondrial fission. Twelve-week-old male Sprague Dawley rats were injected with 50 mg·kg−1·day−1 l-NAME or vehicle (PBS) for 3 days. Aortas were harvested 24 h following the last injection and subjected to SDS-PAGE. Western blotting was done for mitofusin 1, mitofusin 2, optic atrophy 1 (OPA1), fission 1 (Fis1), and dynamin-related protein 1 (DRP-1). A: representative blots and compiled data for mitofusin 1. B: representative blots and compiled data for mitofusin 2. C: representative blots and compiled data for OPA1. D: representative blots and compiled data for Fis1. E: representative blots and compiled data for DRP-1. Graphs shown are means ± SE from 10 animals/group. *P < 0.05, **P < 0.01, and ***P < 0.001
Acute NOS inhibition has minimal effect on autophagy.
Following the decreased fusion and increased fission we observed in response to l-NAME treatment, we hypothesized that an increase in autophagy would take place. We observed a minimally perturbed autophagy profile (Fig. 5). Only BNIP3 decreased 16% (P = 0.0485), whereas Beclin1, p62, and the LC3B2/1 ratio were unchanged with l-NAME treatment.
Fig. 5.
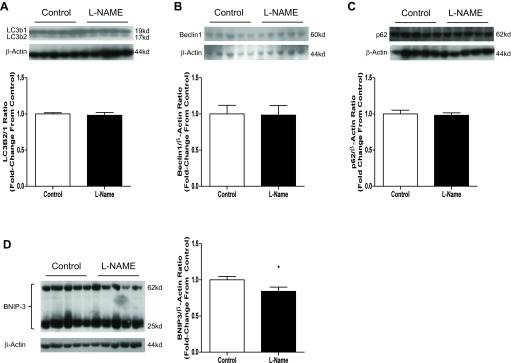
Three-day l-NAME injection has minimal effect on autophagy. Twelve-week-old male Sprague Dawley rats were injected with 50 mg·kg−1·day−1 l-NAME or vehicle (PBS) for 3 days. Aortas were harvested 24 h following the last injection and subjected to SDS-PAGE. Western blotting was done for LC3b, Beclin1, p62, and BCL2/adenovirus E1B 19-kDa protein-interacting protein 3 (BNIP3). A: representative blots of LC3b. LC3b is expressed as a ratio of the 17-kDa fragment to the 19-kDa fragment. B: representative blots and compiled data for Beclin1. C: representative blots and compiled data for p62. D: representative blots and compiled data for BNIP3. Graphs shown are means ± SE from 10 animals/group. *P < 0.05.
l-NAME reverses mitochondrial dynamics in exercise.
eNOS deletion was shown to decrease exercise capacity in mice, secondary to impaired signaling to mitochondrial biogenesis in skeletal muscle (22). We assessed the effect of NOS inhibition on the mitochondrial response to exercise in the vasculature. Figure 6 shows mild elevation of mitochondrial ETC proteins and VDAC1 in response to exercise in control animals [complex I subunit NDUFA9: 25% (NS), complex II subunit SDHA: 41% (NS), complex III core protein: 30% (NS), complex IV subunit 4: 58% (P = 0.0394), complex V α-subunit: NS (Fig. 6A1)]. This response is absent in l-NAME-treated rats, with 6–35% decreases observed in subunits from complexes I-IV (NS, Fig. 6B1). Control animals exhibited an increased fusion profile in response to exercise: mitofusin 1: 25% (NS), mitofusin 2: 87% (P = 0.040, Fig. 6A2), and OPA1: 40% (NS). This adaptation is also lost with l-NAME treatment, with no significant changes in fusion markers observed (mitofusin 2, Fig. 6B2). Last, control animals show a decrease in fission markers in response to exercise: FIS1: 29% (P = 0.0204, Fig. 6A3) and DRP-1: 13% (NS). No changes were observed in l-NAME-treated rats in response to exercise (Fig. 6B3).
Fig. 6.
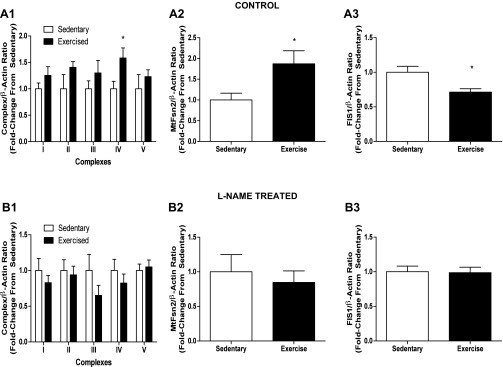
l-NAME treatment perturbs the mitochondrial adaptive response to exercise. Twelve-week-old male Sprague Dawley rats were injected with 50 mg·kg−1·day−1 l-NAME or vehicle (PBS) for 8 days. Cohorts were separated into either exercised or sedentary groups. Exercised animals were run on the treadmill (0% grade) at 15 m/min for 30 min on days 1 and 2 and for 45 min on days 3–8. Injections began on the 1st day of exercise, and injections and exercise continued for 8 days. After the last exercise bout and injection (24 h), animals were killed, and aorta tissue was removed and subjected to SDS-PAGE. A1: compiled data from Western blotting performed for mitochondrial ETC subunits representing complexes I–V. A2: compiled data for mitofusin 2. A3: compiled data for Fis1. B1: compiled data from Western analysis for ETC complex subunits from animals receiving l-NAME injection. B2: compiled data for mitofusin 2. B3: compiled data for Fis1. Graphs shown are means ± SE from 5 animals/group. *P < 0.05.
DISCUSSION
Endothelial dysfunction increases the risk of CVD and all-cause mortality (48, 66). A primary contributor to endothelial dysfunction is eNOS dysfunction. In this manuscript, we demonstrate that eNOS/NOS are potent regulators of basal and exercise-adapted mitochondrial content. We further demonstrate that mitochondrial biogenesis, fission, and fusion are all affected by eNOS deletion or NOS inhibition. These data may have important functional implications for aberrant vasomotion observed in CVD secondary to impaired eNOS/NOS signaling. Vascular eNOS dysfunction is present across the spectrum of vascular disease, including diabetes, hypertension, dyslipidemia, and cigarette smoking, all processes that lead to atherosclerosis (48). Vascular mitochondria are crucial for maintenance of REDOX state, calcium handling, and vascular contractility (9, 54, 40). eNOS function has been shown to be crucial for mitochondrial biogenesis in vivo in many tissues (35). Shear stress, such as that experienced during pulsatile blood flow, regulates vascular tone through induction of eNOS downstream of SIRT and AMPK (7). SIRT-mediated induction of eNOS induces mitochondrial biogenesis in endothelial cells. Insulin acts on eNOS, both through increasing transcription and through posttranslational modification via Akt (67, 31); this regulation is disrupted in insulin resistance and diabetes (31, 20, 38). Vascular ROS has been shown to uncouple eNOS through conversion of tetrahydrobiopterin (BH4) to dihydrobiopterin (BH2) and decreasing the BH4-to-BH2 ratio, leading to aberrant ROS production by eNOS itself (54). Indeed, eNOS-generated ROS is increased with diabetes, hyperlipidemia, hypertension, and tobacco exposure (8, 21, 14).
Processes that contribute to vascular mitochondrial mass and quality, including biogenesis, fission/fusion, and autophagy, have been little studied in the vasculature. We previously reported that vascular mitochondria respond to 8 days of exercise by increasing ETC content (18). This adaptation is not seen in the context of hypertension, metabolic syndrome, and diabetes (18). The role of NO in vascular mitochondrial regulation is little understood. We tested the hypothesis that NOS dysfunction contributes to failed mitochondrial adaptation in the aorta. The data presented here confirm that eNOS and NO signal to PGC-1α and CREB and induce mitochondrial biogenesis in the vasculature. Additionally, we report the novel finding that NO is important for modulation of vascular mitochondrial dynamics and demonstrate that NOS inhibition impairs mitochondrial adaptation to exercise.
To assess the influence of NOS on vascular mitochondrial dynamics, we employed two models of NOS dysfunction. Previous work by Lee-Young et al. in mouse skeletal muscle showed that deletion of eNOS led to decreased mitochondrial ETC content (23). The data shown here suggest that vascular eNOS is critical for mitochondrial biogenesis and also for appropriate mitochondrial turnover. As expected from the work done by Nisoli and colleagues, we observed a decrease in mitochondrial ETC complex content in eNOS−/− mice, a model of chronic endothelial dysfunction. Using 3-day l-NAME treatment allowed us to identify the impact of NOS on vascular mitochondrial content: significant decreases in subunit content from respiratory complexes I, III, and IV. Our work examined the impact of eNOS-specific and NOS inhibition on whole aorta. In the eNOS null mice, the presumed primary site of eNOS (and therefore target of eNOS deletion) is the endothelium. Our data allow us to say that chronic loss of eNOS leads to decreased aortic mitochondrial content. The mitochondrial proteins were measured in aortic lysates which are predominantly smooth muscle cells in origin. We are unable to comment on the contributions of each cell type. Establishment of NO as a regulator of vascular mitochondrial mass and quality is an important step in understanding the cellular cross talk that regulates physiological vasomotion.
In humans, aerobic exercise has been shown to decrease arterial stiffness and increases endothelial-mediated vasodilatation in people with diabetes (2, 26, 24). Studies by Young-Lee and colleagues in mouse skeletal muscle revealed decreased exercise capacity in eNOS−/− mice, coincident with impaired signaling through AMPK in response to exercise (22). Using l-NAME treatment in conjunction with exercise, we observe findings that recapitulate those observed in metabolic syndrome rats: failed mitochondrial protein induction with exercise in l-NAME-treated rats. Of note, both the control and the l-NAME-treated rats completed the same exercise protocol. Decreased mitochondrial protein content with l-NAME is consistent with reports that exercise-induced mitochondrial biogenesis requires NOS and can be blocked by l-NAME (30). Our data, however, contrast with reports by Wadley and McConell in skeletal muscle, where l-NAME interfered with baseline, but not exercise-induced, mitochondrial biogenesis (57). Further work by Wadley and colleagues indicates that neither nNOS nor eNOS are essential for mitochondrial biogenesis in skeletal muscle (56). Taken together, these skeletal muscle studies support a role for NOS in skeletal muscle mitochondrial biogenesis that can be circumvented by other pathways activated by exercise. The data in this manuscript point to a central role for NO in adaptive mitochondrial biogenesis in the vasculature but do not allow us to elucidate the role of nNOS or eNOS individually.
Mitochondrial dysfunction, including excess mitochondrial ROS generation, mutations, and impaired calcium handling, has been observed in aging, diabetes mellitus, tobacco exposure, and hyperlipidemia (42, 9, 53, 40). In addition, diabetes has a consistent effect to increase mitochondrial ROS generation, thereby contributing to microvascular complications (5). Increased production of mitochondrial ROS has been linked to vascular stiffness, as well as events in atherogenesis such as smooth muscle cell migration (59). Vascular-specific MnSOD overexpression is protective against endothelial dysfunction in hyperlipidemia, diabetes, and hypertension (65). Additional published work suggested that loss of MnSOD function resulted in mitochondrial dysfunction (4, 63) and vascular stiffness (50). Our data in clinically relevant models demonstrate loss of MnSOD in both eNOS−/− mice and with l-NAME treatment, suggesting a tight relationship between eNOS, MnSOD, and mitochondrial dysfunction. Our data also show that UCP3 is induced during short-term NOS inhibition. This is consistent with an acute mitochondrial stress in the 3-day l-NAME treatment, contributing to induction of UCP3 induction that will uncouple the oxidative phosphorylation pathway and decrease ROS generation (44). UCP3 may serve a protective role in preventing ROS generation in the face of failed biogenesis and defunct mitochondrial turnover. It is possible that mitochondrial dysfunction is secondary to loss of mitochondrial antioxidant defense or that loss of antioxidant defense is secondary to persistent stress placed on the antioxidant system by excess mitochondrial oxidant stress. Our study design does not permit clarification of whether it is loss of antioxidant defense or abnormal mitochondrial function that is the primary consequence of targeted deletion of eNOS.
Vascular mitochondria regulate many processes beyond energetics, including vascular calcium flux (29), vasomotion (47, 49), and smooth muscle cell quiescence; disruption of mitochondrial dynamic processes is linked to vascular dysfunction (55, 36). Dysregulation of mitochondrial dynamics has been observed in the liver and cardiac muscle in diabetes-mediated stress (64), and hyperglycemia-mediated increase in fission has also been implicated in microvascular renal disease secondary to diabetes (60). Mitochondrial fusion has been shown to maintain polarity of the mitochondrial membrane and calcium regulation (1). Organelle mixing facilitated by mitofusin 2 enables specific modulation and buffering of calcium flux through numerous calcium ion channels (6, 28, 29). Of additional pathophysiological interest, increased fusion and decreased fission are protective against smooth muscle cell proliferation, whereas fission is essential for smooth muscle cell proliferation in intact resistance arteries (6). Interestingly, a feedback loop between mitochondrial polarity and eNOS function has recently been established in the cerebral vasculature (16). Our data are consistent with a bidirectional relationship between eNOS and mitochondrial dynamics.
Previous work by our lab showed an increase in mitochondrial ETC content in response to exercise, whereas a decrease was observed in diabetes mellitus (18). The mitochondrial dynamics involved in the observed mitochondrial dysfunction, if any, remained to be explored. Data in this study show that the homeostatic balance of mitochondrial biogenesis, fission, and fusion are perturbed in the context of reduced NOS function, potentially resulting in an ultimate decline in mitochondrial quantity and quality. At baseline, we observe the fission/fusion balance tipped in favor of fission, and, in the absence of increased autophagy, low-quality, fragmented mitochondria would be expected. In response to exercise, data shown here suggest that NOS dysfunction leads to decreased fusion and increased fission. Fusion has been described as protective of mitochondria in high protein kinase A states, including starvation, preventing degradation by autophagy (13, 37). The apparent shift away from fusion in the context of NOS inhibition may prime high-quality mitochondria for degradation.
In this manuscript, we present novel data in eNOS−/− mice demonstrating the effect of chronic eNOS dysfunction on vascular mitochondria, specifically the reduction of oxidative phosphorylation enzyme content and mitochondrial antioxidant defense. This model represents the prevalent endothelial dysfunction present in many vascular disease states, and our findings in the vasculature are consistent with those seen in skeletal muscle. Additionally, the observed impaired vascular mitochondrial response to exercise in l-NAME-treated rats is consistent with previous reports in the mouse model, which showed failed mitochondrial adaptation to exercise challenge in the skeletal muscle. The eNOS null model has limitations in that the rodent is able to compensate for genetic loss of eNOS with many intracellular and vascular homeostatic mechanisms. To complement this model of chronic endothelial dysfunction, we employed l-NAME, which enabled us to study the dynamic impact of NOS inhibition in a short time period. Although 3 days of l-NAME has its own limitations (this treatment may induce hypertension, which could affect mitochondria), short-term NOS inhibition in this study points to a role for NO in biogenesis and also in mitochondrial dynamics. In summary, acute NOS inhibition by l-NAME results in reduced vascular mitochondrial biogenesis, increased fission in the context of decreased fusion, and a maladaptive autophagy profile. These observations may have important biological relevance to the new field of vascular mitochondrial regulation and the significant sequelae of endothelial dysfunction.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant 1R25-HL-103286-02.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: M.W.M., L.A.K., and L.F.O.-F. performed experiments; M.W.M. and L.F.O.-F. analyzed data; M.W.M., L.A.K., P.A.W., and J.E.B.R. interpreted results of experiments; M.W.M. prepared figures; M.W.M., L.A.K., and J.E.B.R. drafted manuscript; M.W.M., L.A.K., A.C.K., V.B., P.A.W., and J.E.B.R. edited and revised manuscript; M.W.M., L.A.K., A.C.K., V.B., P.A.W., and J.E.B.R. approved final version of manuscript; L.A.K., V.B., P.A.W., and J.E.B.R. conception and design of research.
ACKNOWLEDGMENTS
We thank Sarah Nelson and Zachary Baud for technical assistance with this project.
REFERENCES
- 1. Abhikit S, Bhaskaran R, Narayanasamy A, Chakroborty A, Manickam N, Dixit M, Mohan V, Blasubramanyam M. Hyperinsulinemia-induced vascular smooth muscle cell migration and proliferation is mediated by converging mechanisms of mitochondrial dysfunction and oxidative stress. Mol Cell Biochem 373: 95–105, 2013 [DOI] [PubMed] [Google Scholar]
- 2. Aizawa K, Shoemaker JK, Overend TJ, Petrella RJ. Effects of lifestyle modification on central artery stiffness in metabolic syndrome subjects with pre-hypertension and/or prediabetes. Diabetes Res Clin Pract 83: 249–256, 2009 [DOI] [PubMed] [Google Scholar]
- 3. Anne Stetler R, Leak RK, Gao Y, Chen J. The dynamics of the mitochondrial organelle as a potential therapeutic target. J Cereb Blood Flow Metab 2: 125–137, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, Reuf J, Horaist C, Lebovitz R, Hunter GC, McIntyre K, Runge MS. Mitochondrial integrity and function in atherogenesis. Circulation 106: 544–549, 2002 [DOI] [PubMed] [Google Scholar]
- 5. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 414: 813–820, 2001 [DOI] [PubMed] [Google Scholar]
- 6. Chalmers S, Saunter C, Wilson C, Coats P, Girkin JM, McCarron JG. Mitochondrial motility and vascular smooth muscle proliferation. Arterioscler Thromb Vasc Biol 32: 3000–3011, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Z, Peng IC, Cui X, Li YS, Chien S, Shyy JY. Shear stress, SIRT1, and vascular homeostasis. Proc Natl Acad Sci USA 107: 10268–10273, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cosentino F, Hishikawa K, Katusic ZS, Luscher TF. High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation 96: 25–28, 1997 [DOI] [PubMed] [Google Scholar]
- 9. Crabtree MJ, Smith CL, Lam G, Goligorsky MS, Gross SS. 8-Dihydrobiopterin in endothelial cells determines glucose-elicited changes in NO vs. superoxide production by eNOS. Am J Physiol Heart Circ Physiol 294: H1530–H1540, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dorn GW., 2nd Mitochondrial dynamics in heart disease. Biochim Biophys Acta 1833: 233–241, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Evora PR, Evora PM, Celotto AC, Rodrigues AJ, Joviliano EE. Cardiovascular therapeutics targets on the NO-sGC-cGMP signaling pathway: a critical overview. Curr Drug Targets 13: 1207–14, 2012 [DOI] [PubMed] [Google Scholar]
- 12. Galloway CA, Lee H, Nejjar S, Jhun BS, Yu T, Hsu W, Yoon Y. Transgenic control of mitochondrial fission induces mitochondrial uncoupling and relieves diabetic oxidative stress. Diabetes 61: 2093–2104, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nature Cell Biology 13: 589–598, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heitzer T, Yla-Herttuala S, Luoma J, Kurz S, Munzel T, Just H, Olschewski M, Drexler H. Cigarette smoking potentiates endothelial dysfunction of forearm resistance vessels in patients with hypercholesterolemia: role of oxidized LDL. Circulation 93: 1346–1353, 1996 [DOI] [PubMed] [Google Scholar]
- 15. Huang A, Sun D, Shesely EG, Levee EM, Koller A, Kaley G. Neuronal NOS-dependent dilation to flow in coronary arteries of male eNOS-KO mice. Am J Physiol Heart Circ Physiol 282: H429–H436, 2002 [DOI] [PubMed] [Google Scholar]
- 16. Katakam PVG, Wappler E, Katz P, Rutkai I, Institoris A, Domoki F, Gáspár T, Grovenburg SM, Snipes JA, Busija DW. Depolarization of mitochondria in endothelial cells promotes cerebral vascular vasodilation by activation of nitric oxide synthase. Arterioscler Thromb Vasc Biol 33: 752–759, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 462: 245–253, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Knaub LA, McCune S, Chicco AJ, Miller M, Moore RL, Birdsey N, Lloyd MI, Villarreal J, Keller AC, Watson PA, Reusch JE. Impaired response to exercise intervention in the vasculature in metabolic syndrome. Diab Vasc Dis Res 10: 222–238, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koga Y, Povalko N, Nishioka J, Katayama K, Shuichi Y, Matsuishi T. Molecular pathology of MELAS and l-arginine effects. Biochim Biophys Acta 1820: 608–614, 2012 [DOI] [PubMed] [Google Scholar]
- 20. Kuboki K, Jiang ZY, Takahara N, Ha SW, Igarashi M, Yamauchi T, Feener EP, Herbert TP, Rhodes CJ, King GL. Regulation of endothelial constitutive nitric oxide synthase gene expression in endothelial cells and in vivo: a specific vascular action of insulin. Circulation 101: 676–681, 2000 [DOI] [PubMed] [Google Scholar]
- 21. Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111: 1201–1209, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee-Young RS, Ayala JE, Fueger PT, Mayes WH, Kang L, Wasserman DH. Obesity impairs skeletal muscle AMPK signaling during exercise: role of AMPKα2 in the regulation of exercise capacity in vivo. Int J Obesity 35: 982–989, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee-Young RS, Ayala JE, Hunley CF, James FD, Bracy DP, Kang L, Wasserman DH. Endothelial nitric oxide synthase is central to skeletal muscle metabolic regulation and enzymatic signaling during exercise in vivo. Am J Physiol Regul Integr Comp Physiol 298: R1399–R1408, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Loimaala A, Groundstroem K, Rinne M, Nenonen A, Huhtala H, Parkkari J, Vuori I. Effect of long-term endurance and strength training on metabolic control and arterial elasticity in patients with type 2 diabetes mellitus. Am J Cardiol 103: 972–977, 2009 [DOI] [PubMed] [Google Scholar]
- 25. Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res 100: 460–473, 2007 [DOI] [PubMed] [Google Scholar]
- 26. Madden KM, Lockhart C, Cuff D, Potter TF, Meneilly GS. Short-term aerobic exercise reduces arterial stiffness in older adults with type 2 diabetes, hypertension, and hypercholesterolemia. Diabetes Care 32: 1531–1535, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, Chen Y, Morrow E, Weir EK, Rehman J, Archer SL. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 110: 1484–1497, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McCarron JG, Olson ML, Chalmers S. Mitochondrial regulation of cytosolic Ca(2)(+) signals in smooth muscle. Pflugers Arch 464: 51–62, 2012 [DOI] [PubMed] [Google Scholar]
- 29. McCarron JG, Olson ML, Wilson C, Sandison ME, Chalmers S. Examining the role of mitochondria in Ca(2+) signaling in native vascular smooth muscle. Microcirculation In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McConell GK, Ng GP, Phillips M, Ruan Z, Macaulay SL, Wadley GD. Central role of nitric oxide synthase in AICAR and caffeine-induced mitochondrial biogenesis in L6 myocytes. J Appl Physiol 108: 589–595, 2010 [DOI] [PubMed] [Google Scholar]
- 31. Montagnani M, Chen H, Barr VA, Quon MJ. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser(1179). J Biol Chem 276: 30392–30398, 2001 [DOI] [PubMed] [Google Scholar]
- 32. Nemes A, Geleijnse ML, Sluiter W, Vydt TC, Soliman OI, van Dalen BM, Vletter WB, ten Cate FJ, Smeets HJ, de Coo RF. Aortic distensibility alterations in adults with m.3243A>G MELAS gene mutation. Swiss Med Wkly 139: 117–120, 2009 [DOI] [PubMed] [Google Scholar]
- 33. Nilsson MI, Greene NP, Dobson JP, Wiggs MP, Gasier HG, Macias BR, Shimkus KL, Fluckey JD. Insulin resistance syndrome blunts the mitochondrial anabolic response following resistance exercise. Am J Physiol Endocrinol Metab 299: E466–E474, 2010 [DOI] [PubMed] [Google Scholar]
- 34. Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299: 896–899, 2003 [DOI] [PubMed] [Google Scholar]
- 35. Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 310: 314–317, 2005 [DOI] [PubMed] [Google Scholar]
- 36. Ong S, Hausenloy DJ. Mitochondrial morphology and cardiovascular disease. Cardiovasc Res 88: 16–29, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA 108: 10190–10195, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roberts CK, Vaziri ND, Wang XQ, Barnard RJ. Enhanced NO inactivation and hypertension induced by a high-fat, refined-carbohydrate diet. Hypertension 36: 423–429, 2000 [DOI] [PubMed] [Google Scholar]
- 39. Romanello V, Sandri M. Mitochondrial biogenesis and fragmentation as regulators of muscle protein degradation. Curr Hypertens Rep 12: 433–439, 2010 [DOI] [PubMed] [Google Scholar]
- 40. Sansbury BE, Jones SP, Riggs DW, Darley-Usmar VM, Hill BG. Bioenergetic function in cardiovascular cells: the importance of the reserve capacity and its biological regulation. Chem Biol Interact 191: 288–295, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sesaki H, Jensen RE. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J Cell Biol 147: 699–706, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shen GX. Mitochondrial dysfunction, oxidative stress and diabetic cardiovascular disorders. Cardiovasc Hematol Disord Drug Targets 12: 106–112, 2012 [DOI] [PubMed] [Google Scholar]
- 43. Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, Duess MA, Levit A, Kim B, Hartman ML, Joseph L, Shirihai OS, Vita JA. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 124: 444–453, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Skulachev VP. Uncoupling: new approaches to an old problem of bioenergetics. Biochim Biophys Acta Bioenerg 1363: 100–124, 1998 [DOI] [PubMed] [Google Scholar]
- 45. Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, Kandel ER. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell 87: 1015–1023, 1996 [DOI] [PubMed] [Google Scholar]
- 46. Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell 20: 3525–3532, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sward K, Dreja K, Lindqvist A, Persson E, Hellstrand P. Influence of mitochondrial inhibition on global and local [Ca(2+)](I) in rat tail artery. Circ Res 90: 792–799, 2002 [DOI] [PubMed] [Google Scholar]
- 48. Tabit CE, Chung WB, Hamburg NM, Vita JA. Endothelial dysfunction in diabetes mellitus: molecular mechanisms and clinical implications. Rev Endocr Metab Disord 11: 61–74, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Taggart MJ, Wray S. Hypoxia and smooth muscle function: key regulatory events during metabolic stress. J Physiol 509: 315–325, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. t Hoen PA, Van der Lans CA, Van Eck M, Bijsterbosch MK, Van Berkel TJ, Twisk J. Aorta of ApoE-deficient mice responds to atherogenic stimuli by a prelesional increase and subsequent decrease in the expression of antioxidant enzymes. Circ Res 93: 262–269, 2003 [DOI] [PubMed] [Google Scholar]
- 51. Toime LJ, Brand MD. Uncoupling protein-3 lowers reactive oxygen species production in isolated mitochondria. Free Radic Biol Med 49: 606–611, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27: 433–446, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ungvari Z, Sonntag WE, Csiszar A. Mitochondria and aging in the vascular system. J Mol Med 88: 1021–1027, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci USA 95: 9220–9225, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Viitanen M, Sundstrom E, Baumann M, Poyhonen M, Tikka S, Benbahani H. Experimental studies of mitochondrial function in CADASIL vascular smooth muscle cells. Exp Cell Res 319: 134–143, 2013 [DOI] [PubMed] [Google Scholar]
- 56. Wadley GD, Choate J, McConell GK. NOS isoform-specific regulation of basal but not exercise-induced mitochondrial biogenesis in mouse skeletal muscle. J Physiol 585: 253–262, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wadley GD, McConell GK. Effect of nitric oxide synthase inhibition on mitochondrial biogenesis in rat skeletal muscle. J Appl Physiol 102: 314–320, 2007 [DOI] [PubMed] [Google Scholar]
- 58. Wang H, Liu J, Wu L. Methylglyoxal-induced mitochondrial dysfunction in vascular smooth muscle cells. Biochem Pharmacol 77: 1709–1716, 2009 [DOI] [PubMed] [Google Scholar]
- 59. Wang JN, Shi N, Chen SY. Manganese superoxide dismutase inhibits neointima formation through attenuation of migration and proliferation of vascular smooth muscle cells. Free Radic Biol Med 52: 173–181, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, Chang BH, Schumacker PT, Danesh FR. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab 15: 186–200, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Weber TA, Reichert AS. Impaired quality control of mitochondria: aging from a new perspective. Exp Gerontol 45: 503–511, 2010 [DOI] [PubMed] [Google Scholar]
- 62. Weisiger RA, Fridovich I. Superoxide dismutase. Organelle specificity. J Biol Chem 248: 3582–3592, 1973 [PubMed] [Google Scholar]
- 63. Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ, Richardson A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem 273: 28510–28515, 1998 [DOI] [PubMed] [Google Scholar]
- 64. Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA 103: 2653–2658, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zanetti M, Sato J, Jost CJ, Gloviczki P, Katusic ZS, O'Brien T. Gene transfer of manganese superoxide dismutase reverses vascular dysfunction in the absence but not in the presence of atherosclerotic plaque. Human Gene Therapy 12: 1407–1416, 2001 [DOI] [PubMed] [Google Scholar]
- 66. Zeiher AM, Drexler H, Wollschlager H, Just H. Modulation of coronary vasomotor tone in humans. Progressive endothelial dysfunction with different early stages of coronary atherosclerosis. Circulation 83: 391–401, 1991 [DOI] [PubMed] [Google Scholar]
- 67. Zeng G, Nystrom FH, Ravichandran LV, Cong L, Kirby M, Mostowski H, Quon MJ. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation 101: 1539–1545, 2000 [DOI] [PubMed] [Google Scholar]
- 68. Zhang D, Ma C, Li S, Ran Y, Chen J, Lu P, Shi S, Zhu D. Effect of Mitofusin 2 on smooth muscle cells proliferation in hypoxic pulmonary hypertension. Microvasc Res 84: 286–296, 2012 [DOI] [PubMed] [Google Scholar]