

Abstract
BACKGROUND & AIMS
Hepatocellular carcinoma (HCC) is a heterogeneous tumor that develops via activation of multiple pathways and molecular alterations. It has been a challenge to identify molecular classes of HCC and design treatment strategies for each specific subtype. MicroRNAs (miRNAs) are involved in HCC pathogenesis and their expression profiles have been used to classify cancers. We analyzed miRNA expression in human HCC samples to identify molecular subclasses and oncogenic miRNAs.
METHODS
We performed miRNA profiling of 89 HCC samples using a ligation-mediated amplification method. Subclasses were identified by unsupervised clustering analysis. We identified molecular features specific for each subclass using expression pattern (Affymetrix U133 2.0), DNA change (Affymetrix STY Mapping Array), mutation (CTNNB1), and immunohistochemical (phosphor[p]-Akt, p-IGF-IR, p-S6, p-EGFR, β-catenin) analyses. The roles of selected miRNAs were investigated in cell lines and in an orthotopic model of HCC.
RESULTS
We identified 3 main clusters of HCCs: the Wnt (32 of 89; 36%), interferon-related (29 of 89; 33%), and proliferation (28 of 89; 31%) subclasses. A subset of patients with tumors in the proliferation subclass (8 of 89; 9%) overexpressed a family of poorly characterized miRNAs from chr19q13.42. Expression of miR-517a and miR-520c (from ch19q13.42) increased proliferation, migration, and invasion of HCC cells in vitro. MiR-517a promoted tumorigenesis and metastatic dissemination in vivo.
CONCLUSIONS
We propose miRNA-based classification of 3 subclasses of HCC. Among the proliferation class, miR-517a is an oncogenic miRNA that promotes tumor progression. There is rationale for developing therapies that miRNA 517 for patients with HCC.
Keywords: Liver Cancer, Tumor Profiling, OncomiR, On-cogenesis
Hepatocellular carcinoma (HCC) is the third leading cause of cancer-related death worldwide and the principal cause of mortality among cirrhotic patients.1 Unlike most solid tumors, HCC mortality has increased significantly in the 1990 –2005 period,2 most likely owing to the large number of patients with long-standing hepatitis.3 Only one third of patients are eligible for curative treatments (resection, liver transplantation, local ablation), and the multikinase inhibitor sorafenib represents the only approved systemic agent for advanced cases.4,5
From a molecular perspective, HCC is a highly heterogeneous tumor. Increasing evidence suggests that its complexity and clinical variability mainly depend on different molecular alterations that arise during the development and progression of this disease.6 – 8 Translational studies report aberrant activation of different cellular pathways, including IGF, Ras, Akt/mTOR, and Wnt signaling in HCC.9 –11 Hence, a major clinical challenge is to identify the predominant molecular mechanisms in distinct HCC molecular classes to select the best therapeutic option for each individual on the basis of the genomic background of their tumor.
MicroRNAs (miRNAs) are small noncoding RNAs that regulate gene expression and multiple cellular processes including cell differentiation, stem cell maintenance, and epithelial–mesenchymal transition.12,13 miRNA deregulation represents a common feature in cancer and can be caused by different mechanisms such as amplification/ deletion, chromosomal rearrangements, and epigenetic regulation.14 Depending on the genes targeted, miRNAs can act as tumor suppressors or oncogenes.15 Recent evidence shows abnormal expression of miRNAs in HCC, including up-regulation of miR-21, miR-221, miR-151, and down-regulation of Let-7, miR-29, and miR-122.16 –21 Of note, the expression levels of certain miRNAs (eg, miR-26a) correlate with poor prognosis in HCC.22 Importantly, miRNA profiling represents a powerful tool to accurately differentiate cancers from normal tissue and to classify cancer subtypes.23–26 Particularly in HCC, miRNA profiling will provide new insights into the molecular complexity of this disease and reveal novel therapeutic targets.
Herein, we report a miRNA-based molecular classification of hepatitis C virus (HCV)-related HCC and the discovery of a subset of HCCs overexpressing a family of miRNAs on chr19q13.41. Specifically, we show that miR-517a on the 19q13.41 locus promotes cell proliferation and migration in vitro, and induces tumorigenesis in vivo, pointing out its role as a novel oncomiR in HCC.
Materials and Methods
Human Tissue Samples
Human samples were obtained from the 3 hospitals of the HCC Genomic Consortium: Mount Sinai Clinical Hospital (New York, NY), Istituto Nazionale dei Tumori (Milan, Italy), and Hospital Clinic (Barcelona, Spain). The training set included 89 fresh-frozen HCC samples collected from patients who underwent surgical resection (n = 79) or liver transplantation (n = 10), which previously were analyzed for messenger RNA (mRNA) and genomic copy number profiling.27 Formalin-fixed paraffin-embedded tissues of 165 HCCs (validation set) from patients with different etiologies (HCV, hepatitis B virus [HBV], alcohol, and others) were used to validate the expression of selected miRNAs. Clinical characteristics of HCC patients who underwent liver resection (n = 79 and n = 161 for training and validation sets, respectively) are described in Supplementary Table 1. An informed consent form was signed by each patient and approval from the Institutional Review Board was obtained.
RNA Extraction and miRNA, mRNA Genomic Copy Number Profiling
Total RNA from frozen tissues and cell lines was extracted using Trizol Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. RNA from formalin-fixed paraffin-embedded sections was extracted as previously described.28 RNA was quantified using the NanoDrop ND-1000 spectrometer (NanoDrop, Wilmington, DE). Quality and integrity were measured with a bioanalyzer (Agilent, Palo Alto, CA). The expression levels of 358 miRNAs were determined using a bead-based amplification method as previously described.23,29
Genome-wide mRNA and copy number profiling of the training set were performed as previously described.27 mRNA profiling of the validation set was performed using whole-genome DNA-mediated annealing, selection, extension, and ligation assay.30,31 Human Gene 1.0 ST Array (Affymetrix) was used for mRNA profiling of Huh7 cells.
The dataset is available from NCBI’s Gene Expression Omnibus: accession number GSE20596.
Quantitative Real-Time Polymerase Chain Reaction
Complementary DNA was synthesized using miRNA-specific primers and TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, Austin, TX) according to the manufacturer’s instructions. miR-23 (constant expression among the samples of the training set) was used to normalize the expression levels of miR-517a, miR-520g, and miR-516-5p in the validation set. The ddCt method was used to calculate miRNA expression levels.32 A median relative expression level greater than 10 for all the 3 was used to define subclass C2 in the validation set. Ct values in samples with detectable levels of miR-517a are listed in Supplementary Table 2.
Cell Culture
Huh-7 cells (Riken Bioresource Center) were grown in Dulbecco’s modified Eagle medium supplemented with 10% heat-inactivated fetal bovine serum. SNU-449 cells (American Type Culture Collection) were cultured in RPMI supplemented with 10% heat-inactivated fetal bovine serum. Cells were maintained at 37°C in a 5% CO2 atmosphere.
Treatment With Demethylase and Histone Deacetylase Inhibitor
Huh7 cells were treated with the demethylating agent 5-aza-2′deoxycytidine 3 μmol/L and the histone deacetylase inhibitor 4-phenylbutyric acid 3 mmol/L (Sigma-Aldrich, St. Louis, MO). 5-Aza-2′deoxycytidine was removed after 24 hours whereas 4-phenylbutyric acid was administered daily for 6 days.
Methylation-Specific Polymerase Chain Reaction
Genomic DNA (n = 6 samples in subclass C2 were available) was treated with sodium bisulfite using the EpiTect Bisulfite Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Sequence primers are listed in Supplementary Table 3. Polymerase chain reaction (PCR) details are described in the Supplementary Materials and Methods.
Transfection With Synthetic miRNAs
Details about transfection with synthetic miRNAs are provided in the Supplementary Materials and Methods.
Proliferation, Migration, and Invasion Assays
Cells were labeled for 3 hours with 1 μCi/mL 3H-thymidine, fixed in 1 N hydrochloride acid, and lysed in 0.25 N NaOH 24, 48, and 72 hours after transfection. Thymidine incorporation was measured in a scintillation counter. Migration and invasion assays were performed using Transwell chambers (8-μmol/L pore size; BD Bioscience, Bedford, MA), as previously described.33,34
Wound-Healing Assay
Cells were grown to confluence and serum-starved for 24 hours. After pretreatment with mitomycin C for 3 hours to inhibit proliferation, cell monolayers were wounded with a sterile tip and maintained in serum-free medium for 24 hours. Images were taken after 0 and 24 hours after wounding on an inverted microscope (×100). Distance measurements between the 2 edges were performed using ImageJ software (National Institutes of Health, Bethesda, MD).
miRNA Stable Transfection
The generation of miR-517a stably transfected cells are described in the Supplementary Materials and Methods.
Orthotopic Model of HCC
Luciferase-tagged Huh7 cells expressed with miR-517a or control vector were injected into the left liver lobe of nude BALB/C mice (6 – 8 weeks of age). A detailed description of the method is reported in the Supplementary Materials and Methods. This study was performed in accordance with the guidelines and regulations from the Institutional Animal Care and Use Committee.
In Vivo Imaging System
Mice were anesthetized with isoflurane/oxygen, injected intraperitoneally with luciferin (150 mg/kg body weight), and placed on the imaging stage. Mice were imaged 1 day after surgery and once a week for 9 consecutive weeks using the In Vivo Imaging System (Xenogen Corp, Alameda, CA). Photons emitted from luciferase-expressing cells were quantified using Living Image software c (Xenogen Corp) and used as an indicator of tumor growth. At week 10 mice were killed and ex vivo imaging of collected organs was performed.
Histology
Liver tumors and secondary organs (spleen, stomach, lung, colon, and kidneys) from mice included in the study were excised, fixed in 4% paraformaldehyde solution, and embedded in paraffin. Slides (5 μm) were stained with H&E for histologic evaluation.
Bioinformatic Analysis
Unsupervised hierarchical clustering was performed using dChip software (http://biosun1.harvard.edu/complab/dchip/). A subclass mapping algorithm was used to evaluate similarities in the global gene-expression profile between the training and validation sets.35 The University of California Santa Cruz Genome Bioinformatics Genome Browser database (www.genome.ucsc.edu) was used to search for CpG islands in the area surrounding the chr19q13.41 locus. MethylPrimer software (http://www.urogene.org//methprimer) was used to design primers for methylation-specific PCR. Gene Set Enrichment Analysis (GSEA) was used to assess dysregulation of molecular pathways and association with previously reported molecular subclasses of HCC.36 The Ingenuity Pathway Analysis system (Ingenuity Systems; available: www.ingenuity.com) was used to analyze signal pathway networks and canonic pathways from microarray data. The Ingenuity Pathway Analysis generates interactions between genes and proteins based on findings reported in the literature. Differential gene expression analysis for the training and validation sets was performed using the Comparative Marker Selection module of the GenePattern analysis toolkit37 with a significance threshold of false discovery rate less than 0.05. Analysis of dysregulated genes in the cell line experiment was performed using Cyber-T software (http://cybert.microarray.ics.uci.edu/)38 with a significance threshold of posterior probability of differential expression greater than 0.90 and a P value of less than .01. In silico prediction analysis for miR-517a genes was performed using TargetScan (www.targetscan.org), MirBase Sequence Database (www.http://www.ebi.ac.uk), and the DIANA-MicroT algorithm version 3.0 (http://diana.cslab.ece.ntua.gr/microT).
Statistical Analysis
Comparisons between groups were performed using either the t test or the nonparametric Mann–Whitney test for continuous variables, and the Fisher exact test for comparison of proportions. The Pearson coefficient was used to assess correlations between variables. Survival data from the animal study were plotted with Kaplan–Meier curves and significance was calculated using the log-rank test. All analyses were performed using the SPSS package (SPSS 15.0, Chicago, IL) and R statistical package (www.r-project.org).
Results
iRNA-Based Molecular Classification of HCC
The expression levels of 358 human miRNAs were investigated in 89 HCV-related HCC samples (training set) collected from patients who underwent liver resection or liver transplantation in the 3 hospitals of the HCC Genomic Consortium: Mount Sinai School of Medicine (New York, NY), Istituto Nazionale dei Tumori (Milan, Italy), and Hospital Clinic (Barcelona, Spain). Unsupervised hierarchical clustering of miRNA data identified 3 main clusters of HCC, named clusters A (32 of 89; 36%), B (29 of 89; 32%), and C (28 of 89; 31%). The last cluster was divided further into 3 subclusters with clearly distinct miRNA expression patterns: C1 (15 of 89; 17%), C2 (8 of 89; 9%), and C3 (5 of 89; 6%) (Figure 1A). To define each subclass, we performed a comprehensive genomic analysis by integrating miRNA data with gene expression analysis, copy number changes, and assessment of cellular pathway activation by immunohistochemical and mutation analyses previously performed.27 Interestingly, each subclass was associated to distinct molecular alterations (Figure 1C). Cluster A was enriched in samples with β-catenin gene mutations (20 of 32; 62.5%; P < .001) and positive β-catenin immunostaining (16 of 32; 50%; P <.05), indicating aberrant activation of the β-catenin/Wnt canonical signaling pathway. GSEA was performed to find significant enrichments of gene signatures from databases of public domain. As expected, in cluster A we found enrichment of signatures previously associated with β-catenin mutations: CTNNB1 class by Chiang et al,27 G5-6 by Zucman-Rossi,39 S3 class by Hoshida et al,40 and a significant association with a gene signature predictive of good survival by Lee et al41 (false discovery rate [FDR] < 0.25) (Supplementary Table 4). Of note, low serum levels of α-fetoprotein were associated to tumors classified in cluster A (median serum level, 13 ng/mL; interquartile range, 6.25–23 ng/mL; P < .05). Cluster B was enriched in samples characterized by expression of interferon-response–related genes (P < .01).27 Interestingly, these tumors displayed a smaller size (median size, 2.8 cm in diameter) compared with the other HCCs (median size, 4.4 cm in diameter) (P < .01).
Figure 1.

miRNA-based molecular classification of HCC. (A) Heat map showing unsupervised hierarchical clustering of 89 HCV-related HCCs based on the expression levels of 358 human miRNAs. MiRNAs are in row, samples are in column. High and low expression levels are represented in red and blue, respectively. Molecular subclasses (A, B, C1, C2, C3) are indicated by colored bars. Up-regulation of C19MC family in subclass C2 is shown. Immunostaining for pEGFR, pAkt, pS6, pIGF1R, β-catenin localization, and the mutation status of TP53 and CTNNB1 genes are indicated in different colors (grey, positive; black, negative; white, missing value). Overlaps with transcriptomic-based molecular classes previously reported by our group are shown. Different colors correspond to distinct molecular classes. Chiang et al’s27 classification includes CTNNB1 class (blue), proliferation class (green), interferon-related class (pink), the Poly7 (polysomy of chromosome 7) class (orange), and unannotated samples (brown). Hoshida et al’s40 classification includes S1 class (blue), S2 class (green), and S3 class (yellow). (B) Summary of the proposed miRNA-based molecular classification of HCC. AFP, α-fetoprotein; VI, vascular invasion.
Cluster C was enriched by samples characterized by positive staining for the phosphorylated forms of IGF-1R (P < .01) and Akt (P < .05), indicating abnormal activation of IGF and mTOR/Akt/phosphatidylinositol (PI)3K pathways in these tumors. C2 tumors showed significant up-regulation (median fold change, 8.8; FDR < 0.001) of 23 miRNAs (miR-517a, miR-517b, miR-517c, miR-520g, miR-520h, miR-519b, miR-519d, miR-516-5p, miR-519a, miR-520c, miR-520b, miR-520f, miR-526b*, miR-524*, miR-516-1*, miR-526b, miR-519e, miR-512-3p, miR-522, miR-526a, miR-518f*, miR-518b, and miR-525) belonging to the largest miRNA cluster ever reported (~100 Kb), located on chr19q13.41 (also referred to as chromosome 19 miRNA cluster, C19MC) (Table 1).42 Among them, miR-517 was the most up-regulated miRNA (fold change, 23.78; FDR < 0.001). Similarly, overexpression of another cluster of miRNAs located in the imprinted DLK-DIO3 region on chromosome 14 (14q32.2-32.31)43 was observed in subclass C3 (median fold change, 6.3; FDR < 0.02) (Supplementary Table 5). GSEA analysis revealed enrichment of gene signatures associated to proliferative pathways (Zucman-Rossi’s G123,39 Chiang et al’s27 proliferation) and Lee et al’s41 poor survival in class C1 (FDR < 0.25). Interestingly, C2 tumors showed an enrichment of a Met-positivity signature (FDR < 0.1), which is associated to oxidative stress responses as well as cell motility, cytoskeletal organization, and angiogenesis.44 C3 tumors were associated to molecular signatures of aggressive tumors with poor prognosis including EpCAM-positivity,45 Hoshida et al’s40 S2, Lee et al’s41 poor survival, Zucman-Rossi’s G123,39 Chiang et al’s27 proliferation, and Kaposi-Novak et al’s44 Met signatures (FDR < 0.25). Molecular correlations showed that C3 tumors were characterized by p53 mutation (3 of 5; 60%; P < .05) and pAkt staining (4 of 5; 80%; P < .05). From a clinical perspective, C1 tumors were associated with vascular invasion (P < .05) and C2 HCCs with poor differentiation degree (P < .05). C3 subclass was characterized by high levels of α-fetoprotein (median serum levels, 22 × 103 ng/mL; interquartile range, 202–346 × 103 ng/mL; P < .001), presence of vascular invasion (P < .05), and large tumor size (median size, 7.5 cm; P < .05).
Table 1.
C19MC miRNAs Up-Regulated in Subclass C2
| Probe ID | miRNA | FDR | Fold change |
|---|---|---|---|
| JLA180 | miR-517a, b, c | 0.00 | 23.78 |
| JLA176 | miR-520g, h | 0.00 | 17.13 |
| JLA161 | miR-519b | 0.00 | 15.61 |
| JLA29 | miR-519d | 0.00 | 14.89 |
| JLA27 | miR-516-5p | 0.00 | 13.81 |
| JLA7 | miR-519a | 0.00 | 12.44 |
| JLA17 | mir-520c, b, f | 0.00 | 12.13 |
| JLA160 | miR-526b* | 0.00 | 10.98 |
| JLA170 | miR-524* | 0.00 | 8.79 |
| JLA71 | mir-516-1* | 0.00 | 7.47 |
| JLA159 | miR-526b | 0.00 | 6.93 |
| JLA154 | miR-519e | 0.00 | 6.51 |
| JLA150 | miR-512-3p | 0.00 | 6.13 |
| JLA2 | miR-522 | 0.00 | 5.56 |
| JLA164 | miR-526a,518f* | 0.00 | 4.51 |
| JLA28 | miR-518b | 0.00 | 4.32 |
| JLA162 | miR-525 | 0.00 | 3.80 |
Finally, we analyzed the expression of previously reported dysregulated miRNAs in our cohort of samples. Markedly, we found that miR-26a and miR-26b were down-regulated in C2 (FDR < 0.001) and C3 (FDR < 0.05) subclasses (Supplementary Figure 1A) and their low expression levels correlated with poor survival (Supplementary Figure 1B), in agreement with previous findings.22
Transcriptomic Characterization of C19MC-Expressing HCCs
For a more in-depth analysis we focused on subclass C2, which was characterized by high expression levels of C19MC miRNAs, a poorly characterized and primate-specific family of miRNAs. The Ingenuity Pathway Analysis software identified several canonical pathways significantly related to up-regulated genes including macropinocytosis, focal adhesion kinase signaling and nuclear factor-κB pathway among others (Supplementary Figure 2A), whereas few biological processes were related to down-regulated genes (Supplementary Figure 2B). A network-based analysis revealed a central regulating role of ERK/MEK signaling, connecting dysregulated genes in C2 subclass (Supplementary Figure 3).
Validation of C19MC miRNA Expression in an Independent Cohort of HCC
We selected 3 miRNAs representative of 3 subfamilies of the cluster (miR-517a, miR-520g, and miR-516-5p) as markers of C2 subclass, and analyzed their expression levels in an independent set of 165 formalin-fixed paraffin-embedded HCCs by quantitative real-time PCR. We observed that 22 HCCs (13%) had median fold changes greater than 2 of all 3 miRNAs compared with other HCCs (Figure 2A). Among them, 7 (4%) were characterized by very high expression (fold changes, >10) and were considered as C2 subclass. No significant association between C19MC miRNA expression and etiology was found. Gene expression patterns associated with the C2 (C19MC) classification was significantly similar between the training and validation sets, supporting the conclusion that the 3 miRNA-based classification of subclass C2 recapitulated the same biological entity of HCC tumors (Figure 2B) (P < .01).
Figure 2.
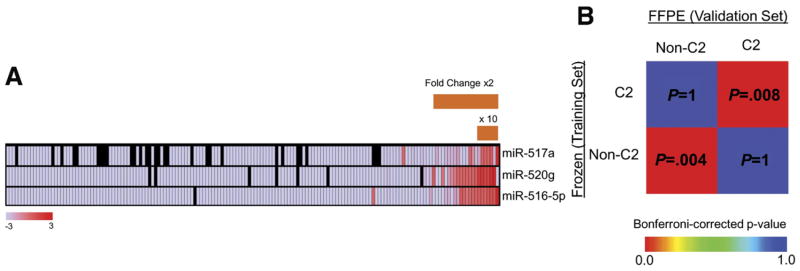
Validation of subclass C2 in an independent set of HCC. (A) Relative expression levels of miR-517a, miR-520g, and miR-516-5p were used as molecular surrogates of subclass C2 and evaluated in 165 formalin-fixed paraffin-embedded (FFPE) HCCs. High and low expression levels are indicated in gradient scale boxes. Black boxes, missing values. (B) Subclass association matrix for subclass C2 and other classes (non-C2) in the training (frozen tissues) and validation (FFPE sections) sets. Subclasses C2 in both HCC sets characterized by different tissue origin were significantly associated.
Mechanisms of C19CM miRNAs Overexpression
Next, we sought to determine which molecular mechanisms could be involved in the aberrant overexpression of C19MC miRNAs in subclass C2 of our training set. Only 1 of 8 samples (12.5%) showed genomic copy number gains of C19MC (mean copy numbers in 1 Mb surrounding 19q13.41 region, 2.97) (Figure 3A, sample 8). The presence of a CpG island located 15 kb upstream of the C19MC locus suggested that epigenetic mechanisms could be implicated in the regulation of 19q13.41 miRNAs expression (Figure 3B). To show the functional significance of the methylation status of this CpG island in the regulation of C19MC miRNAs we exposed the C19MC-negative Huh7 cells to the demethylating agent 5-aza-2′deoxycytidine and the histone deacetylase inhibitor 4-phenylbutyric acid, and evaluated miR-517a levels by quantitative real-time PCR. Interestingly, miR-517a expression was massively increased after treatment (fold change, 51; P < .001) (Figure 3C), along with demethylation of CG sites in the CpG island upstream of the cluster (Supplementary Figure 3). Consistent with this finding, we detected the presence of un-methylated sequences of the CpG island upstream of the C19MC cluster in 4 of 6 (67%) human samples belonging to subclass C2 (Figure 3D).
Figure 3.

Molecular mechanisms involved in C19MC miRNAs overexpression. (A) Inferred copy number of chr19 for each sample in subclass C2. C19MC locus is indicated by an arrow. One sample (indicated by a red box) showed copy gain in the q arm of chr19 (inferred copy numbers in the 1 Mb surrounding 19q13.41 region, 2.97). (B) Genomic localization of C19MC miRNAs on chr19. A CpG island is located 15 kb upstream of the cluster. (C) Expression level of miR-517a after treatment with 5-aza-2′deoxycytidine (5-AZA-CDR) and 4-phenylbutyric acid (PBA) in Huh7 cells compared with untreated cells. Results are expressed as fold changes ± standard error of the mean. (D) Methylation-specific PCR of the CpG island located upstream of the C19MC locus. Presence of PCR product indicates methylated (lane M) or unmethylated (lane U) alleles. Huh7 (DNA from Huh7 without bisulfite conversion), negative control; SsI-Huh7 (enzymatically methylated DNA), positive control. HCC: subclass C2 (n = 6 samples); normal, 1 representative normal liver.
Oncogenic Activity of miR-517a and miR-520c In Vitro
To test whether members of the C19MC family showed a functional role in HCC, we investigated the role of C19MC miRNAs in the Huh7 cells, which derive from a well-differentiated and nonmetastatic human HCC46 and SNU-449 cell line, which has been established by a single HCC with perinodular extensions. We focused on miR-517a and miR-520c, representing 2 different sub-families within the C19MC cluster. Both miRNAs were expressed at very low levels in Huh7 and SNU-449 cells (Supplementary Figure 5A).
Interestingly, enhanced expression of either miR-517a or miR-520c but not a control oligonucleotide promoted cell proliferation (P < .05) (Figure 4A) and conferred a potent migratory (P < .001) and invasive phenotype (P < .05) to Huh7 cells across Transwell chambers uncoated or coated with Matrigel, respectively (Figure 4B and C). In addition, expression of both miRNAs increased the migratory capability of Huh7 cells in a wound-healing assay (P < .002) (Figure 4D). Similar results were obtained using SNU-449 cells, confirming the oncogenic role of miR-517a and miR-520c (Supplementary Figure 5B–E).
Figure 4.

miR-517a and miR-520c promote cell proliferation, migration, and invasion in vitro. (A) Proliferation assay after transfection with control oligonucleotide, miR-517a, or miR-520c in Huh7 cells. Results are expressed as a percentage of 3H-thymidine incorporation (counts per million) ± standard error of the mean compared with control. (B) Transwell migration assay. Cells that migrated from the upper well of a Transwell chamber into the lower well were stained and counted. Magnification, ×100. (C) Cell invasion assays. Inserts coated with Matrigel were used to investigate the invasive potential of miR-517a and miR-520c transfected cells. Magnification, ×100. (D) Wound-healing assay. Cell monolayers were scratched and images were taken 0 and 24 hours after wounding. Results are expressed as distance (in μm) between the 2 edges ± standard error of the mean. Magnification, ×100.
Biological and Clinical Relevance of miR-517a Overexpression
For further analyses, miR-517a was selected because of the novelty and magnitude of its expression in HCC. To assess the biological relevance of miR-517a expression, we generated a stably miR-517a-transduced Huh7 cell line using a lentiviral vector system. Microarray analysis of miR-517a– expressing Huh7 cells was performed to interrogate the effects of miR-517 overexpression on the global molecular status of HCC cells. We found 433 up-regulated and 153 down-regulated genes in Huh7 cells transduced with miR-517a compared with control vector (P < .005) (Supplementary Tables 6 and 7). Ingenuity Pathway Analysis of microarray data revealed the presence of 2 main networks induced by miR-517a (Supplementary Figure 6), one related to the nuclear factor-κB complex and the other associated with the MAPK/ERK signaling. DNA replication and cell cycle represented the most relevant biological functions induced in miR-517a–transduced cells (FDR < 0.001) (Supplementary Table 8).
To identify potential target genes of miR-517a we performed an in silico prediction analysis using TargetScan, MirBase Sequence Database, and the DIANA-MicroT algorithm version 3.0. Among these genes only 6 were down-regulated (CACNA2D3, CTLA4, GPR20, OR4X1, SLC7A3, and TNIP1) and 3 were up-regulated (IFI35, MCTS1, and NRCAM) in miR-517a–transduced cells. Interestingly, GSEA showed enrichment of the 3 up-regulated genes in C2-tumors (FDR < 0.2).
From a clinical perspective, high levels of miR-517a were associated with the presence of satellite nodules around the main tumor both in the training and validation sets (P < .05).
miR-517a Promotes Tumorigenesis and Metastatic Dissemination In Vivo
Finally, we aimed to investigate whether miR-517a might drive tumorigenesis in vivo using a bioluminescence imaging system to allow sensitive detection of tumor growth in an orthotopic model of HCC. Importantly, we found that miR-517a strongly promoted the tumorigenic potential of Huh7 cells in vivo by inducing the development of aggressive liver tumors in 7 of 10 (70%) mice compared with control vector (1 of 11; 9%) (P < .001) (Figure 5A and B). As a consequence, mice injected with miR-517a– expressing cells showed reduced survival compared with control mice (P < .05) (Figure 5C). Interestingly, miR-517a promoted the formation of different size nodules, indicating intrahepatic dissemination from the primary site of injection (Figure 5D). Ex vivo analysis of dissected organs showed the presence of extrahepatic metastases to secondary organs (lungs and kidneys) in 4 mice injected with miR-517a–transduced cells (4 of 10; 40%) (Figure 6A). Histologic evaluation showed the presence of vascular invasion in liver sections collected from miR-517a tumors and confirmed the presence of multiple metastatic nodules in the lung and perirenal soft tissue (Figure 6B).
Figure 5.

miR-517a promotes tumorigenesis in vivo. (A) Luciferase-tagged Huh7 cells transduced with miR-517a or control vector were injected into the left lobe of the liver of nude mice. Representative images taken at day 1, and at 2, 4, 6, and 8 weeks are displayed. (B) Incidence of liver tumors in mice injected with Huh7 transduced with miR-517a or control vector. (C) Kaplan–Meier curves representing the decrease in survival of mice injected with miR-517a–expressing Huh7 compared with control-Huh7. (D) Multiple tumor nodules were observed in liver of mice administered with miR-517a–transduced Huh7.
Figure 6.
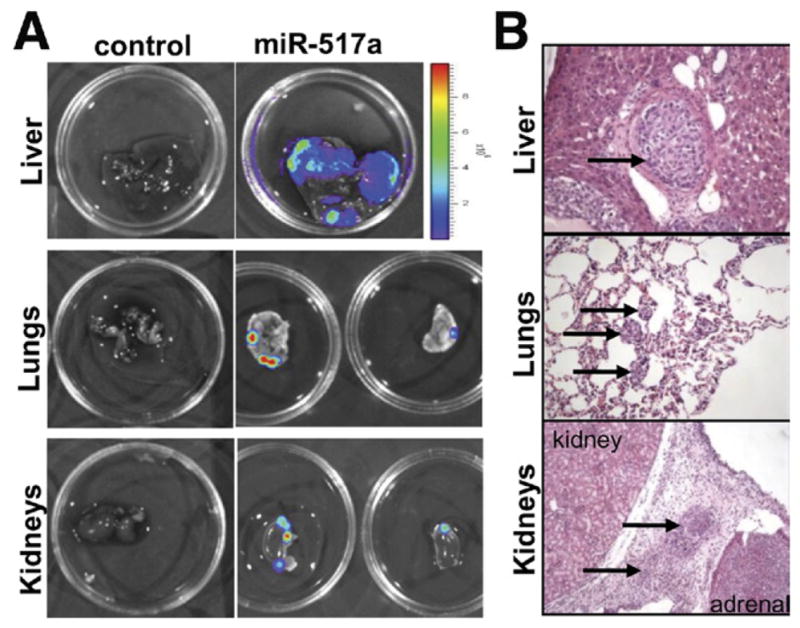
miR-517a induces metastatic dissemination of Huh7 cells in vivo. (A) Bioluminescent imaging of dissected organs showed the presence of large liver tumors and metastases in lungs and kidneys of mice injected with miR-517a– expressing cells. Representative images are shown. (B) Histologic analysis showed the presence of vascular invasion and metastases in distant organs in mice administered with miR-517a– expressing Huh7.
Discussion
This study provides compelling evidence that miR-517a, which belongs to the large C19MC miRNA cluster, acts as a novel oncomiR in HCC. Previous studies showed that C19MC miRNAs are expressed in undifferentiated tissues including placenta, human embryonic, and hematopoietic stem cells.47– 49 In particular, the expression of different members of the C19MC cluster (miR-520g, miR-518b, miR-518c, miR-512-3p, miR-524*) has been shown to be strongly down-regulated during differentiation of hematopoietic embryonic stem cells, indicating their role in stem cell maintenance.47 Recently, Li et al50 reported overexpression of C19MC miRNAs in a subgroup of aggressive brain tumors owing to genomic amplification of the chr19q13.41 locus. Furthermore, chromosomal rearrangements involving the 19q13.41 locus were reported in a subgroup of thyroid adenomas.51 In our study we observed that genomic amplification poorly contributed to the overexpression of C19MC miRNAs in HCC, whereas aberrant hypomethylation was more frequent. Therefore, our analysis confirms previous findings indicating that epigenetic changes could regulate the expression of C19MC miRNAs.52,53
Importantly, the study by Huang et al33 showed that overexpression of miR-520c was sufficient to promote invasion and metastasis of breast cancer cells, indicating that members of the C19MC family could be involved in cell motility. Consistent with this finding, our data indicate that enhanced expression of either miR-517a or miR-520c promotes a more malignant phenotype of HCC cells. To further confirm this finding, we tested the function of miR-517a in vivo using an orthotopic mouse model of HCC to more closely mimic HCC in human beings. The major utility of orthotopic cancer models are their ability to recapitulate the metastatic potential and patterns seen in human cancer. Previous studies have suggested that Huh7 cells do not induce the development of liver tumors in vivo,54 a finding that we also observed in our control animals. In contrast, however, overexpression of miR-517a in orthotopic Huh7 cells greatly promoted their tumorigenic potential and induced the development of large and invasive liver tumors. Although specific target genes for miR-517a still need to be discovered, our bioinformatic analysis revealed that miR-517a induced perturbations of the MAPK and nuclear factor-κB pathways, in line with the findings reported by Luo et al.55 Interestingly, macropinocytosis represented the most significant biological process associated to up-regulated genes in C2 tumors. The process of macropinocytosis contributes to both growth and motile processes of cells,56 providing further evidence linking miR-517a with cell proliferation and migration. Altogether, our results indicate that miR-517a is involved in HCC pathogenesis and likely represents a promising molecular target for new therapies. Although miR-517a–overexpressing HCCs represent only a small percentage of cases (approximately 9%–10% of cases), strategies targeting miR-517a could prove beneficial and optimal in terms of personalized management of patients.
Recent reports suggest that miRNA-based therapy for the treatment of cancer is feasible and potentially effective. MiR-26a and Let-7 have been identified as promising candidates because of their relevance in cancer pathogenesis.19,57 The continuous discovery of novel oncomiRs and tumor suppressor miRNAs represent an exciting and relevant area of progress. Chemically modified cholesterol-conjugated oligonucleotides, termed antagomirs, have been developed to inhibit the expression of oncogenic miRNAs, and their use currently is under investigation in preclinical studies.58 To date, preliminary animal studies have indicated that miRNA-based therapy is safe and nontoxic, although further evaluation is required before assessing its safety in human beings.59 In this context, our study provides a clear rationale for testing antago-mir-based approaches targeting miR-517a and other potential oncogenic members of the C19MC family in pre-clinical models of HCC. (Supplementary Figure 4).
Supplementary Material
Acknowledgments
The authors would like to thank Brian Brown and Jun Lu for their helpful discussions and Judit Peix for generous help.
Funding
Josep Llovet has received grants from the National Institutes of Health–NIDDK 1R01DK076986-01, FP7-2010-Health European Commission HEPTROMIC grant (proposal no: 259744-2), National Institute of Health (Spain) grant I+D Program (SAF-2007-61898), and the Samuel Waxman Cancer Research Foundation; Dr Llovet and the HCC Consortium are recipients of the AACR-Landon Innovator International award; Vincenzo Mazzaferro is partially sponsored by the Italian Association for Cancer Research and by a grant from the Italian National Ministry of Health; Scott Friedman has grants from the National Institutes of Health (1RO1DK37340 and 1RO1DK56621); Sara Toffanin is supported by a fellowship from the National Cancer Institute (Milan, Italy); Anja Lachenmayer is supported by a fellowship from the German Research Foundation; Augusto Villanueva is a recipient of a Sheila Sherlock fellowship (European Association for the Study of the Liver); and Jordi Bruix has a grant from the Instituto de Salud Carlos III (PI 08/0146).
Abbreviations used in this paper
- Akt
____________
- C19MC
chr19q13.41
- CpG
___________
- FDR
false discovery rate
- GSEA
Gene Set Enrichment Analysis
- IGF
___________________
- •••
microRNA cluster
- miRNA
microRNA
- PCR
polymerase chain reaction
Footnotes
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi: 10.1053/j.gastro.2011.02.009.
Sara Toffanin was responsible for the conception and design of the study, generation, collection, assembly, analysis, and/or interpretation of data, and drafting or revision of the manuscript; Yujin Hoshida, Anja Lachenmayer, Augusto Villanueva, Laia Cabellos, Beatriz Minguez, Radoslav Savic, Stephen Ward, Swan Thung, Derek Chiang, Clara Alsinet, Victoria Tovar, Sasan Roayaie, Myron Schwartz, Jordi Bruix, Samuel Waxman, Scott L. Friedman, Todd Golub, and Vincenzo Mazzaferro were responsible for generation, collection, assembly, analysis, and/or interpretation of data; and Josep Llovet was responsible for the conception and design of the study, generation, collection, assembly, analysis, and/ or interpretation of data, drafting or revision of the manuscript, and approval of the final version of the manuscript.
Transcript profiling: the dataset is available from NCBI’s Gene Expression Omnibus: accession number GSE20596.
Conflicts of interest
The authors disclose no conflicts.
References
- 1.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 3.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 4.Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 5.Lang L. FDA approves sorafenib for patients with inoperable liver cancer. Gastroenterology. 2008;134:379. doi: 10.1053/j.gastro.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 6.Villanueva A, Newell P, Chiang DY, et al. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis. 2007;27:55–76. doi: 10.1055/s-2006-960171. [DOI] [PubMed] [Google Scholar]
- 7.Zucman-Rossi J, Laurent-Puig P. Genetic diversity of hepatocellular carcinomas and its potential impact on targeted therapies. Pharmacogenomics. 2007;8:997–1003. doi: 10.2217/14622416.8.8.997. [DOI] [PubMed] [Google Scholar]
- 8.Hoshida Y, Toffanin S, Lachenmayer A, et al. Molecular classification and novel targets in hepatocellular carcinoma: recent advancements. Semin Liver Dis. 2010;30:35–51. doi: 10.1055/s-0030-1247131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Villanueva A, Chiang DY, Newell P, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135:1972–1983. doi: 10.1053/j.gastro.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newell P, Toffanin S, Villanueva A, et al. Ras pathway activation in hepatocellular carcinoma and anti-tumoral effect of combined sorafenib and rapamycin in vivo. J Hepatol. 2009;51:725–733. doi: 10.1016/j.jhep.2009.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tovar V, Alsinet C, Villanueva A, et al. IGF activation in a molecular subclass of hepatocellular carcinoma and pre-clinical efficacy of IGF-1R blockage. J Hepatol. 2010;52:550–559. doi: 10.1016/j.jhep.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 13.Lewis BP, Shih IH, Jones-Rhoades MW, et al. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 14.Calin GA, Sevignani C, Dumitru CD, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen CZ. MicroRNAs as oncogenes and tumor suppressors. N Engl J Med. 2005;353:1768–1771. doi: 10.1056/NEJMp058190. [DOI] [PubMed] [Google Scholar]
- 16.Meng F, Henson R, Wehbe-Janek H, et al. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647– 658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pineau P, Volinia S, McJunkin K, et al. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:264–269. doi: 10.1073/pnas.0907904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding J, Huang S, Wu S, et al. Gain of miR-151 on chromosome 8q24. 3 facilitates tumour cell migration and spreading through downregulating RhoGDIA. Nat Cell Biol. 2010;12:390–399. doi: 10.1038/ncb2039. [DOI] [PubMed] [Google Scholar]
- 19.Viswanathan SR, Powers JT, Einhorn W, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41:843– 848. doi: 10.1038/ng.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiong Y, Fang JH, Yun JP, et al. Effects of microRNA-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology. 2010;51:836– 845. doi: 10.1002/hep.23380. [DOI] [PubMed] [Google Scholar]
- 21.Coulouarn C, Factor VM, Andersen JB, et al. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene. 2009;28:3526–3536. doi: 10.1038/onc.2009.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ji J, Shi J, Budhu A, et al. MicroRNA expression, survival, and response to interferon in liver cancer. N Engl J Med. 2009;361:1437–1447. doi: 10.1056/NEJMoa0901282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834– 838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 24.Rosenfeld N, Aharonov R, Meiri E, et al. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol. 2008;26:462– 469. doi: 10.1038/nbt1392. [DOI] [PubMed] [Google Scholar]
- 25.Murakami Y, Yasuda T, Saigo K, et al. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene. 2006;25:2537–2545. doi: 10.1038/sj.onc.1209283. [DOI] [PubMed] [Google Scholar]
- 26.Ladeiro Y, Couchy G, Balabaud C, et al. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology. 2008;47:1955–1963. doi: 10.1002/hep.22256. [DOI] [PubMed] [Google Scholar]
- 27.Chiang DY, Villanueva A, Hoshida Y, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68:6779– 6788. doi: 10.1158/0008-5472.CAN-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoshida Y, Villanueva A, Kobayashi M, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359:1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mi S, Lu J, Sun M, et al. MicroRNA expression signatures accurately discriminate acute lymphoblastic leukemia from acute myeloid leukemia. Proc Natl Acad Sci U S A. 2007;104:19971–19976. doi: 10.1073/pnas.0709313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan JB, Yeakley JM, Bibikova M, et al. A versatile assay for high-throughput gene expression profiling on universal array matrices. Genome Res. 2004;14:878– 885. doi: 10.1101/gr.2167504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.April C, Klotzle B, Royce T, et al. Whole-genome gene expression profiling of formalin-fixed, paraffin-embedded tissue samples. PLoS One. 2009;4:e8162. doi: 10.1371/journal.pone.0008162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Llovet JM, Chen Y, Wurmbach E, et al. A molecular signature to discriminate dysplastic nodules from early hepatocellular carcinoma in HCV cirrhosis. Gastroenterology. 2006;131:1758–1767. doi: 10.1053/j.gastro.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 33.Huang Q, Gumireddy K, Schrier M, et al. The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat Cell Biol. 2008;10:202–210. doi: 10.1038/ncb1681. [DOI] [PubMed] [Google Scholar]
- 34.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682– 688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 35.Hoshida Y, Brunet JP, Tamayo P, et al. Subclass mapping: identifying common subtypes in independent disease data sets. PLoS ONE. 2007;2:e1195. doi: 10.1371/journal.pone.0001195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reich M, Liefeld T, Gould J, et al. GenePattern 2. 0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 38.Baldi P, Long AD. A Bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes. Bioinformatics. 2001;17:509–519. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- 39.Boyault S, Rickman DS, de Reynies A, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42–52. doi: 10.1002/hep.21467. [DOI] [PubMed] [Google Scholar]
- 40.Hoshida Y, Nijman SM, Kobayashi M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385–7392. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee JS, Chu IS, Heo J, et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40:667– 676. doi: 10.1002/hep.20375. [DOI] [PubMed] [Google Scholar]
- 42.Bentwich I, Avniel A, Karov Y, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;37:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- 43.Glazov EA, McWilliam S, Barris WC, et al. Origin, evolution, and biological role of miRNA cluster in DLK-DIO3 genomic region in placental mammals. Mol Biol Evol. 2008;25:939–948. doi: 10.1093/molbev/msn045. [DOI] [PubMed] [Google Scholar]
- 44.Kaposi-Novak P, Lee JS, Gomez-Quiroz L, et al. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest. 2006;116:1582–1595. doi: 10.1172/JCI27236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamashita T, Forgues M, Wang W, et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008;68:1451–1461. doi: 10.1158/0008-5472.CAN-07-6013. [DOI] [PubMed] [Google Scholar]
- 46.Nakabayashi H, Taketa K, Miyano K, et al. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 1982;42:3858–3863. [PubMed] [Google Scholar]
- 47.Bar M, Wyman SK, Fritz BR, et al. MicroRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem Cells. 2008;26:2496–2505. doi: 10.1634/stemcells.2008-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laurent LC, Chen J, Ulitsky I, et al. Comprehensive microRNA profiling reveals a unique human embryonic stem cell signature dominated by a single seed sequence. Stem Cells. 2008;26:1506–1516. doi: 10.1634/stemcells.2007-1081. [DOI] [PubMed] [Google Scholar]
- 49.Ren J, Jin P, Wang E, et al. MicroRNA and gene expression patterns in the differentiation of human embryonic stem cells. J Transl Med. 2009;7:20. doi: 10.1186/1479-5876-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li M, Lee KF, Lu Y, et al. Frequent amplification of a chr19q13. 41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell. 2009;16:533–546. doi: 10.1016/j.ccr.2009.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rippe V, Dittberner L, Lorenz VN, et al. The two stem cell MicroRNA gene clusters C19MC and miR-371-3 are activated by specific chromosomal rearrangements in a subgroup of thyroid adenomas. PLoS One. 2010;5:e9485. doi: 10.1371/journal.pone.0009485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsai KW, Kao HW, Chen HC, et al. Epigenetic control of the expression of a primate-specific microRNA cluster in human cancer cells. Epigenetics. 2009;4:587–592. doi: 10.4161/epi.4.8.10230. [DOI] [PubMed] [Google Scholar]
- 53.Zhang L, Volinia S, Bonome T, et al. Genomic and epigenetic alterations deregulate microRNA expression in human epithelial ovarian cancer. Proc Natl Acad Sci U S A. 2008;105:7004–7009. doi: 10.1073/pnas.0801615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Labonte P, Kadhim S, Bowlin T, et al. Inhibition of tumor growth with doxorubicin in a new orthotopically implanted human hepatocellular carcinoma model. Hepatol Res. 2000;18:72– 85. doi: 10.1016/s1386-6346(99)00087-x. [DOI] [PubMed] [Google Scholar]
- 55.Luo SS, Ishibashi O, Ishikawa G, et al. Human villous trophoblasts express and secrete placenta-specific microRNAs into maternal circulation via exosomes. Biol Reprod. 2009;81:717–729. doi: 10.1095/biolreprod.108.075481. [DOI] [PubMed] [Google Scholar]
- 56.Dharmawardhane S, Schurmann A, Sells MA, et al. Regulation of macropinocytosis by p21-activated kinase-1. Mol Biol Cell. 2000;11:3341–3352. doi: 10.1091/mbc.11.10.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kota J, Chivukula RR, O’Donnell KA, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Toffanin S, Villanueva A, Llovet JM. miRNA delivery: emerging therapy for hepatocellular carcinoma. Gastroenterology. 2010;138:1202–1204. doi: 10.1053/j.gastro.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 59.Wang V, Wu W. MicroRNA-based therapeutics for cancer. BioDrugs. 2009;23:15–23. doi: 10.2165/00063030-200923010-00002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.